Abstract
Cyclooxygenase-2 (COX-2) is implicated in the development of chronic inflammatory diseases. Recently, pyridazine derivatives have emerged as a novel prototype to develop COX-2 inhibitors. Accordingly, some pyridazine-based COX-2 inhibitors are reported herein. The reaction of aldehyde 3 and different hydrazines yielded the corresponding hydrazones. The hydrazones were further derivatized to the title compounds, which were assessed for COX-1 and COX-2 inhibitory action, gastric ulcerogenic effects, and lipid peroxidation properties. Molecular docking studies and determination of the physicochemical parameters were also carried out. The allocated structures of the reported compounds were coherent with their spectroscopic data. The compounds 9a (IC50 = 15.50 nM, 114.77%), 9b (IC50 = 17.50 nM, 101.65%), 12 (IC50 = 17.10 nM, 104.03%), 16b (IC50 = 16.90 nM, 105.26%), and 17 (IC50 = 17.70 nM, 100.5%) displayed better COX-2 inhibition than celecoxib (IC50 = 17.79 nM, 100%). These outcomes were harmonious with the molecular docking studies of 9a, 9b, 12, 16b, and 17. These compounds also displayed comparable onset and the duration of action concerning celecoxib and indomethacin in the in vivo studies. No ulcerogenic effects were observed for 9a and 12, whereas 9b, 16b, and 17 showed an insignificant ulcerogenic effect compared to celecoxib. The compounds 9a, 9b, 12, 16b, and 17 displayed a better lipid peroxidation profile than celecoxib and indomethacin. The compounds 9a (%ABS = 84.09), 9b (%ABS = 84.09), 12 (%ABS = 66.87), 16b (%ABS = 75.02), and 17 (%ABS = 81.42) also displayed appreciable calculated absorption compared to celecoxib (%ABS = 82.09). The compounds 9a, 9b, 11, 16b, and 17 have been recognized and postulated as non-ulcerogenic COX-2 inhibitors with promising physicochemical parameters and gastric safety profile. These compounds may be useful candidates to combat diseases caused by higher levels of COX-2.
1. Introduction
Inflammation is a self-protective reaction of human body tissues towards injurious stimuli like infection, irritants, poisonous substances, irradiation, and tissue injury [1]. The inflammation can be divided into acute inflammation and chronic inflammation. The symptoms of acute inflammation comprise of swelling, heat, immobility, redness, and pain, which may last for several days [2]. However, persistent inflammation for a longer time or chronic inflammation may lead to the development of diseases like gout, ankylosing spondylitis, osteoarthritis, rheumatoid arthritis, Alzheimer’s disease, ulcerative colitis, depression, epilepsy, irritable bowel diseases, kidney injury, cancer, asthma, hepatitis, pancreatitis, and atherosclerosis [1,3]. According to the published data, 40% and 61% of the population of the age > 60 years in the UK and Saudi Arabia, respectively, are suffering from arthritis [4]. It is estimated that about 1/4th of the adults in the USA will be affected by osteoarthritis by 2030 [5]. Non-steroidal anti-inflammatory drugs (NSAIDs) are regularly prescribed for chronic inflammatory diseases. NSAIDs inhibit the cyclooxygenase enzyme (COX). COX is accountable for transforming arachidonic acid into prostaglandins, which initiate the inflammatory events in a cell [1,2]. The constitutive COX-1 is responsible for the maintenance functions of the cell, including the safety of the gastric mucosa, aggregation of platelets, and control of the renal blood flow [6,7]. COX-2 cannot be detected in healthy cells. However, this inducible enzyme is produced intracellularly after harmful stimuli. It is responsible for the development of inflammatory events in a cell, which ultimately lead to the development of inflammatory diseases [1,3]. The commonly used NSAIDs cause ulcerogenic effects after prolonged use because they inhibit both COX-2 and COX-1 [6,7]. The ulcerogenic effect of NSAIDs is credited to the inhibition of COX-1, and the anti-inflammatory action is credited to the inhibition of COX-2 [8,9]. Based on this understanding, celecoxib, rofecoxib, and etoricoxib were developed as specific COX-2 inhibitors [10,11,12]. However, cerebrovascular risk and cardiac toxicity have been reported as adverse effects of some particular COX-2 inhibitors at the standard dose, for example, rofecoxib [12,13]. Accordingly, medicinal chemists are looking forward to developing new anti-inflammatory agents, which lack the aforementioned adverse effects and have a promising gastric safety profile [14].
Pyridazine, a famous diazine ring, is part of many pharmacodynamic agents like indolidan (antihypertensive), bemoradan (antihypertensive), levosimendan (congestive heart failure), pimobendan (congestive heart failure), milrinone (cardiotonic), minaprine (antidepressant), imazodan (PDE3 inhibitor), zardaverine (PDE3 inhibitor), and olaparib (anticancer) [15,16,17,18,19]. Emorfazone (Pentoil), an analgesic and anti-inflammatory pyridazine derivative, is in clinical use in Japan and is claimed to lack gastric side effects [19]. Similarly, Zomipirac has been derivatized to a pyridazine derivative, which had a COX-1/COX-2 selectivity index > 1500 [20,21]. Novel targets for developing anti-inflammatory agents bearing the pyridazine scaffold along with the general chemistry, mechanism of action, and the structure–activity relationship (SAR) have also been reported [19]. Ulcerogenic effects or gastrointestinal bleeding are the most frequently encountered adverse effects of the clinically used NSAIDs [13]. As per the recent publications, the pyridazine moiety is a new template for developing COX-2 inhibitors [15,16,17,18,19]. The possibility of the various structural modifications in the pyridazine ring, and the literature revealing the COX-2 inhibitory potential of the pyridazine nucleus [15,16,17,18,19,20,21] makes it a promising scaffold to develop non-ulcerogenic COX-2 inhibitors. As per the published reports, incorporation of the thiazole moiety and the 4-thiazolidinone moiety in a compound imparts selectivity towards COX-2 inhibition and also improves its gastric safety profile [22,23,24,25,26,27,28]. Accordingly, as an extension of our work to develop superior anti-inflammatory compounds [29,30,31,32], we report herein novel pyridazine-based thiazole and 4-thiazolidinone derivatives as cyclooxygenase-2 inhibitors with an improved gastric safety profile.
2. Results and Discussion
2.1. Chemistry
The intermediates 2, 3, 4b, and 4c were reported in our previous publication [30]. The intermediates 1, 5a, 5b, and 13 are available commercially. The condensation of aldehyde 3 with hydrazine hydrate, thiosemicarbazide, and phenylthiosemicarbazide afforded hydrazone 4a and the thiosemicarbazone derivatives 4b,c, respectively (Scheme 1).
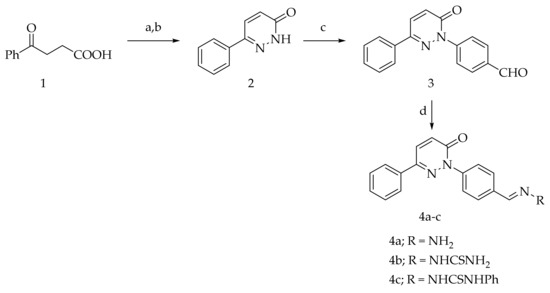
Scheme 1.
Preparation of 4a–c (Reagents and conditions: (a) NH2NH2·H2O, EtOH, reflux; (b) Br2/AcOH, (c) 4-fluorobenzaldehyde, DMSO/K2CO3, reflux; (d) RNH2, EtOH, few drops AcOH, reflux).
The cycloalkylation of the thiocarbamoyl group of 4b with the ethyl α-chloroacetate (5a) and ethyl α-chloropropionate (5b) in glacial acetic acid comprising a catalytic amount of the fused sodium acetate at the reflux temperature afforded the corresponding 4-thiazolidinone derivatives, 6a and 6b, respectively (Scheme 2). The formation of 6a and 6b is expected to proceed through the initial S-alkylation via the loss of sodium chloride followed by the intramolecular cyclization with the elimination of ethanol. The Hantzsch reaction of 4b with the ethyl α-chloro acetoacetate in acetic acid in the presence of sodium acetate led to the formation of 4-methyl-thiazole derivative 8 (Scheme 2). The possibility of compound 7 was excluded based on the spectral analyses. The treatment of 4b with 4-substituted phenacyl bromides in refluxing ethanol in the presence of anhydrous sodium acetate produced the corresponding thiazole derivatives, 9a and 9b (Scheme 2). The cyclization reaction of 4b with dimethyl acetylenedicarboxylate in methanol provided the 4-thiazolidinone derivative 10 (Scheme 2).
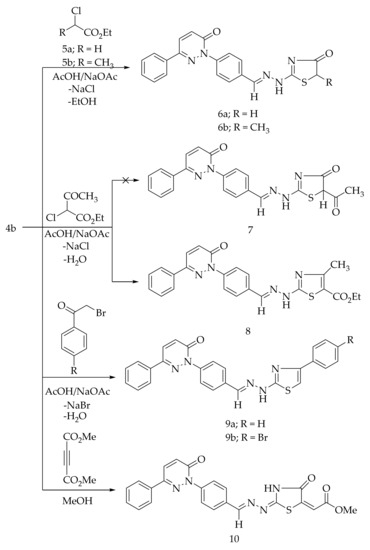
Scheme 2.
Preparation of 6a,b, 8, 9a,b, and 10.
The condensation of the active methylene group of 6a with electrophilic dimethylformamide-dimethylacetal (DMF-DMA) in dry dioxane afforded 5-dimethylaminomethylidine derivative 11 (Scheme 3). Similarly, the treatment of 6a with isatin in dioxane comprising a catalytic amount of piperidine provided compound 12 (Scheme 3). The treatment of 6a with α-cinnamonitriles in dioxane containing a catalytic amount of piperidine furnished the benzylidene derivatives 16a,b, wherein other possible structures 15a,b were ruled out based on the spectral data. Another synthetic route of compounds 16a,b was achieved via the Knoevenagel condensation of compounds 6a with the corresponding aromatic aldehydes (Scheme 3).
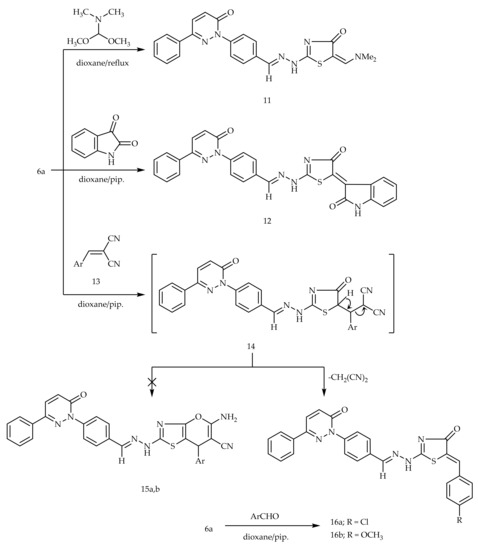
Scheme 3.
Preparation of 11, 12, and 16a,b.
The cyclocondensation of the thiocarbamoyl group of 4c with the ethyl α-chloroacetate in acetic acid containing a catalytic amount of the anhydrous sodium acetate provided 4-thiazolidinone derivative 17 (Scheme 4).
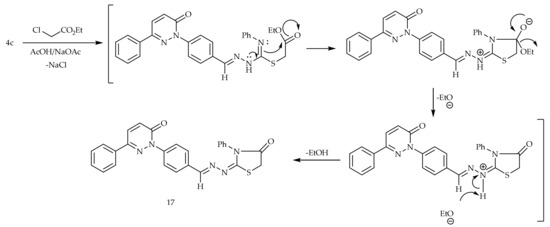
Scheme 4.
Preparation of 17.
Finally, the reaction of 4c with dimethyl acetylenedicarboxylate in methanol at the reflux temperature afforded the compound 18 (Scheme 5). The formation of 18 is expected to proceed via the nucleophilic addition of S-atom to the sp-carbon of the dimethyl acetylenedicarboxylate to generate the intermediate A. The intermediate A after the intramolecular cyclization through the nucleophilic amino group of the thiosemicarbazone yielded 18.
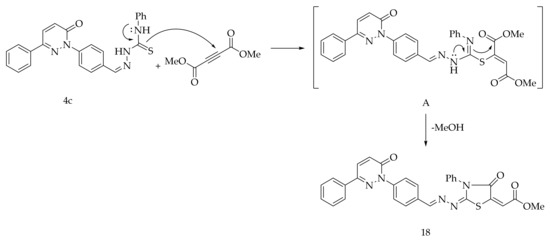
Scheme 5.
Preparation of 18.
There is a possibility of at least four or more geometrical isomers for some of the compounds, for example, 10, 11, 12, 16a, 16b, 17, and 18. However, these isomers may be able to interconvert. Therefore, we have not mentioned the E or Z configuration in the nomenclature of our compounds. The structures of 4a, 6a, 6b, 8, 9a, 9b, 10, 11, 12, 16a, 16b, 17, and 18 were proven on the basis of their spectroscopical data. The detailed spectroscopical data are provided in the experimental part.
2.2. Biological Activity
2.2.1. In Vitro COX Inhibitory Action
The compounds 4a, 6a, 6b, 8, 9a, 9b, 10, 11, 12, 16a, 16b, 17, and 18 were examined as COX-1 and COX-2 inhibitors along with indomethacin and celecoxib. It was performed by the 10-fold dilution technique utilizing test packs of the human COX-1/COX-2 (Cayman Chemicals, 560131, Ann Arbor, MI, USA) [30]. Indomethacin and celecoxib were used as standard drugs. Indomethacin is an ulcerogenic non-specific COX-1 and COX-2 inhibitor, whereas celecoxib is considered as a non-ulcerogenic-specific COX-2 inhibitor [6,10].
It is a well known fact that the inhibition of COX-1 is mainly responsible for the ulcerogenic effect of NSAIDs like indomethacin [6,7]. It is also well documented that specific COX-2 inhibitors like celecoxib are potent anti-inflammatory agents and possess a better gastric safety profile because they do not inhibit COX-1 [10]. Therefore, for a better comparison, the %COX-1 inhibition of indomethacin was normalized to 100% for COX-1, and the %COX-2 inhibition of celecoxib was normalized to 100% for COX-2 (Table 1). The selectivity index of celecoxib was also normalized to 100%. All the compounds comprising celecoxib (IC50 = 320 nM) displayed greater IC50 against COX-1, when compared to indomethacin (IC50 = 220 nM) (Table 1, Figure 1). This result points out that our compounds should have a better gastric safety profile than indomethacin [8,9]. Our belief is further strengthened by the fact that our compounds showed better inhibition of COX-2 in contrast to COX-1. The compounds 9a (IC50 = 15.50 nM, 114.77%), 9b (IC50 = 17.50 nM, 101.65%), 12 (IC50 = 17.10 nM, 104.03%), 16b (IC50 = 16.90 nM, 105.26%), and 17 (IC50 = 17.70 nM, 100.5%) demonstrated better COX-2 inhibition than celecoxib (IC50 = 17.79, 100%). The selectivity index (SI) of 9a (SI = 21.29, 118.40%) and 16b (SI = 18.63, 103.61%) was superior to celecoxib (SI = 17.98, 100%). The SI of 9b (SI = 15.71, 87.37%), 12 (SI = 17.25, 95.93%), and 17 (SI = 16.10, 89.54%) was also comparable to celecoxib (SI = 17.98, 100%). Based on the data mentioned above, 9a, 9b, 12, 16b, and 17 were chosen for the in vivo anti-inflammatory activity.

Table 1.
In vitro cyclooxygenase inhibitory effect (N = 3, Mean ± SD) of 4a, 6a, 6b, 8, 9a, 9b, 10, 11, 12, 16a, 16b, 17, and 18.
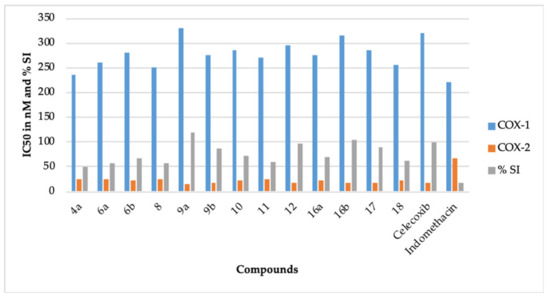
Figure 1.
The IC50 (nM) and the % selectivity index (SI) of compounds 4a, 6a, 6b, 8, 9a, 9b, 10, 11, 12, 16a, 16b, 17, 18, celecoxib, and indomethacin.
The novel pyridazine derivatives can be categorized as thiazole derivatives (8, 9a, and 9b) and 4-thiazolidinone derivatives (6a, 6b, 10, 11, 12, 16a, 16b, 17, and 18). It is apparent from Table 1 that the thiazole derivative 9a (4-phenyl thiazole group) was more potent than thiazole derivative 9b (4-bromophenyl thiazole group). This reflects that the incorporation of bromine in the structure of 9a decreases its COX-2 inhibitory potential. This result is in concurrence with the earlier report [22]. However, the COX-2 inhibitory potential of the corresponding chrolo, fluoro, iodo, and nitro derivatives of 9b should also be assessed for a better understanding of this observation. The thiazole derivative 8 (4-methyl-thiazole-5-carboxylate group) displayed a further decrease in the COX-2 inhibitory potential. This also indicates that the incorporation of methyl and carboxylate groups in the structure of 9a decreases its COX-2 inhibitory activity.
In case of 4-thiazolidinone derivatives, the COX-2 inhibitory activity increases as 11 < 6a < 18 < 6b < 16a < 10 < celecoxib < 17 < 12 < 16b. The presence of the 5-dimethylaminomethylidene group (11), unsubstituted 4-thiazolidinone ring (6a), 5-methoxycarbonylmethylidene-3-phenyl group (18), methyl group (6b), 4-chlorophenylmethylidene group (16a), and 5-methoxycarbonylmethylidene group (10) at position 5 of the 4-thiazolidinone ring provides compounds with lesser or average COX-2 inhibitory action. However, when the chlorine of the 16a is replaced with a methoxy group, a potent COX-2 inhibitor 16b is obtained. This observation indicates that the presence of the electron donor group in these types of compounds increases the COX-2 inhibitory action. This reflection is also in concurrence with the earlier reports that the presence of the electron donor group may increase the COX-2 inhibitory action of thiazolidinone ring-bearing compounds [22,24]. The 2,3-disubstituted-4-thiazolidinone derivative (17) has been recognized as a potent inhibitor of COX-2. Some earlier reports also support this fact [26,28]. We also believe that this kind of other 2,3-disubstituted derivatives may provide potent COX-2 inhibitors. The isatin-bearing compound 12 also provided a potent inhibitor of COX-2. The incorporation of the isatin moiety is reported to potentiate the COX-2 inhibitory activity of a compound [33]. Recently, we reported the isomeric 4-thiazolidinone-bearing pyridazine derivatives, which had a methylidine linker joining the phenyl ring and the 4-thiazolidinone ring [30]. The presently reported 4-thiazolidinone-bearing pyridazine derivatives contain a methylidene hydrazinyl linker between the phenyl ring and the 4-thiazolidinone ring. A comparison of the COX-2 inhibitory activity among these isomeric compounds reveals that the incorporation of a methylidene hydrazinyl linker provides potent COX-2 inhibitors. This observation is in concurrence with our earlier report that states that compounds bearing a hydrazine moiety display higher COX-2 inhibition [30].
2.2.2. In Vivo Anti-Inflammatory Activity
The compounds 9a, 9b, 12, 16b, and 17 were chosen for the in vivo anti-inflammatory action because they displayed better COX-2 inhibition and lesser COX-1 inhibition. It was performed by the carrageenan-induced rat paw edema method [30,34]. A total of eight groups of rats were utilized, wherein each group comprised of six rats. The compounds were administered orally (10 mg/kg) (Table 2, Figure 2).
Table 2.
In vivo anti-inflammatory activity of 9a, 9b, 12, 16b, and 17.
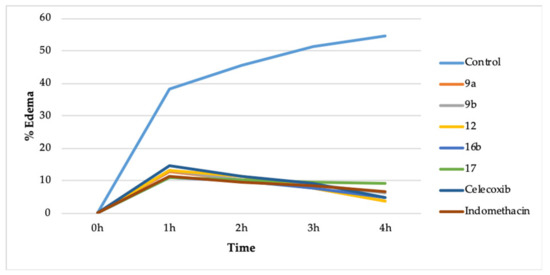
Figure 2.
In vivo action of 9a, 9b, 12, 16b, 17, celecoxib, and indomethacin.
The compounds 9a (3.76%), 12 (3.70%), and 16b (4.85%) reduced the % edema more than celecoxib (4.92%) and indomethacin (6.60%) after 4 h of drug administration. The compound 9b (6.39%) reduced the % edema more than indomethacin (6.60%) but less than celecoxib (4.92%) after 4 h of drug administration. However, compound 17 (9.16%) reduced the % edema less than indomethacin (6.60%) and celecoxib (4.92%). The compounds 9a, 9b, 12, 16b, and 17 also have a comparable onset of action plus duration of action concerning celecoxib and indomethacin (Table 2, Figure 2).
Accordingly, it was determined that the novel pyridazine-based thiazole and 4-thiazolidinone derivatives are active lead compounds to develop future COX-2 inhibitors [22,24,26,28].
2.2.3. Ulcerogenic Activity
The compromising gastric safety profile of the existing NSAIDs is a concern [6,7]. The ulcerogenic effects are more pronounced in non-specific COX inhibitors like indomethacin, whereas specific COX-2 inhibitors like celecoxib lack this side effect [6,10]. Accordingly, 9a, 9b, 12, 16b, and 17 were evaluated for their gastric safety profile. It was performed by the indomethacin-induced gastric erosion method [30]. A total of eight groups of rats were utilized, wherein each group comprised of six rats. The compounds were administered orally (10 mg/kg) (Table 3, Figure 3).
Table 3.
The gastric safety profile of 9a, 9b, 12, 16b, and 17.
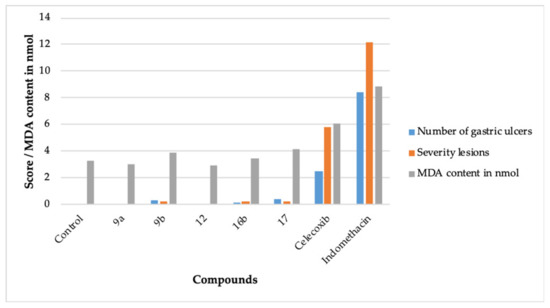
Figure 3.
The ulcerogenic and lipid peroxidation activity effect of 9a, 9b, 11, 15b, 17, celecoxib, and indomethacin.
The compounds 9a and 12 did not produce any ulcerogenic impact. The compounds 9b, 16b, and 17 also exhibited insignificant ulcerogenic effects. The negligible ulcerogenic effect produced by 9a, 9b, 12, 16b, and 17 is attributed to their higher COX-2 inhibitory potential plus their lesser potential to inhibit COX-1 (Table 1) [8,9]. It is well established that the presence of the -COOH group in NSAIDs aids in their ulcerogenic effect [6]. Another prospect of the insignificant ulcerogenic effect produced by 9a, 9b, 12, 16b, and 17 may be the absence of the -COOH moiety in their structure.
2.2.4. Lipid Peroxidation Studies
The lipid peroxidation inhibitory activity of a compound makes it less ulcerogenic [35,36]. The lipid peroxidation in tissue increases the malondialdehyde (MDA) content in the tissue [35]. A decrease in the MDA content is a measure to assess the non-ulcerogenic effect of a compound. Accordingly, the compounds 9a, 9b, 12, 16b, and 17 were evaluated for their lipid peroxidation effects [35,36] (Table 3, Figure 3). The compounds 9a, 9b, 12, 16b, and 17 displayed a better lipid peroxidation profile than celecoxib and indomethacin, which provides concurrence to the ulcerogenic activity data (Table 3).
2.3. Molecular Modelling
Molecular docking studies of 9a, 9b, 12, 16b, and 17 were carried out to validate the results of their biological activities [30,37,38,39] (Table 4).
Table 4.
Molecular docking data of 9a, 9b, 12, 16b, and 17.
The molecular docking data of celecoxib was described in our earlier publication [30]. The docking score (DS in kcal/mol) of compounds 9a (DS = −10.8), 9b (DS = −10.2), 12 (DS = −10.5), 16b (DS = −10.6), and 17 (DS = −9.5) was superior to celecoxib (DS = −9.4) (Table 4). The superior docking scores of 9a, 9b, 12, 16b, and 17 support the in vitro COX-2 inhibitory analysis results of 9a (IC50 = 15.50 nM, 114.77%), 9b (IC50 = 17.50 nM, 101.65%), 12 (IC50 = 17.10 nM, 104.03%), 16b (IC50 = 16.90 nM, 105.26%), and 17 (IC50 = 17.70 nM, 100.5%), and celecoxib (IC50 = 17.79 nM, 100%). Table 4 also mentions the hydrogen bonding pattern of celecoxib, 9a, 9b, 12, 16b, and 17 with the COX-2 protein. We also consider that the superior COX-2 inhibitory action of celecoxib, 9a, 9b, 12, 16b, and 17 is attributed to their hydrogen bonding interactions [30].
2.4. Physicochemical Parameters
The molecular properties of 9a, 9b, 12, 16b, and 17 were determined by the available online software, Molinspiration [40]. The calculated absorption (%ABS), number of H-bond acceptors (nON), number of H-bond donors (nOHNH), number of rotatable bonds (nrotb), and the topological polar surface area (tPSA) are provided in Table 5.
Table 5.
The physicochemical parameters of 9a, 9b, 12, 16b, 17, celecoxib, and indomethacin.
The compounds 9a, 12, 16b, and 17 passed Lipinski’s rule [40,41]. This rule defines the molecular properties of a drug, which governs its pharmacokinetic performance, for example, the absorption of a drug. It also helps to evaluate a compound’s ability to be orally active based on its molecular properties [40,41], which are mentioned in Table 5. To pass Lipinski’s rule, a compound should not have more than one violation of the calculated properties. As per a recent report, Lipinski’s rule of five does not have any significant deficiency in defining the druggability of a compound and is still useful today [42]. It can be observed that compound 9a displayed a better calculated absorption (%ABS = 84.09) than celecoxib (%ABS = 82.09) (Figure 4). The compound 9b (%ABS = 84.09) also displayed a better calculated absorption than celecoxib. The compound 9b did not pass Lipinski’s rule. However, there are many clinically used natural compounds and drugs that violate Lipinski’s rule [42]. The compound 12 (%ABS = 66.87), compound 16b (%ABS = 75.02), and compound 17 (%ABS = 81.42) also exhibited appreciable calculated absorption. These observations revealed that the compounds 9a, 9b, 12, 16b, and 17 possess not only better COX-2 inhibitory action than indomethacin and celecoxib but also possess a promising pharmacokinetic/physicochemical profile.
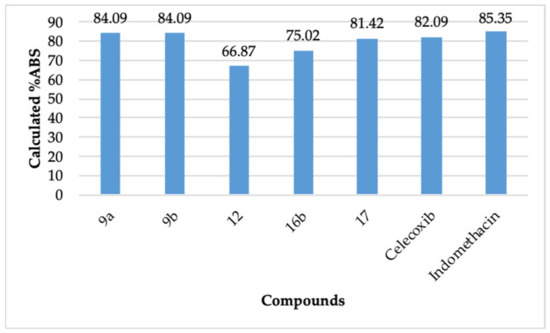
Figure 4.
Calculated degree of absorption (%ABS) of 9a, 9b, 12, 16b, 17, celecoxib, and indomethacin.
3. Materials and Methods
The uncorrected melting points, FTIR spectra (KBr), NMR spectra (1H-NMR & 13C-NMR), mass spectra, and elemental analyses were obtained by Gallenkamp apparatus (MFB-595- 010M; Weiss Gallenkamp, London, UK), Shimadzu 440 spectrometer (Shimadzu, Tokyo, Japan), Varian Gemini 500 MHz spectrometer (Palo Alto, CA, USA), GCMS/QP 1000 Ex mass spectrometer (70 eV) (Shimadzu, Tokyo, Japan), and the VARIO El Elementer apparatus (Langenselbold, Germany), respectively. The melting points are mentioned in °C, IR signals are mentioned as (KBr, ν in cm−1), 1H-NMR signals are mentioned as (500 MHz, DMSO-d6; δ in ppm), 13C-NMR signals are mentioned as (125 MHz, DMSO-d6, δ in ppm), and the mass peaks (MS) are represented as (m/z). The intermediates 2, 3, 4b, and 4c were reported in our previous publications [30,43]. SciFinder was used to ascertain the novelty of 4a, 6a, 6b, 8, 9a, 9b, 10, 11, 12, 16a, 16b, 17, and 18. The preparation methods of 4a, 6a, 6b, 8, 9a, 9b, 10, 11,12, 16a, 16b, 17, and 18 are depicted in Scheme 1, Scheme 2, Scheme 3, Scheme 4 and Scheme 5.
3.1. Chemistry
General preparation of 4a–c
A mixture comprising 3 (3.62 mmoles), hydrazine hydrate or thiosemicarbazide or phenylthiosemicarbazide (3.62 mmoles), AcOH (5 mL), and EtOH (50 mL) was refluxed at 80 °C for 3–5 h. The separated precipitate was filtered and recrystallized by MeOH. The compounds 4b and 4c were reported in our previous publications [30].
2-(4-(Hydrazineylidenemethyl)phenyl)-6-phenylpyridazin-3(2H)-one (4a): Yield (75%); M.P.: 235–236 °C; IR: 3371 & 3216 (NH2), 3063, 1662 (C=O); 1H-NMR: 6.98 (s, 2H, NH2), 7.14 (d, 1H, Ar-H), 7.42 (q, 3H, Ar-H), 7.61 (dd, 4H, Ar-H), 7.79 (s, 1H, azomethine-H), 7.88 (d, 2H, Ar-H), 8.05 (d, 1H, Ar-H); 13C-NMR: 159.06 (C=O), 144.59, 140.95, 137.56, 136.59, 134.64, 131.45, 131.27, 129.99, 129.37 (2C), 128.45 (2C), 126.46 (2C), 126.08 (2C), 125.52 (2C); Mass: 290 (M+, 100% base peak); Anal. Calcd. for C17H14N4O: C, 70.33; H, 4.86; N, 19.30. Found: C, 70.22; H, 4.70; N, 19.24.
General synthesis of 6a, 6b, 7, 9a, and 9b
A mixture comprising 4b (10 mmoles), ethyl 2-chloroacetate (10 mmoles) or ethyl 2-chloropropanoate (10 mmoles) or ethyl 2-chloro-3-oxobutanoate (10 mmoles) or 2-bromo-1-phenylethan-1-one (10 mmoles) or 2-bromo-1-(4-bromophenyl)ethan-1-one (10 mmoles) and CH3COONa (20 mmoles) in 40 mL of AcOH was heated to 125 °C for 4–6 h. The precipitate was collected and recrystallized by dioxane.
2-(2-(4-(6-Oxo-3-phenylpyridazin-1(6H)-yl)benzylidene)hydrazineyl)thiazol-4(5H)-one (6a): Yield (65%); M.P.: 250–252 °C; IR: 3150 (NH), 2942, 2850, 1720 & 1665 (C=O), 1584; 1H-NMR: 3.92 (s, 2H, -S-CH2-), 7.20 (d, 2H, Ar-H), 7.48 (q, 3H, Ar-H), 7.80 (d, 2H, Ar-H), 7.89 (d, 2H, Ar-H), 8.14 (d, 2H, Ar-H), 8.48 (s, 1H, azomethine-H), 12.12 (s, 1H, NH); 13C-NMR: 170.43 (C=O), 159.06 (C=O), 155.87, 154.58, 144.88, 143.59, 134.57, 134.15, 131.64, 130.14 (2C), 129.43 (2C), 128.25 (2C), 126.56 (2C), 126.34 (2C), 33.54 (S-CH2-); Mass: 389 (M+), 274 (100% base peak); Anal. Calcd. for C20H15N5O2S: C, 61.68; H, 3.88; N, 17.98. Found: C, 61.47; H, 3.76; N, 17.89.
5-Methyl-2-(2-(4-(6-oxo-3-phenylpyridazin-1(6H)-yl)benzylidene)hydrazineyl)thiazol-4(5H)-one (6b): Yield (67%): M.P.: 285–286 °C; IR: 3115 (NH), 3059, 1714 & 1668 (C=O), 1595; 1H-NMR: 1.57 (d, 3H, CH3), 4.22 (q, 1H, CH), 7.22 (d, 1H, Ar-H), 7.46 (q, 3H, Ar-H), 7.80 (d, 2H, Ar-H), 7.94 (dd, 4H, Ar-H), 8.14 (d, 1H, Ar-H), 8.48 (s, 1H, azomethine-H), 12.02 (s, 1H, NH); 13C-NMR: 174.21 (C=O), 159.06 (C=O), 155.91, 153.47, 144.89, 143.61, 134.58, 134.15, 131.65, 130.14 (2C), 129.43 (2C), 128.26 (2C), 126.57 (2C), 126.35 (2C), 42.71 (-S-CH2-), 19.20 (-CH3); Mass: 403 (M+), 274 (100% base peak); Anal. Calcd. for C21H17N5O2S: C, 62.52; H, 4.25; N, 17.36. Found: C, 62.50; H, 4.22; N, 17.35.
Ethyl 4-methyl-2-(2-(4-(6-oxo-3-phenylpyridazin-1(6H)-yl)benzylidene)hydrazineyl) thiazole-5-carboxylate (8): Yield (74%): M.P.: 245–246 °C; IR: 3115 (NH), 3059, 1715 & 1665 (C=O), 1595; 1H-NMR: 1.43 (s, 3H, CH3), 2.26 (s, 3H, CH3), 4.20 (q, 2H, CH2), 7.24 (d, 2H, Ar-H), 7.49–7.60 (m, 4H, Ar-H), 8.05 (m, 4H, Ar-H), 8.20 (d, 1H, Ar-H), 8.49 (s, 1H, azomethine-H), 9.77 (s, 1H, NH); 13C-NMR: 170.51 (C=O), 167.15 (C=O), 160.95, 160.02, 159.13, 157.72, 145.10, 143.47, 135.19, 134.58, 134.45, 133.39, 131.78, 131.14, 130.98, 130.19, 129.47, 128.93, 126.65, 126.19, 116.91, 60.89 (-O-CH2-), 17.27 (-CH3), 14.75 (Ester -CH3); Mass: 459 (M+), 274 (100% base peak); Anal. Calcd. for C24H21N5O3S: C, 62.73; H, 4.61; N, 15.24. Found: C, 62.70; H, 4.55; N, 15.24.
6-Phenyl-2-(4-((2-(4-phenylthiazol-2-yl)hydrazinylidene)methyl)phenyl)pyridazin-3(2H)-one (9a): Yield (72%): M.P.: 280–281 °C; IR: 3115 (NH), 3059, 1666 (C=O), 1595; 1H-NMR: 7.04–7.50 (m, 9H, Ar-H + thiazole-H), 7.79–8.11 (m, 9H, Ar-H + azomethine-H), 12.28 (s, 1H, NH); 13C-NMR: 168.60 (C=O), 159.07, 144.82, 142.46, 140.67, 135.13, 134.64, 134.44, 132.37, 131.60, 131.52, 130.10, 129.43, 129.08 (2C), 128.02 (2C), 126.79 (2C), 126.55 (2C), 126.34 (2C), 126.0 (2C), 104.33; Mass: 449 (M+), 77 (100% base peak); Anal. Calcd. for C26H19N5OS: C, 69.47; H, 4.26; N, 15.58. Found: C, 69.45; H, 4.25; N, 15.55.
2-(4-((2-(4-(4-Bromophenyl)thiazol-2-yl)hydrazineylidene)methyl)phenyl)-6-phenylpyridazin-3(2H)-one (9b): Yield (70%): M.P.: 261–262 °C; IR: 3115 (NH), 3059, 1670 (C=O), 1595; 1H-NMR: 7.20–8.13 (m, 17H, Ar-H + thiazole-H5 + azomethine-H), 12.31 (s, 1H, NH); 13C-NMR: 168.78 (C=O), 159.08, 149.92, 144.83, 142.53, 141.64, 140.87, 134.66, 134.36, 132.02, 131.62, 131.51, 130.11, 129.44, 128.59 (2C), 128.04, 127.91 (2C), 126.84, 126.57 (2C), 126.34, 126.15, 121.03, 105.24; Mass: 527 (M+), 529 (M+ + 2), 274 (100% base peak); Anal. Calcd. for C26H18BrN5OS: C, 59.10; H, 3.43; N, 13.25. Found: C, 59.11; H, 3.42; N, 13.21.
Synthesis of Methyl2-(4-oxo-2-((4-(6-oxo-3-phenylpyridazin-1(6H)-yl)benzylidene)hydrazineylidene) -thiazolidin-5-ylidene)acetate (10). A mixture comprising 4b (5 mmoles), dimethyl acetylene dicarboxylate (5 mmol), and MeOH (30 mL) was reacted at 80 °C for 1 h. The resultant residue was cooled at 25 °C. The solid was filtered, and recrystallized by dioxane. Yield (75%): M.P.: 290–291 °C; IR: 3183 (NH), 2963, 2776, 1724 (C=O), 1710 & 1678 (C=O), 1614; 1H-NMR: 3.77 (s, 3H, OMe), 6.69 (s, 1H, CH=C), 7.24–7.39 (m, 8H, Ar-H), 7.75–7.92 (m, 3H, Ar-H), 8.34 (s, 1H, azomethine-H), 12.29 (hump, 1H, NH); 13C-NMR: 175.93 (C=O), 167.57 (C=O), 161.05 (C=O), 159.43, 159.13, 157.51, 155.24, 145.11, 143.50, 134.58, 134.32, 132.42, 132.30, 131.78, 130.19, 129.48, 129.04, 128.71, 126.66, 126.12, 122.12, 121.93, 56.37 (-OCH3); Mass: 459 (M+), 274 (100% base peak); Anal. Calcd. for C23H17N5O4S: C, 60.12; H, 3.73; N, 15.24. Found: C, 60.10; H, 3.69; N, 15.22.
Synthesis of 5-((dimethylamino)methylene)-2-(2-(4-(6-oxo-3-phenylpyridazin-1(6H)-yl) benzylidene) hydrazineyl) thiazol-4(5H)-one (11). A mixture comprising 6a (0.01 mole), dimethylformamide-dimethylacetal (DMF-DMA) (0.01 moles) and dioxane (30 mL) was refluxed for 4 h. The resultant residue was cooled at 25 °C. The solid was isolated, and recrystallized with EtOH-dioxane mixture (1:1). Yield (70%): M.P.: 284–285 °C; IR: 3183 (NH), 2963, 2776, 1724 & 1678 (C=O), 1614; 1H-NMR: 2.94 & 3.13 (2s, 6H, N(CH3)2), 7.18 (d, 1H, Ar-H), 7.49 (brs, 3H, Ar-H + methine-H), 7.74 (d, 3H, Ar-H), 7.94 (q, 3H, Ar-H), 8.12 (d, 2H, Ar-H), 8.25 (s, 1H, azomethine-H), 12.34 (s, 1H, NH); 13C-NMR: 172.72 (C=O), 167.35 (C=O), 159.07, 157.10, 144.91, 144.85, 144.59, 143.74, 143.33, 134.57, 134.47, 134.04, 131.63, 130.14, 129.43, 128.38, 128.06, 127.69, 126.55, 126.35, 126.27, 42.37 (2C, 2CH3); Mass: 444 (M+), 274 (100% base peak); Anal. Calcd. for C23H20N6O2S: C, 62.15; H, 4.54; N, 18.91. Found: C, 62.13; H, 4.52; N, 18.91.
Synthesis of 2-(2-(4-(6-oxo-3-phenylpyridazin-1(6H)-yl)benzylidene)hydrazineyl)-5- (2-oxoindolin-3-ylidene) thiazol-4(5H)-one (12). A mixture comprising 6a (0.01 mole), indoline-2,3-dione (0.01 mole), piperidine (0.5 mL), and dioxane (40 mL) was refluxed for 2 h. The solid was filtered hot, and recrystallized with dioxane. Yield (77%); M.P.: 255–256 °C; IR: 3169 & 3149 (NH), 3054, 2973, 1723 (C=O), 1705 & 1672 (C=O), 1613; 1H-NMR: 6.76 (d, 1H, Ar-H), 6.93 (d, 1H, Ar-H), 7.05 (d, 2H, Ar-H), 7.22 (d, 2H, Ar-H), 7.50 (brs, 3H, Ar-H), 7.87, 7.92 (2brs, 5H, Ar-H), 8.18 (d, 1H, Ar-H), 8.36 (s, 1H, azomethine-H), 11.20, 12.51 (2s, 2H, 2NH); 13C-NMR: 175.43 (C=O), 168.22 (C=O), 161.15 (C=O), 159.53, 159.23, 157.51, 155.34, 145.12, 143.60, 134.58, 134.38, 132.33, 132.24, 131.68, 130.29, 129.48 (2C), 129.14 (2C), 128.71 (2C), 126.67 (3C), 126.22 (2C), 122.14, 121.92; Mass: 518 (M+), 274 (100% base peak); Anal. Calcd. for C28H18N6O3S: C, 64.86; H, 3.50; N, 16.21. Found: C, 64.70; H, 3.37; N, 16.10.
General Synthesis of 16a and 16b
A mixture comprising 6a (10 mmoles), the requisite cinnamonitrile (10 mmoles), piperidine (0.5 mL), and dioxane (40 mL) was refluxed for 4–6 h. The solid produced was collected and recrystallized using dioxane. Alternatively, 16a and 16b can also be prepared by reacting equimolar quantities of 6a with appropriate benzaldehyde.
5-(4-Chlorobenzylidene)-2-(2-(4-(6-oxo-3-phenylpyridazin-1(6H)-yl)benzylidene)-hydra-zinyl)thiazol-4(5H)-one (16a): Yield (81%): M.P.: 297–298 °C; IR: 3160 (NH), 2940, 1729 & 1670 (C=O), 1619; 1H-NMR: 7.24 (d, 1H, Ar-H), 7.51 (brs, 3H, Ar-H), 7.84 (d, 2H, Ar-H), 7.97 (brs, 6H, Ar-H + benzylidene-H), 8.14, 8.24 (2d, 4H, Ar-H), 8.40 (s, 1H, azomethine-H), 10.79 (s, 1H, NH); 13C-NMR: 180.80 (C=O), 176.51 (C=O), 174.15, 159.08, 156.90, 149.16, 144.77, 142.32, 140.81, 136.49, 135.88 (2C), 134.93 (2C), 134.67 (2C), 129.43 (4C), 126.55 (3C), 126.53 (2C), 120.69, 120.30; Mass: 511 (M+), 510 (100% base peak); Anal. Calcd. for C27H18ClN5O2S: C, 63.34; H, 3.54; N, 13.68. Found: C, 63.25; H, 3.48; N, 13.54.
5-(4-Methoxybenzylidene)-2-(2-(4-(6-oxo-3-phenylpyridazin-1(6H)-yl)benzylidene)hydra-zineyl)thiazol-4(5H)-one (16b): Yield (83%): M.P.: >300 °C; IR: 3164 (NH), 1700 & 1665 (C=O), 1613; 1H-NMR: 3.91 (s, 3H, OCH3), 6.94 (d, 2H, Ar-H), 7.24 (d, 2H, Ar-H), 7.50–7.58 (2brs, 5H, Ar-H), 7.80–8.12 (3brs, 6H, Ar-H + CH-benzylidene), 8.18 (d, 1H, Ar-H), 8.50 (s, 1H, azomethine-H), 11.12 (s, 1H, NH); 13C-NMR: 168.27 (C=O), 166.11 (C=O), 165.48, 159.09, 155.46, 155.17, 144.76, 134.65, 131.61 (2C), 130.98 (2C), 130.11 (2C), 129.44 (3C), 129.16 (3C), 127.21 (2C), 126.55 (3C), 126.16 (2C), 55.68 (-OCH3); Mass: 507 (M+), 506 (100% base peak); Anal. Calcd. for C28H21N5O3S: C, 66.26; H, 4.17; N, 13.80. Found: C, 66.15; H, 4.15; N, 13.74.
Synthesis of 2-((4-(6-oxo-3-phenylpyridazin-1(6H)-yl)benzylidene)hydrazineylidene)-3-phenyl thiazolidin-4-one (17). A mixture comprising 4c (10 mmoles), ethyl 2-chloroacetate (10 mmoles), CH3COONa (20 mmoles), and AcOH (30 mL) was heated at 125 °C for 4 h. The produced solid was isolated and recrystallized by dioxane. Yield (72%): M.P.: 283–284 °C; IR: 3172 (NH), 2953, 2756, 1730 & 1699 (C=O), 1621; 1H-NMR: 4.13 (s, 2H, CH2), 7.19 (d, 2H, Ar-H), 7.41-7.54 (m, 7H, Ar-H), 7.78 (d, 1H, Ar-H), 7.87 (d, 2H, Ar-H), 7.93 (d, 2H, Ar-H), 8.13 (d, 2H, Ar-H), 8.40 (s, 1H, azomethine-H); 13C-NMR: 172.53 (C=O), 165.0 (C=O), 159.06, 157.36, 144.88, 135.51, 134.55, 131.63, 130.16, 129.59 (2C), 129.43 (2C), 129.19 (2C), 128.76 (2C), 128.59 (2C), 128.37 (2C), 126.56 (2C), 126.39 (2C), 32.85 (-S-CH2-); Mass: 465 (M+), 274 (100% base peak); Anal. Calcd. for C26H19N5O2S: C, 67.08; H, 4.11; N, 15.04. Found: C, 67.0; H, 4.03; N, 14.98.
Synthesis of Methyl2-(4-oxo-2-((4-(6-oxo-3-phenylpyridazin-1(6H)-yl)benzylidene)hydrazinylidene) -3-phenylthiazolidin-5-ylidene)acetate (18). A mixture comprising 4c (5 mmoles), dimethyl acetylenedicarboxylate (5 mmol), and MeOH (30 mL) was heated to 80 °C for 1 h. The resultant mixture was cooled to 25 °C. The solid separated was collected and recrystallized by ethanol-dimethylformamide (DMF) mixture (1:1). Yield (77%): M.P.: 277–278 °C; IR: 2953, 2756, 1730 (C=O), 1699 & 1670 (C=O), 1621; 1H-NMR: 3.84 (s, 3H, OMe), 6.84 (s, 1H, methine-H), 7.21 (d, 1H, Ar-H), 7.47–7.59 (m, 8H, Ar-H), 7.82 (d, 2H, Ar-H), 7.92 (d, 4H, Ar-H), 8.18 (d, 1H, Ar-H), 8.55 (s, 1H, azomethine-H); 13C-NMR: 169.90 (C=O), 166.48 (C=O), 159.42 (C=O), 157.13, 156.46, 144.94, 142.13, 134.54, 133.44, 131.67, 130.18 (2C), 129.64 (3C), 129.44 (3C), 128.80 (3C), 128.70 (2C), 126.58 (3C), 126.50 (2C), 53.08 (-OCH3); Mass: 535 (M+), 274 (100% base peak); Anal. Calcd. for C29H21N5O4S: C, 65.04; H, 3.95; N, 13.08. Found: C, 65.0; H, 3.91; N, 13.02.
3.2. Biological Activity
3.2.1. In Vitro COX-1 and COX-2 Inhibition Assay
The compounds 4a, 6a, 6b, 8, 9a, 9b, 10, 11, 12, 16a, 16b, 17, and 18 were exposed to their COX-1/COX-2 inhibitory action by a 10-fold dilution strategy (1–10−4 μg/mL) utilizing dimethylsulfoxide (DMSO) [30]. The test packs of the human COX-1/COX-2 were obtained from Cayman Chemicals (560131, Ann Arbor, MI, USA). The supplier’s instructions were followed to prepare the reagents as well as to perform the experiment. Briefly, samples (20 μL), the COX-1 and COX-2 enzyme (10 μL), and heme (10 μL) were mixed with the buffer solution (160 μL), which was supplied with the kits. The resultant combination was incubated at 37 °C in a water bath for 10 min, and arachidonic acid (10 μL) was added to initiate the COX reaction. A saturated solution of stannous chloride (30 μL) was added after 2 min to halt the COX reaction. The resultant mixture was incubated at ambient temperature for 5 min. The Prostaglandin-2α (PGF2α) developed after the COX reaction was measured through ELISA. The samples were shifted to a 96-well plate and incubated for 18 h at 25 °C. The plate was washed to get rid of the unbound components. The Ellman’s reagent (200 μL), containing the acetylcholine substrate, was mixed and further incubated at 25 °C for 1–1.5 h till the absorbance (410 nm) of the Bo well was in the range of 0.3 to 0.8 A.U. The plate was read through the ELISA reader. The IC50 values of COX-1 and COX-2 were determined by the regression analysis.
3.2.2. In Vivo Anti-Inflammatory Activity
The compounds 9a, 9b, 12, 16b, and 17 were evaluated for anti-inflammatory action utilizing Wistar rats (130–150 g) by following the rat paw edema method [30,34]. The animal approval (IAEC/KSOP/E/18/12) was obtained from the CPCSEA. A total of eight groups of rats were utilized, wherein each group comprised of 6 rats. The compounds (test group), celecoxib (standard group), and indomethacin (standard group) were administered orally (10 mg/kg) as a 10% Tween-80 solution. Saline solution (1 mL) was given to the control group. The carrageenan solution (1%, 0.1 mL) was administered by injection after 1 h in the right hind paw of the rats in the test, standard, and the control groups. The volume of the paws was calculated after the carrageenan injection at 0, 1, 2, 3, and 4 h by a plethysmometer. The % edema was calculated as follows:
3.2.3. Gastric Ulcerogenic Activity
The eight groups of rats were fasted for 18 h, wherein each group consisted of 6 rats. The compounds (9a, 9b, 12, 16b, and 17), celecoxib, and indomethacin were given orally (10 mg/kg) as 10% Tween-80 solution. Saline solution (1 mL) was given to the control group. The animals were sacrificed after 4 h to isolate their stomachs. A longitudinal cut was made along the greater curvature, and the presence of ulcers was evaluated. The counting of ulcers was marked as 0 (no ulcer) to 5 (≥ 3 ulcers) [30].
3.2.4. Lipid Peroxidation Inhibitory Activity
It was carried out on compounds 9a, 9b, 12, 16b, 17, celecoxib, and indomethacin after the ulcerogenic activity [35,36]. The gastric mucosa (100 mg) was scraped with two glass slides and homogenized in ice-cold potassium chloride (1.8 mL of 1.15% KCl). Sodium dodecyl sulphate (0.2 mL of 8.1% SDS), acetate buffer (pH = 3.5, 1.5 mL), and thiobarbituric acid (1.5 mL of 0.8% TBA) was mixed with the homogenate, and the mixture was heated to 95 °C for 1 h. The obtained mixture was extracted with a 5-mL mixture (15:1 v/v) of n-butanol and pyridine and centrifuged at 4000 rpm for 10 min. The supernatant liquid was separated, and its absorbance was measured at 532 nm using a spectrophotometer. A standard linear curve (absorbance vs. concentration in nM) was prepared using MDA tetrabutylammonium salt. The lipid peroxidation was calculated from the standard curve. The results are expressed as nmol MDA 100 mg−1 tissues.
3.3. Molecular Docking
The molecular docking of 9a, 9b, 12, 16b, 17, and celecoxib was performed by the method reported in our earlier report [30]. This study was conducted on the HP® computer system (Intel® core i5-4570 CPU, 3.20 GHz, Window 10). ChemDraw was used to design the compounds, wherein ChemBio3D Ultra was used to generate 3-D conformations. The 3-D structure of COX-2 protein (PDB entry 5KIR) was retrieved from PDB [37,38,39]. The Auto Dock and the Auto Dock Vina were utilized to generate the data. The detailed procedure was demonstrated in our earlier report [30].
3.4. Physicochemical Parameters
The concept of “Lipinski’s rule” [40,41] was applied to 9a, 9b, 12, 16b, and 17. The computational molecular properties like the molecular weight, number of H-bond acceptors and donors, log P, number of rotatable-bonds, and molecular polar surface area were determined by the available online software, Molinspiration [40,41]. The degree of absorption (%ABS) was computed as follows [44,45]:
%ABS = 109 − (0.345 × tPSA)
3.5. Statistical Analysis
It was performed by the SPSS software. The p values, N values, mean values, and standard deviation values are mentioned at the desirable places of the manuscript.
4. Conclusions
Five compounds, 9a, 9b, 12, 16b, and 17, demonstrated superior COX-2 inhibition than celecoxib. These compounds had a similar onset/duration of action to celecoxib. The compounds 9a and 12 were devoid of any ulcerogenic effect, whereas 9b, 16b, and 17 showed insignificant ulcerogenic effects. The compounds 9a, 9b, 12, 16b, and 17 also displayed a better lipid peroxidation profile than indomethacin and celecoxib. They also demonstrated a considerable calculated absorption. The compounds 9a, 9b, 11, 16b, and 17 are thus recognized and postulated as non-ulcerogenic COX-2 inhibitors with promising physicochemical parameters and gastric safety profile. These compounds may be useful candidates to combat diseases caused by higher levels of COX-2 like gout, ankylosing spondylitis, osteoarthritis, rheumatoid arthritis, Alzheimer’s disease, ulcerative colitis, depression, epilepsy, irritable bowel diseases, kidney injury, cancer, asthma, hepatitis, pancreatitis, and atherosclerosis.
Supplementary Materials
The Supplementary Materials are available online.
Author Contributions
Conceptualization, A.K., A.D., H.K.T., and M.I.; Data curation, A.K. and H.K.T.; Formal analysis, A.K., M.I., and M.A.B.; Investigation, A.K., H.K.T., M.I., and M.A.B.; Methodology, A.K., A.D., H.K.T., and M.I.; Project administration, A.K.; Resources, A.D., H.K.T., M.I., and M.A.B.; Software, A.K., M.I., and M.A.B.; Supervision, A.D., and H.K.T.; Validation, A.K., A.D., and H.K.T.; Visualization, A.K.; Writing—original draft, A.K., H.K.T., M.I., and M.A.B.; Writing—review and editing, A.D., and M.A.B. All authors have read and agreed to the published version of the manuscript.
Funding
This study received no external funding.
Acknowledgments
The authors thank Apeejay Stya University, Northern Border University, and Prince Sattam Bin Abdulaziz University for providing facilities and technical support to perform this research work.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2017, 9, 7204–7218. [Google Scholar] [CrossRef]
- Varela, M.L.; Mogildea, M.; Moreno, I.; Lopes, A. Acute Inflammation and Metabolism. Inflammation 2018, 41, 1115–1127. [Google Scholar] [CrossRef]
- Nasef, N.A.; Mehta, S.; Ferguson, L.R. Susceptibility to chronic inflammation: An update. Arch. Toxicol. 2017, 91, 1131–1141. [Google Scholar] [CrossRef]
- Al Rashoud, A.S.; Abboud, R.J.; Wang, W.; Wigderowitz, C. Efficacy of low-level laser therapy applied at acupuncture points in knee osteoarthritis: A randomised double-blind comparative trial. Physiotherapy 2014, 100, 242–248. [Google Scholar] [CrossRef]
- Lee, A.S.; Ellman, M.B.; Yan, D.; Kroin, J.S.; Cole, B.J.; Van Wijnen, A.J.; Im, H.J. A current review of molecular mechanisms regarding osteoarthritis and pain. Gene 2013, 527, 440–447. [Google Scholar] [CrossRef]
- Sehajpal, S.; Prasad, D.N.; Singh, R.K. Prodrugs of Non-steroidal Anti-inflammatory Drugs (NSAIDs): A Long March Towards Synthesis of Safer NSAIDs. Mini Rev. Med. Chem. 2018, 18, 1199–1219. [Google Scholar] [CrossRef]
- Radi, Z.A.; Khan, K.N. Cardio-renal safety of non-steroidal anti-inflammatory drugs. J. Toxicol. Sci. 2019, 44, 373–391. [Google Scholar] [CrossRef]
- Korbecki, J.; Baranowska-Bosiacka, I.; Gutowska, I.; Chlubek, D. Cyclooxygenase pathways. Acta Biochim. Pol. 2014, 61, 639–649. [Google Scholar] [CrossRef]
- Tortorella, M.D.; Zhang, Y.; Talley, J. Desirable Properties for 3rd Generation Cyclooxygenase-2 Inhibitors. Mini Rev. Med. Chem. 2016, 16, 1284–1289. [Google Scholar] [CrossRef]
- Kumar, V.; Kaur, K.; Gupta, G.K.; Gupta, A.K.; Kumar, S. Developments in synthesis of the anti-inflammatory drug, celecoxib: A review. Recent Pat. Inflamm. Allergy Drug Discov. 2013, 7, 124–134. [Google Scholar] [CrossRef]
- Martina, S.D.; Vesta, K.S.; Ripley, T.L. Etoricoxib: A highly selective COX-2 inhibitor. Ann. Pharmacother. 2005, 39, 854–862. [Google Scholar] [CrossRef]
- Burnier, M. The safety of rofecoxib. Expert Opin. Drug Saf. 2005, 4, 491–499. [Google Scholar] [CrossRef]
- Fanelli, A.; Ghisi, D.; Aprile, P.L.; Lapi, F. Cardiovascular and cerebrovascular risk with nonsteroidal anti-inflammatory drugs and cyclooxygenase 2 inhibitors: Latest evidence and clinical implications. Ther. Adv. Drug Saf. 2017, 8, 173–182. [Google Scholar] [CrossRef]
- Carullo, G.; Galligano, F.; Aiello, F. Structure-activity relationships for the synthesis of selective cyclooxygenase 2 inhibitors: An overview (2009–2016). MedChemComm 2016, 8, 492–500. [Google Scholar] [CrossRef]
- Khalil, N.A.; Ahmed, E.M.; Mohamed, K.O.; Nissan, Y.M.; Zaitone, S.A. Synthesis and biological evaluation of new pyrazolone-pyridazine conjugates as anti-inflammatory and analgesic agents. Bioorg. Med. Chem. 2014, 22, 2080–2089. [Google Scholar] [CrossRef]
- Saeed, M.M.; Khalil, N.A.; Ahmed, E.M.; Eissa, K.I. Synthesis and anti-inflammatory activity of novel pyridazine and pyridazinone derivatives as non-ulcerogenic agents. Arch. Pharm. Res. 2012, 35, 2077–2092. [Google Scholar] [CrossRef]
- Bingham, S.; Beswick, P.J.; Bountra, C.; Brown, T.; Campbell, I.B.; Chessell, I.P.; Clayton, N.; Collins, S.D.; Davey, P.T.; Goodland, H.; et al. The cyclooxygenase-2 inhibitor GW406381X [2-(4-ethoxyphenyl)-3-[4-(methylsulfonyl)phenyl]-pyrazolo[1,5-b]pyridazine] is effective in animal models of neuropathic pain and central sensitization. J. Pharmacol. Exp. Ther. 2005, 312, 1161–1169. [Google Scholar] [CrossRef]
- Ahmed, E.M.; Kassab, A.E.; El-Malah, A.A.; Hassan, M.S.A. Synthesis and biological evaluation of pyridazinone derivatives as selective COX-2 inhibitors and potential anti-inflammatory agents. Eur. J. Med. Chem. 2019, 171, 25–37. [Google Scholar] [CrossRef]
- Singh, J.; Sharma, D.; Bansal, R. Pyridazinone: An attractive lead for anti-inflammatory and analgesic drug discovery. Future Med. Chem. 2017, 9, 95–127. [Google Scholar] [CrossRef]
- Luong, C.; Miller, A.; Barnett, J.; Chow, J.; Ramesha, C.; Browner, M.F. Flexibility of the NSAID binding site in the structure of human cyclooxygenase-2. Nat. Struct. Biol. 1996, 3, 927–933. [Google Scholar] [CrossRef]
- Fabiola, G.; Damodharan, L.; Pattabhi, V.; Nagarajan, K. Cyclooxygenase-2—An attractive target for fruitful drug design. Curr. Sci. 2001, 80, 26–34. [Google Scholar]
- Liaras, K.; Fesatidou, M.; Geronikaki, A. Thiazoles and Thiazolidinones as COX/LOX Inhibitors. Molecules 2018, 23, 685. [Google Scholar] [CrossRef] [PubMed]
- Abdellatif, K.R.; Abdelgawad, M.A.; Elshemy, H.A.; Alsayed, S.S. Design, synthesis and biological screening of new 4-thiazolidinone derivatives with promising COX-2 selectivity, anti-inflammatory activity and gastric safety profile. Bioorg. Chem. 2016, 64, 1–12. [Google Scholar] [CrossRef]
- Kaminskyy, D.; Kryshchyshyn, A.; Lesyk, R. 5-Ene-4-thiazolidinones—An efficient tool in medicinal chemistry. Eur. J. Med. Chem. 2017, 140, 542–594. [Google Scholar] [CrossRef]
- Omar, Y.M.; Abdu-Allah, H.H.M.; Abdel-Moty, S.G. Synthesis, biological evaluation and docking study of 1,3,4-thiadiazole-thiazolidinone hybrids as anti-inflammatory agents with dual inhibition of COX-2 and 15-LOX. Bioorg. Chem. 2018, 80, 461–471. [Google Scholar] [CrossRef]
- Tripathi, A.C.; Gupta, S.J.; Fatima, G.N.; Sonar, P.K.; Verma, A.; Saraf, S.K. 4-Thiazolidinones: The advances continue. Eur. J. Med. Chem. 2014, 72, 52–77. [Google Scholar] [CrossRef]
- Unsal-Tan, O.; Ozadali, K.; Piskin, K.; Balkan, A. Molecular modeling, synthesis and screening of some new 4-thiazolidinone derivatives with promising selective COX-2 inhibitory activity. Eur. J. Med. Chem. 2012, 57, 59–64. [Google Scholar] [CrossRef]
- Verma, A.; Saraf, S.K. 4-thiazolidinone—A biologically activescaffold. Eur. J. Med. Chem. 2008, 43, 897–905. [Google Scholar] [CrossRef]
- El-Feky, S.A.; Imran, M.; Nayeem, N. Design, Synthesis, and Anti-Inflammatory Activity of Novel Quinazolines. Orient. J. Chem. 2017, 33, 707–716. [Google Scholar] [CrossRef]
- Khan, A.; Diwan, A.; Thabet, H.K.; Imran, M. Synthesis of novel N-substitutedphenyl-6-oxo-3-phenyl pyridazine derivatives as cyclooxygenase-2 inhibitors. Drug Dev. Res. 2020, 2020, 1–12. [Google Scholar] [CrossRef]
- El-Feky, S.A.; Abd El-Samii, Z.K.; Osman, N.A.; Lashine, J.; Kamel, M.A.; Thabet, H.K. Synthesis, molecular docking and anti-inflammatory screening of novel quinoline incorporated pyrazole derivatives using the Pfitzinger reaction II. Bioorg. Chem. 2015, 58, 104–116. [Google Scholar] [CrossRef]
- El-Feky, S.A.; Hamdy, K.T.; Mustafa, T.U. Synthesis, molecular modeling and anti-inflammatory screening of novel fluorinated quinoline incorporated benzimidazole derivatives using the Pfitzinger reaction. J. Fluor. Chem. 2014, 161, 87–94. [Google Scholar] [CrossRef]
- Matheus, M.E.; ViolanteFde, A.; Garden, S.J.; Pinto, A.C.; Fernandes, P.D. Isatins inhibit cyclooxygenase-2 and inducible nitric oxide synthase in a mouse macrophage cell line. Eur. J. Pharmacol. 2007, 556, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Winter, C.A.; Risley, E.A.; Nuss, G.W. Carrageenin-induced edema in hind paw of the rat as an assay for antiiflammatory drugs. Proc. Soc. Exp. Biol. Med. 1962, 111, 544–547. [Google Scholar] [CrossRef]
- Ohkawa, H.; Ohishi, N.; Yagi, K. Assay for lipid peroxides in animaltissues by thiobarbituric acid reaction. Anal. Biochem. 1979, 95, 351–358. [Google Scholar] [CrossRef]
- Banerjee, A.G.; Das, N.; Shengule, S.A.; Sharma, P.A.; Srivastava, R.S.; Shrivastava, S.K. Design, synthesis, evaluation and molecular modelling studies of some novel 5,6-diphenyl-1,2,4-triazin-3(2H)-ones bearing five-member heterocyclic moieties as potential COX-2 inhibitors: A hybrid pharmacophore approach. Bioorg. Chem. 2016, 69, 102–120. [Google Scholar] [CrossRef] [PubMed]
- Orlando, B.J.; Malkowski, M.G. Crystal structure of rofecoxib bound to human cyclooxygenase-2. Acta Crystallogr. F Struct. Biol. Commun. 2016, 72, 772–776. [Google Scholar] [CrossRef]
- Pérez-Villanueva, J.; Yépez-Mulia, L.; González-Sánchez, I.; Palacios-Espinosa, J.F.; Soria-Arteche, O.; Sainz-Espuñes, T.D.R.; Cerbón, M.A.; Rodríguez-Villar, K.; Rodríguez-Vicente, A.K.; Cortés-Gines, M.; et al. Synthesis and Biological Evaluation of 2H-Indazole Derivatives: Towards Antimicrobial and Anti-Inflammatory Dual Agents. Molecules 2017, 22, 1864. [Google Scholar] [CrossRef]
- Jiao, J.; Yang, Y.; Wu, Z.; Li, B.; Zheng, Q.; Wei, S.; Wang, Y.; Yang, M. Screening cyclooxygenase-2 inhibitors from Andrographis paniculata to treat inflammation based on bio-affinity ultrafiltration coupled with UPLC-Q-TOF-MS. Fitoterapia 2019, 137, 104259. [Google Scholar] [CrossRef]
- Molinspiration Cheminformatics. Available online: www.molinspiration.com/services/properties.html (accessed on 25 February 2020).
- Lipinski, C.A.; Lombardo, L.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estime solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Benet, L.Z.; Hosey, C.M.; Ursu, O.; Oprea, T.I. BDDCS, the Rule of 5 and drugability. Adv. Drug Deliv. Rev. 2016, 101, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Shakeel, F.; Imran, M.; Haq, N.; Alshehri, S.; Anwer, M.K. Synthesis, Characterization and Solubility Determination of 6-Phenyl-pyridazin-3(2H)-one in Different Pharmaceutical Solvents. Molecules 2019, 24, 3404. [Google Scholar] [CrossRef] [PubMed]
- Amin, N.H.; Mohammed, A.A.; Abdellatif, K.R. Novel4-methylsulfonylphenylderivatives as NSAIDS with preferentialCOX-2inhibition. Future Med. Chem. 2018, 10, 53–70. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Abraham, M.H.; Lee, J. Rate-limited steps of human oral absorption and QSAR studies. Pharm. Res. 2002, 19, 1446–1457. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).