
Recent Advances in the Chemical Synthesis and Evaluation of Anticancer Nucleoside Analogues


Abstract
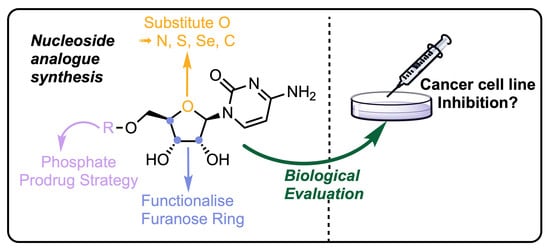
1. Introduction
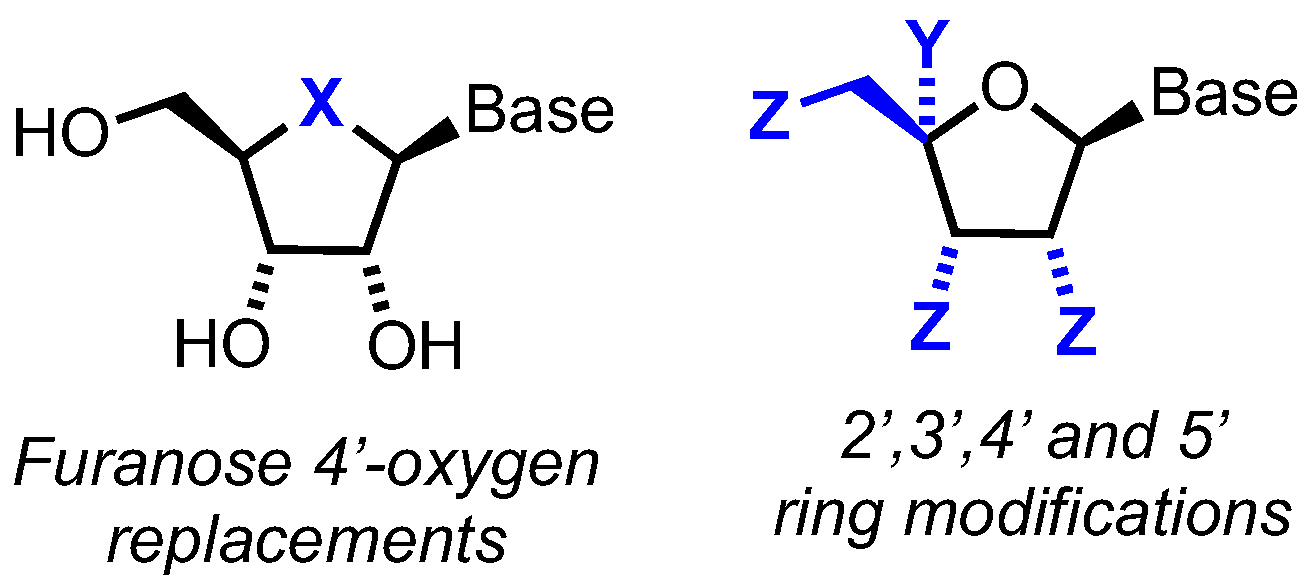
2. Furanose Oxygen Atom Replacements
2.1. Azanucleosides
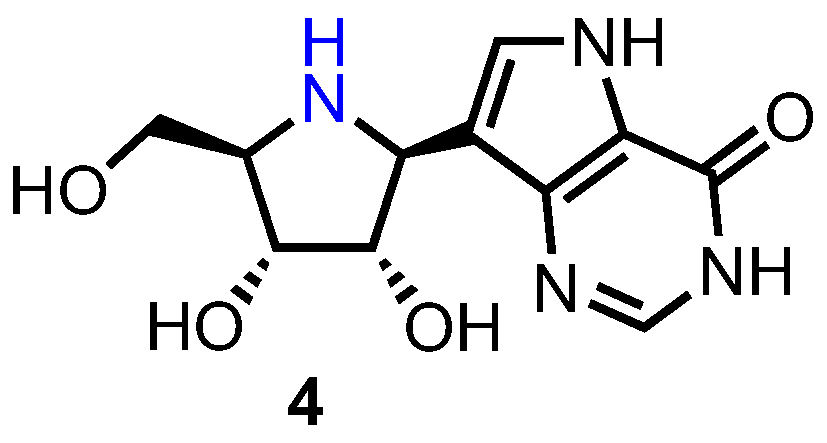
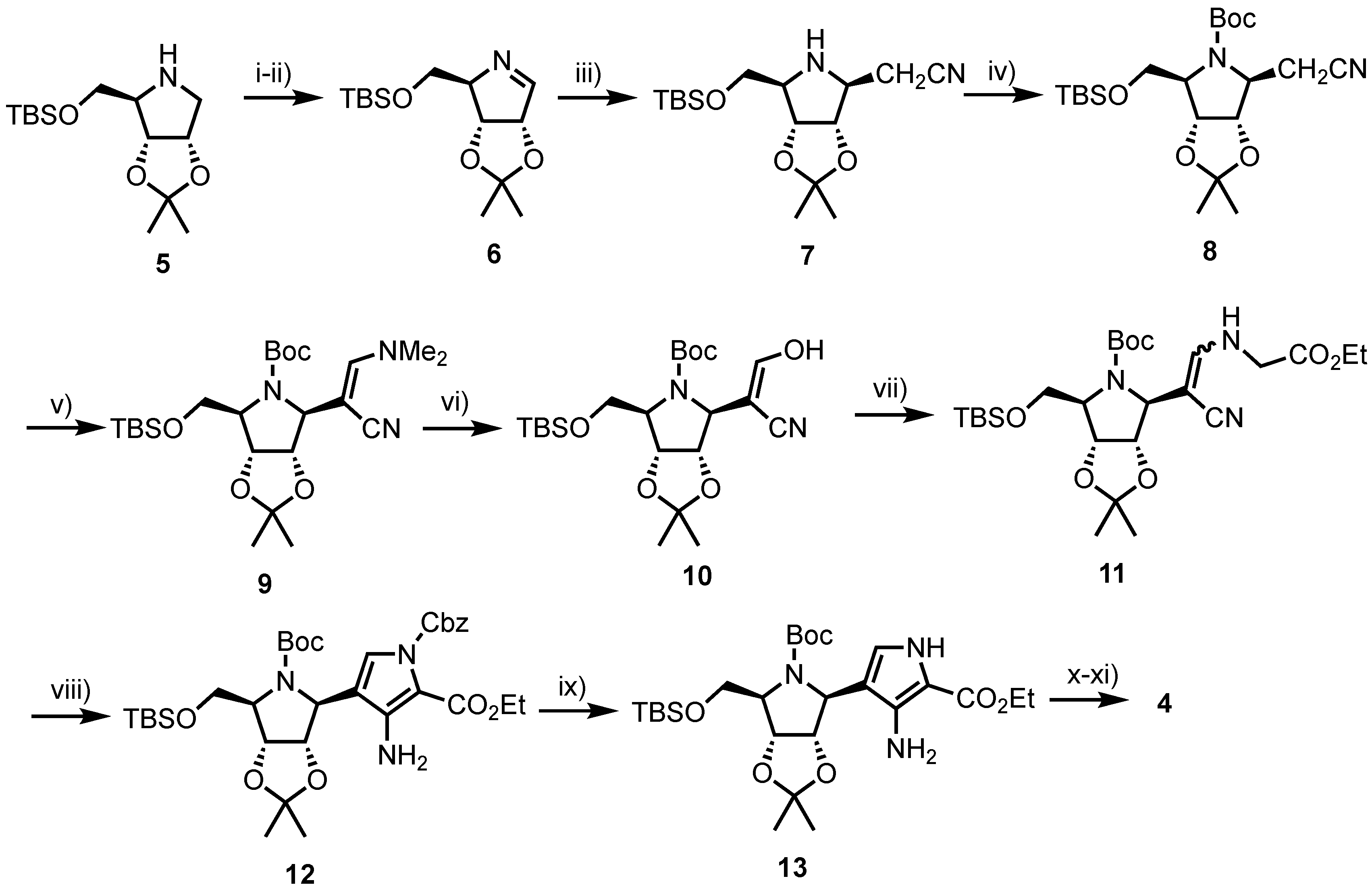
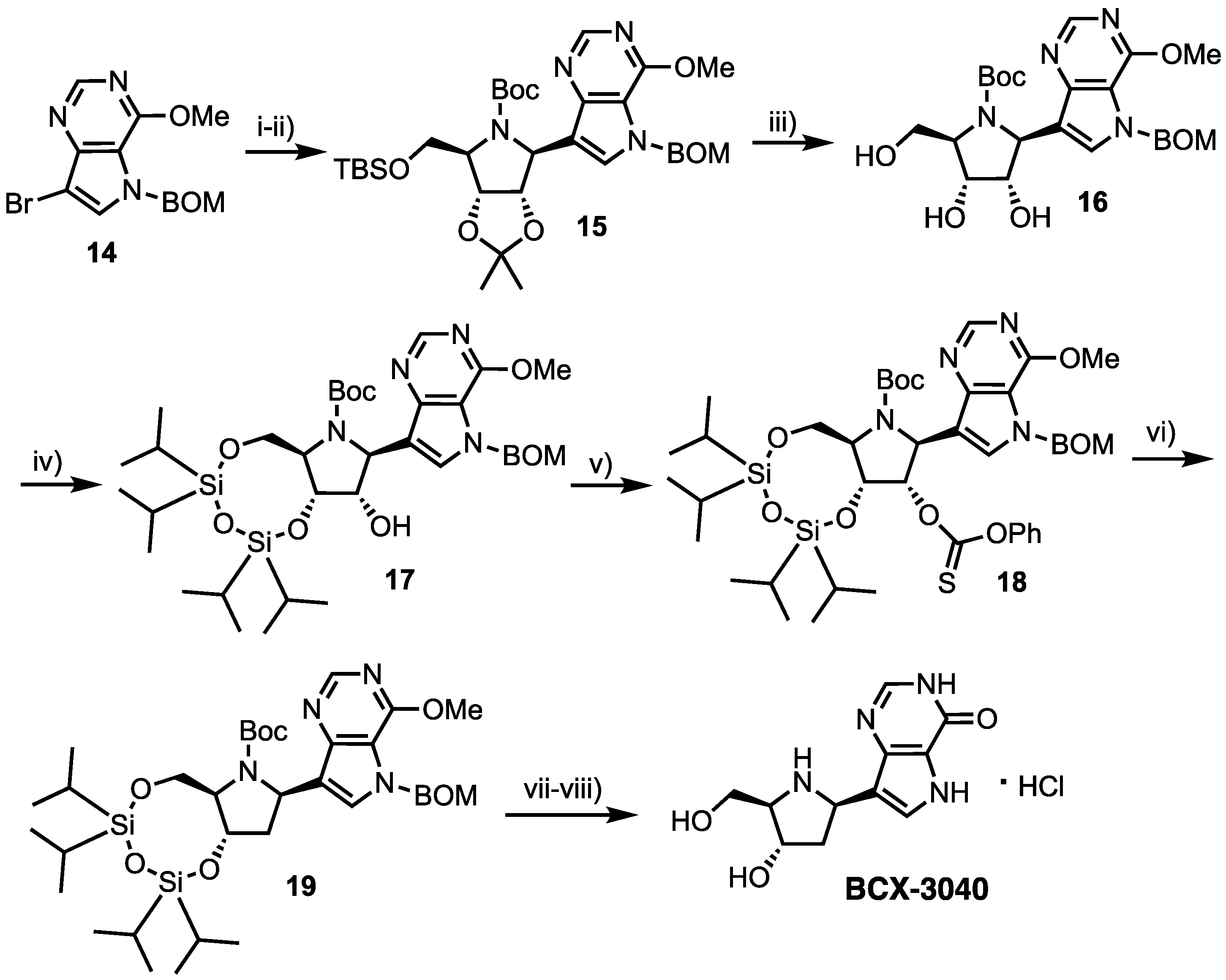
Development of Forodesine
2.2. Thionucleosides
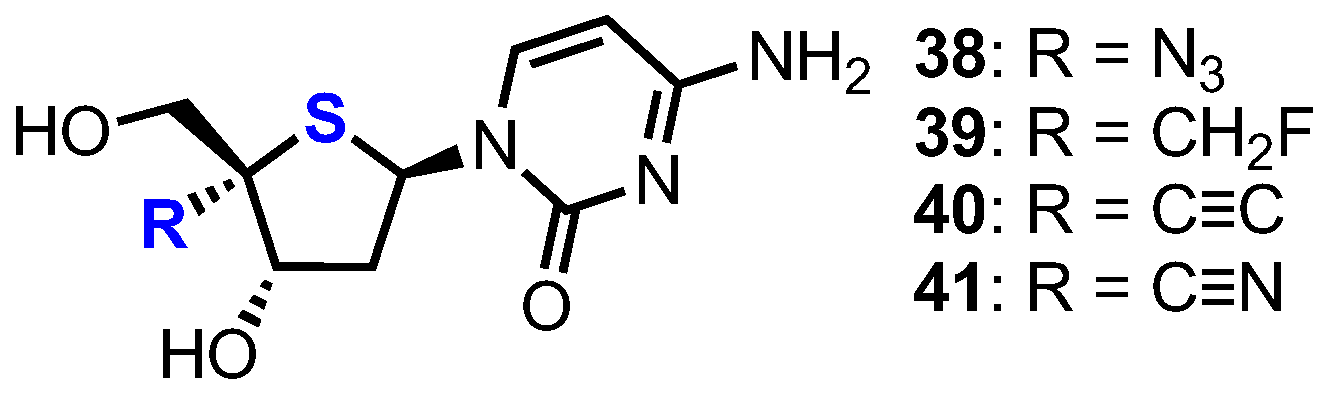
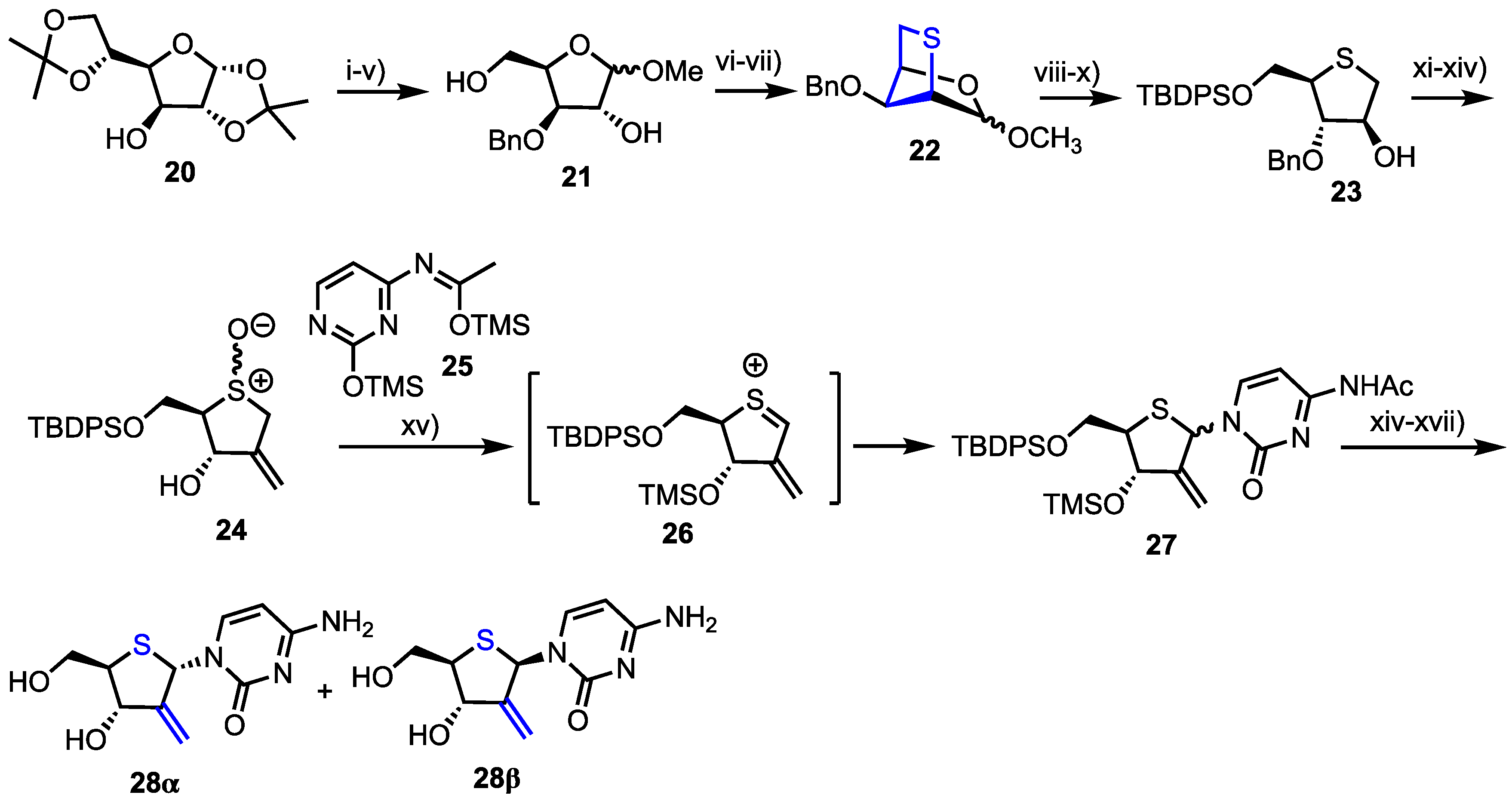
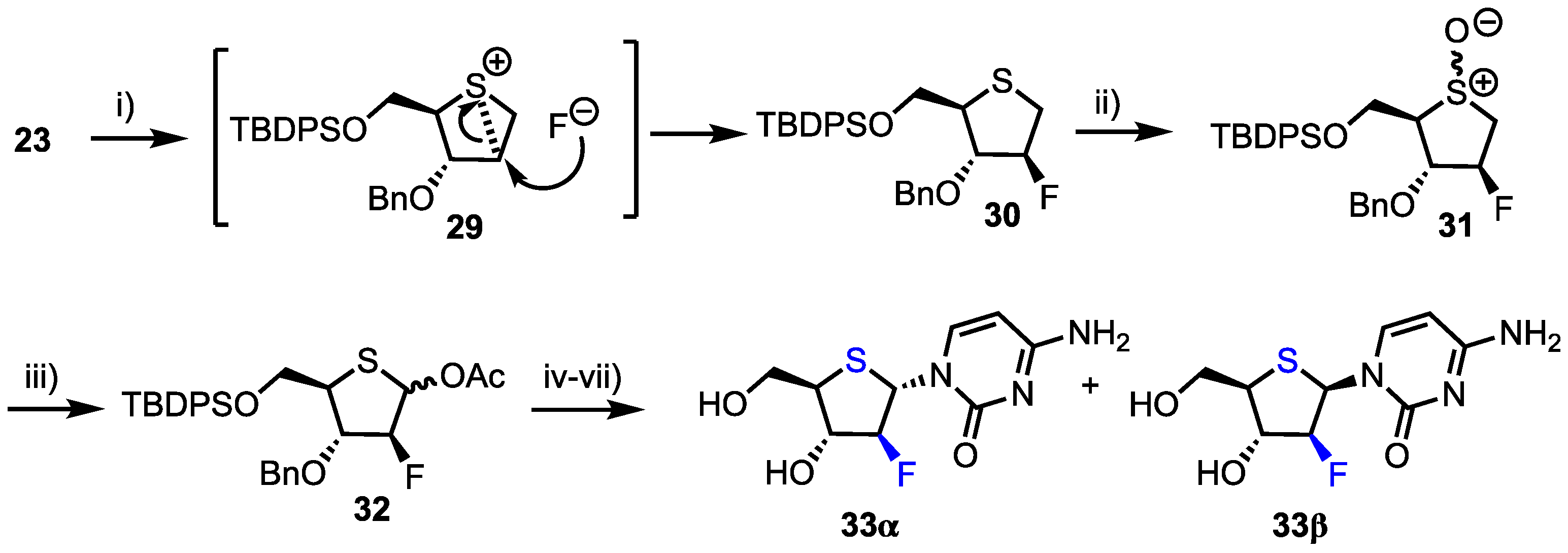
2.2.1. 2′-Modfied Thionucleosides
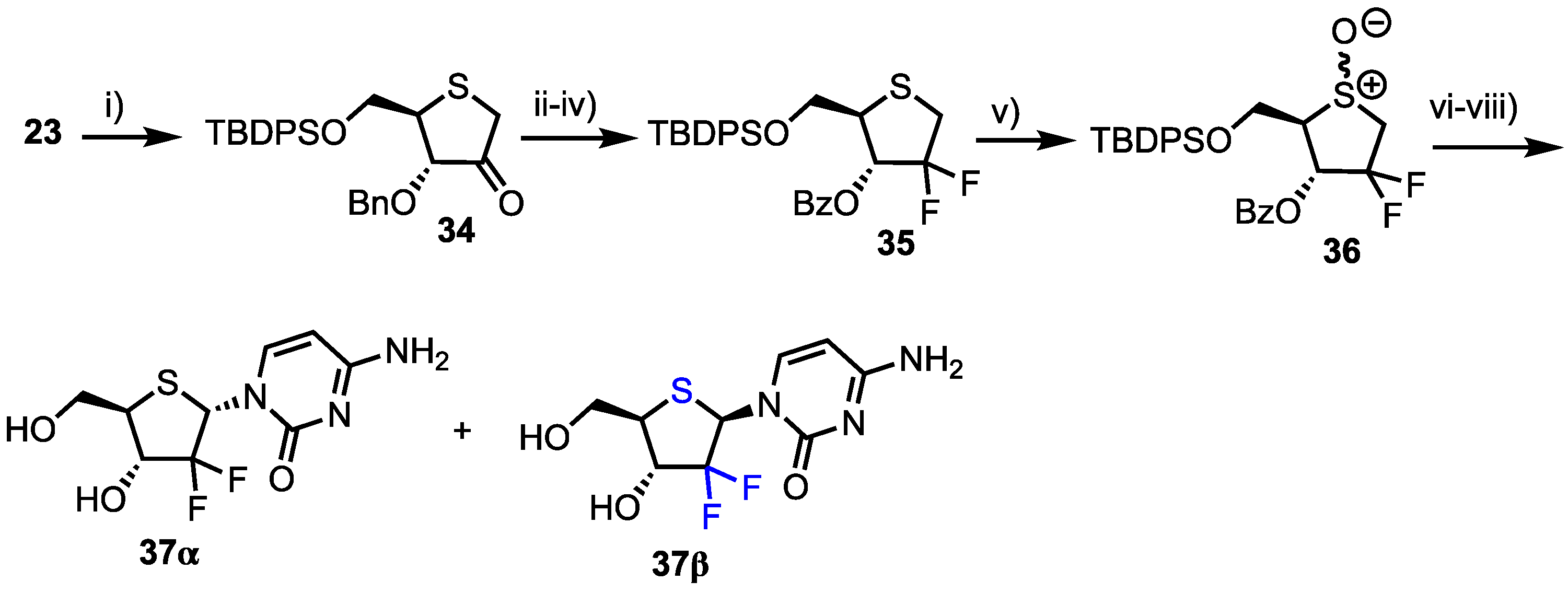
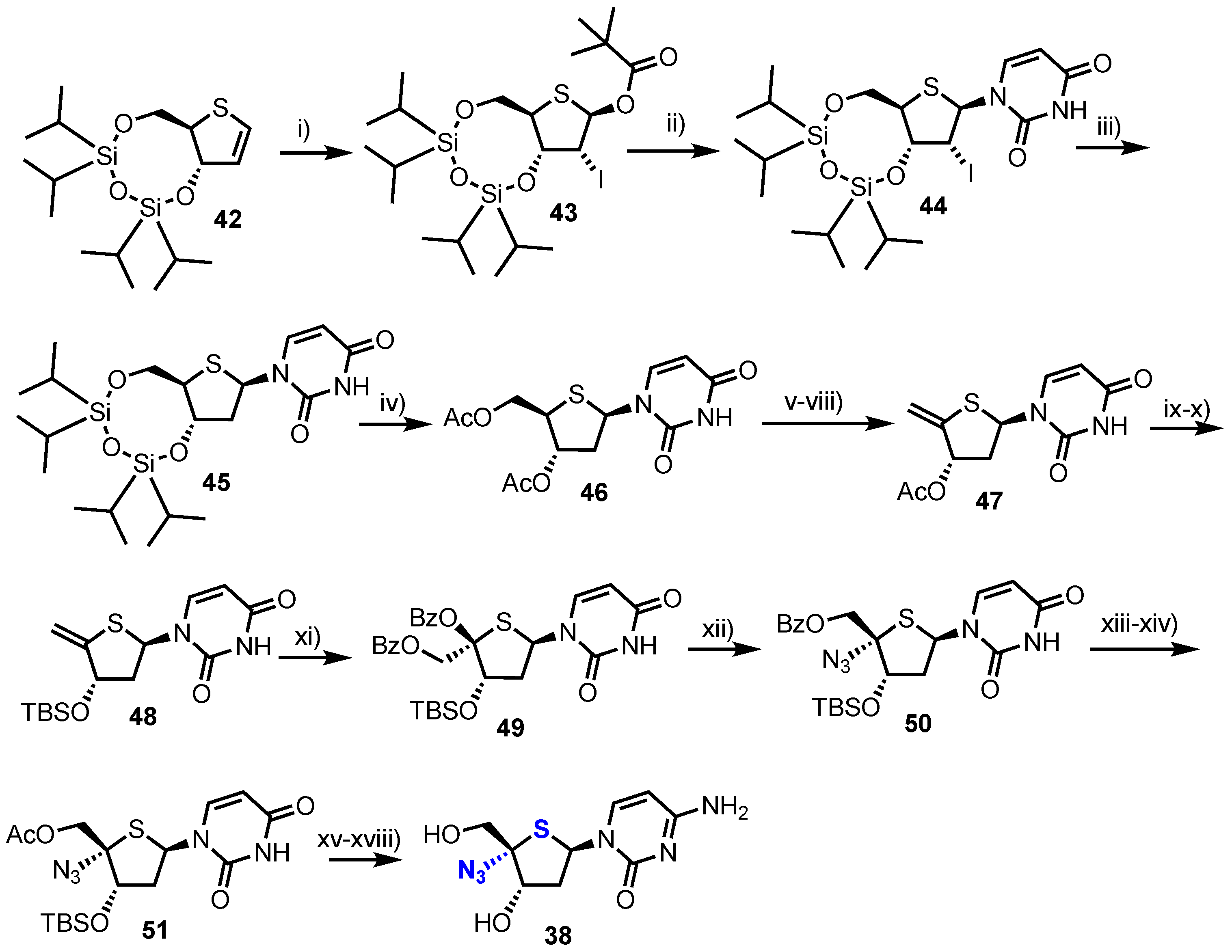
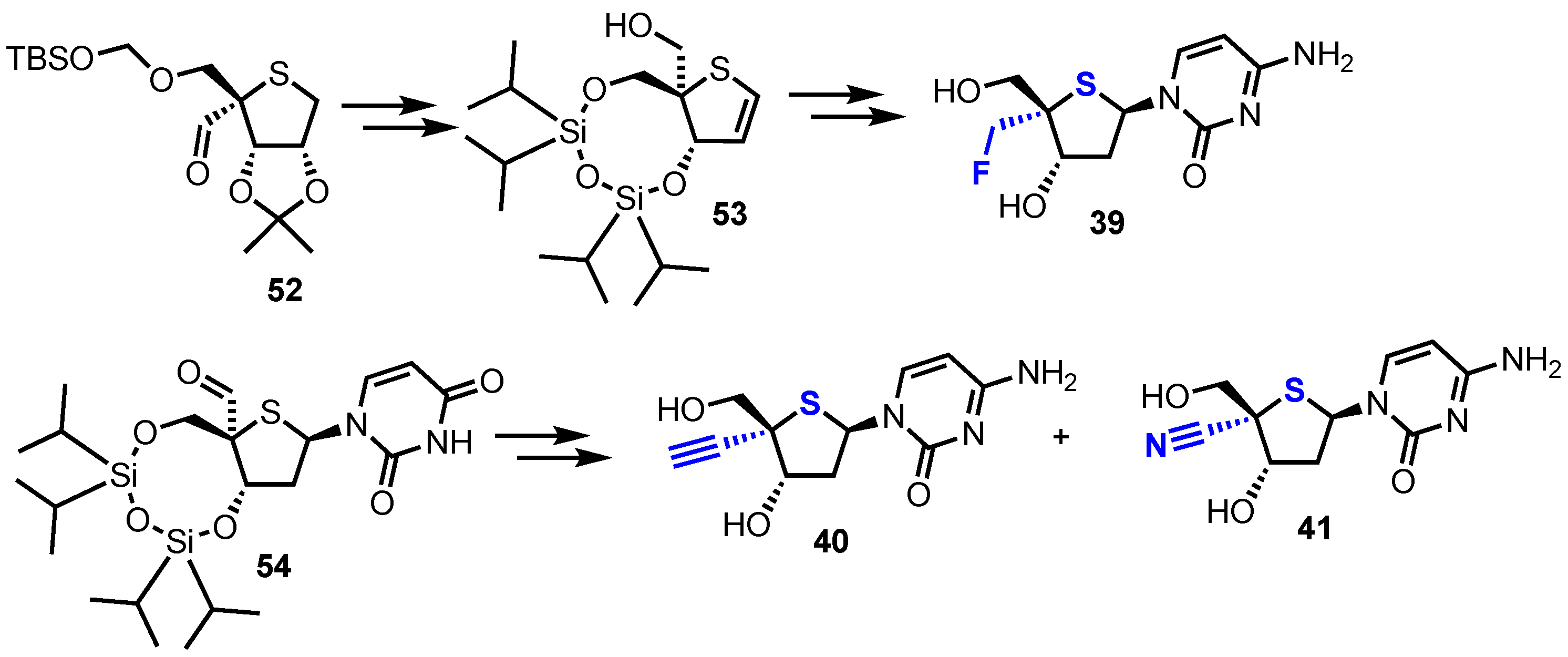
2.2.2. 4′-Modfied-2′-deoxythionucleosides
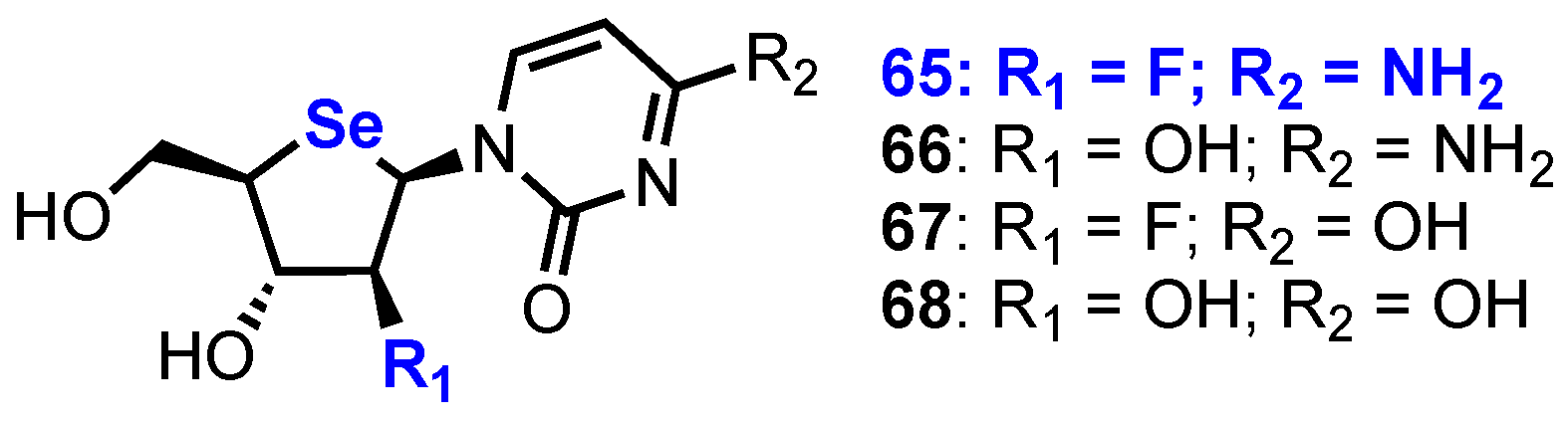
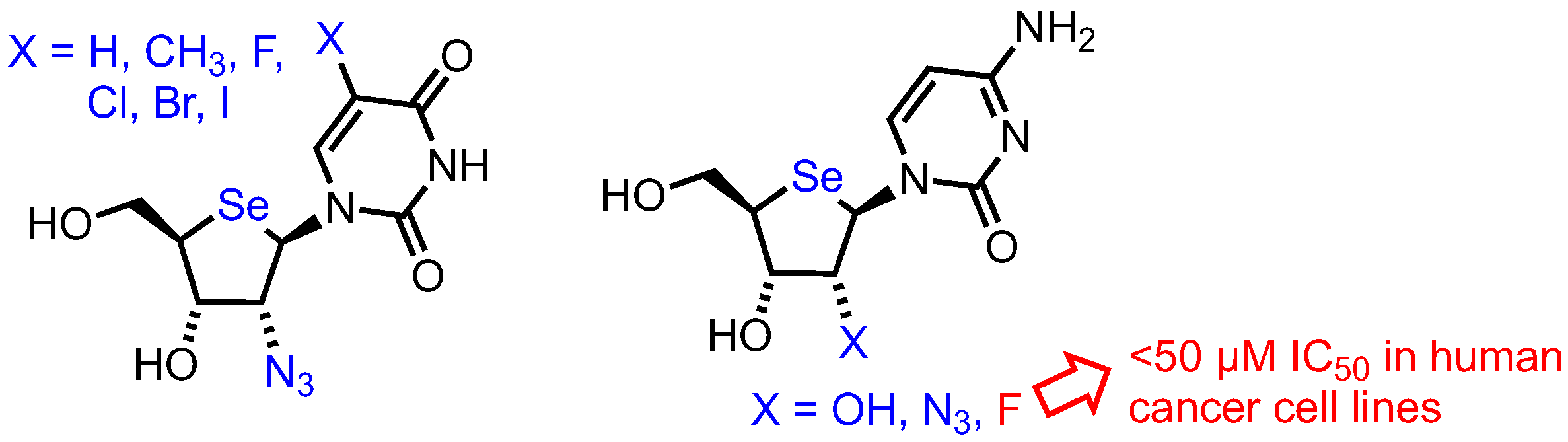
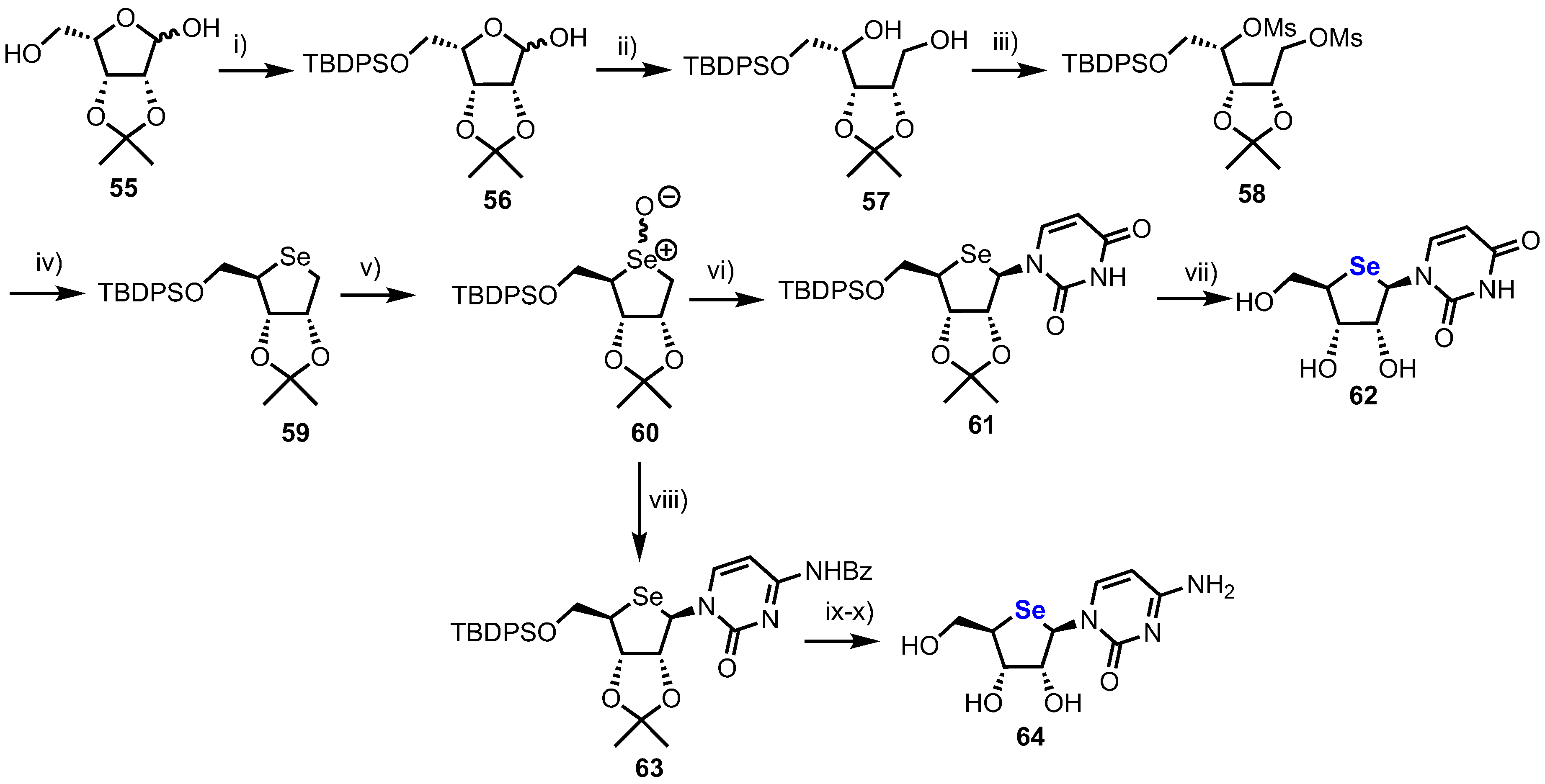
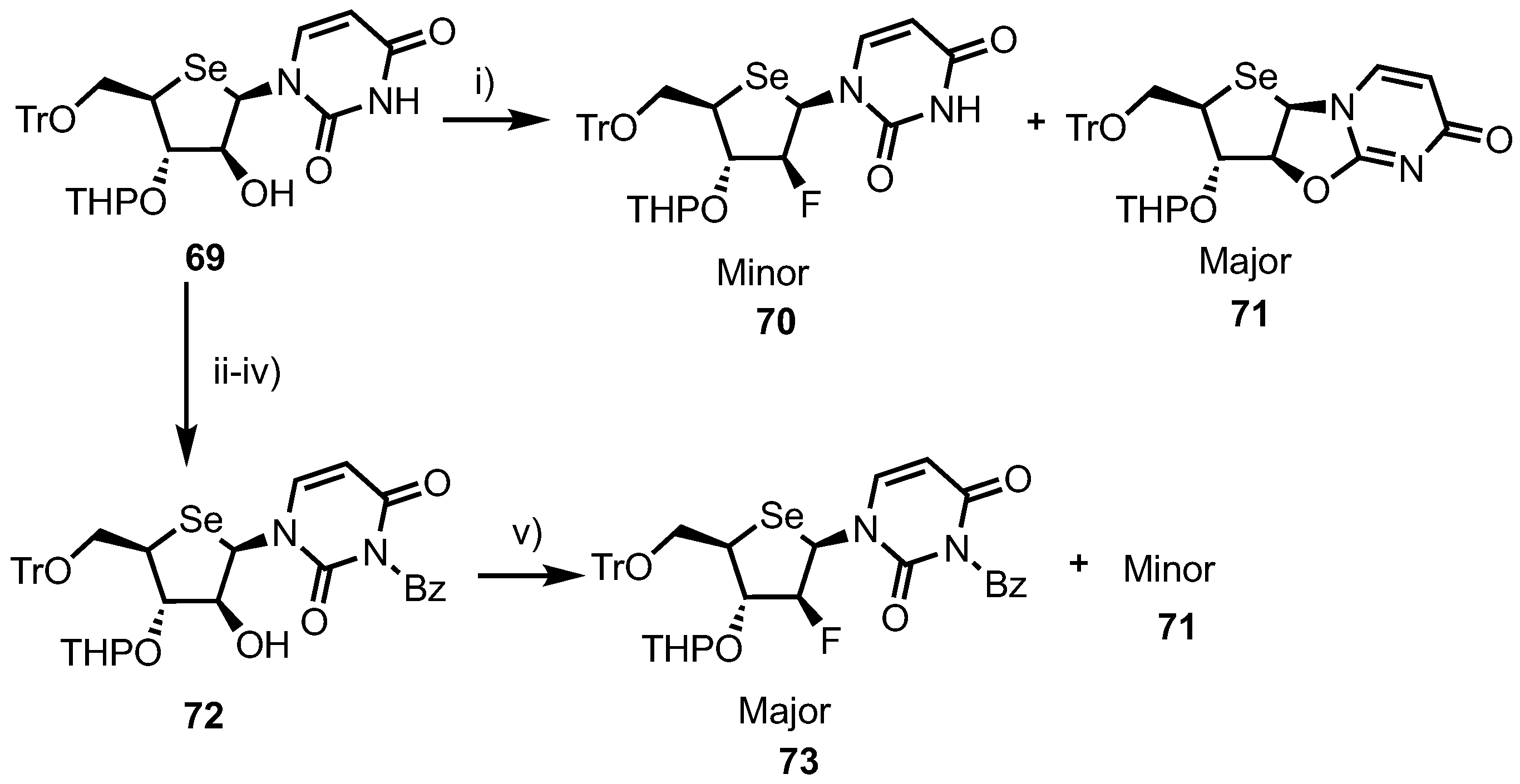
2.3. Selenonucleosides
2’-Substituted-4’-selenoribofuranosyl Pyrimidines
2.4. Carbocyclic Nucleosides
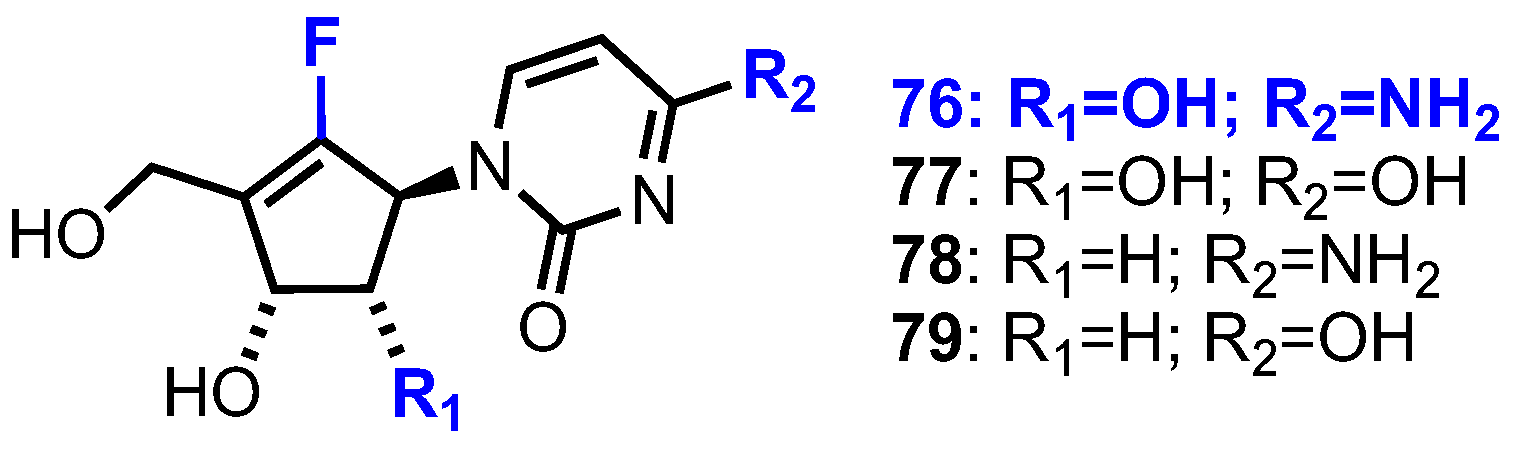
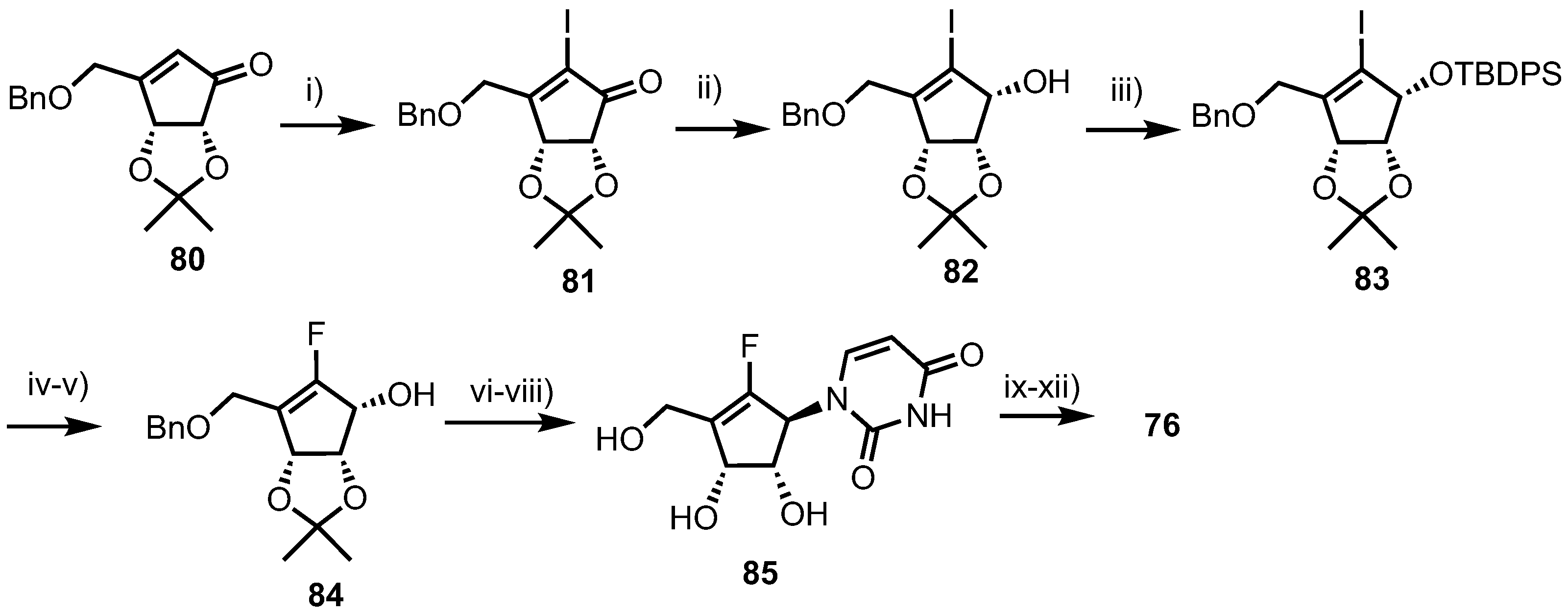
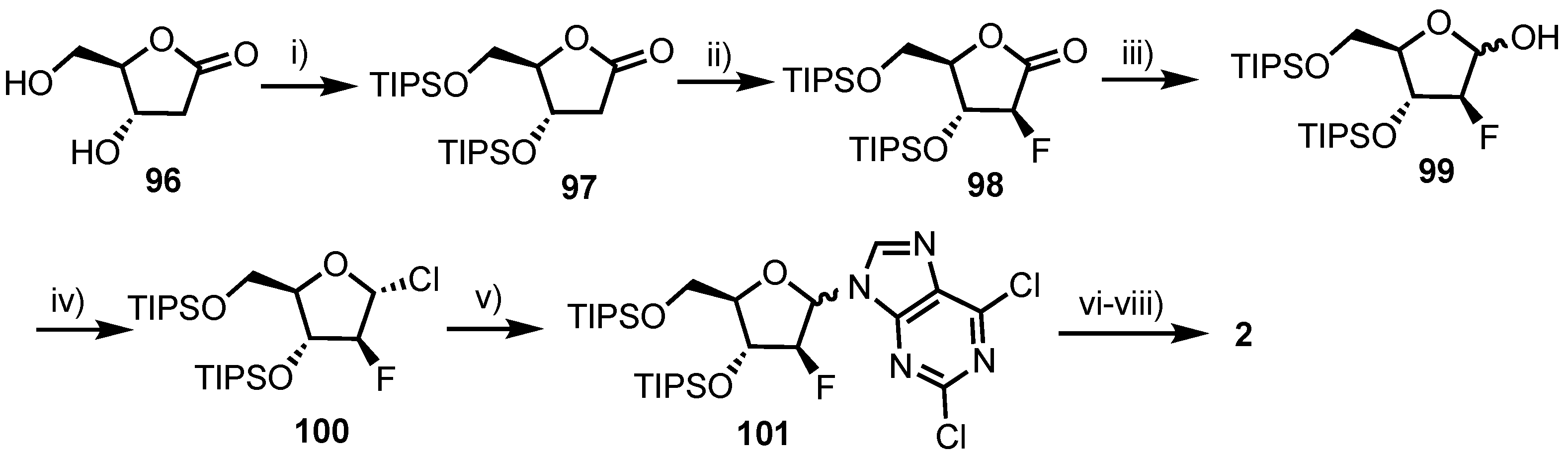
2.4.1. Fluorinated Derivatives of Neplanocin A
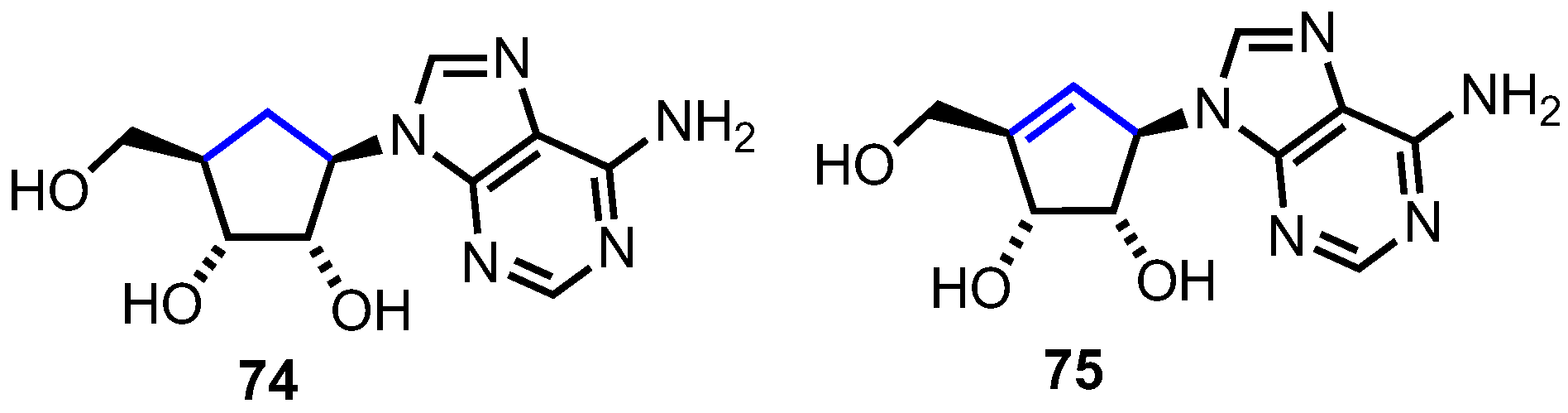
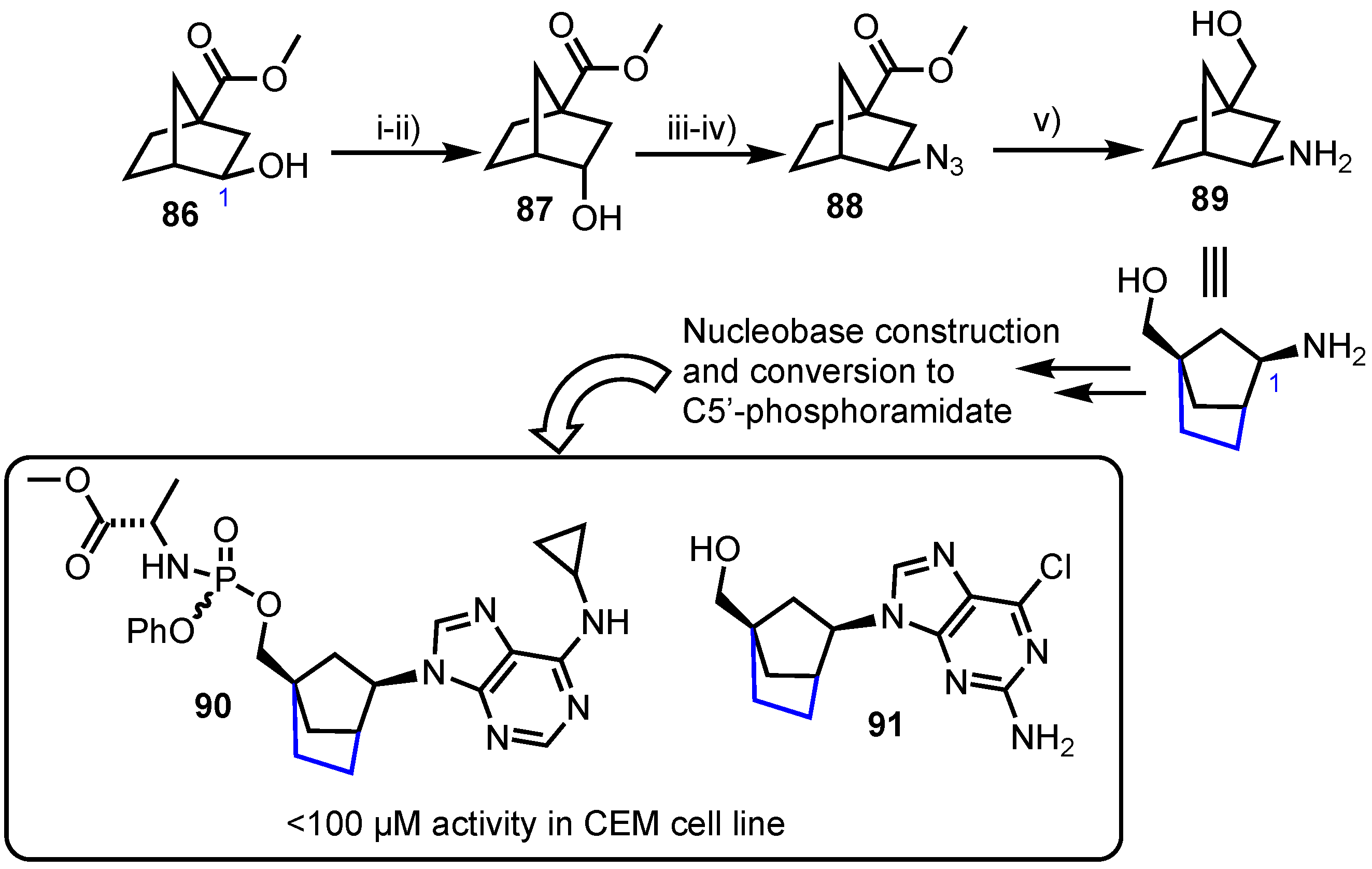
2.4.2. Norbornane-Derived (C2’,C4’-bridged) Carbocyclic Nucleosides
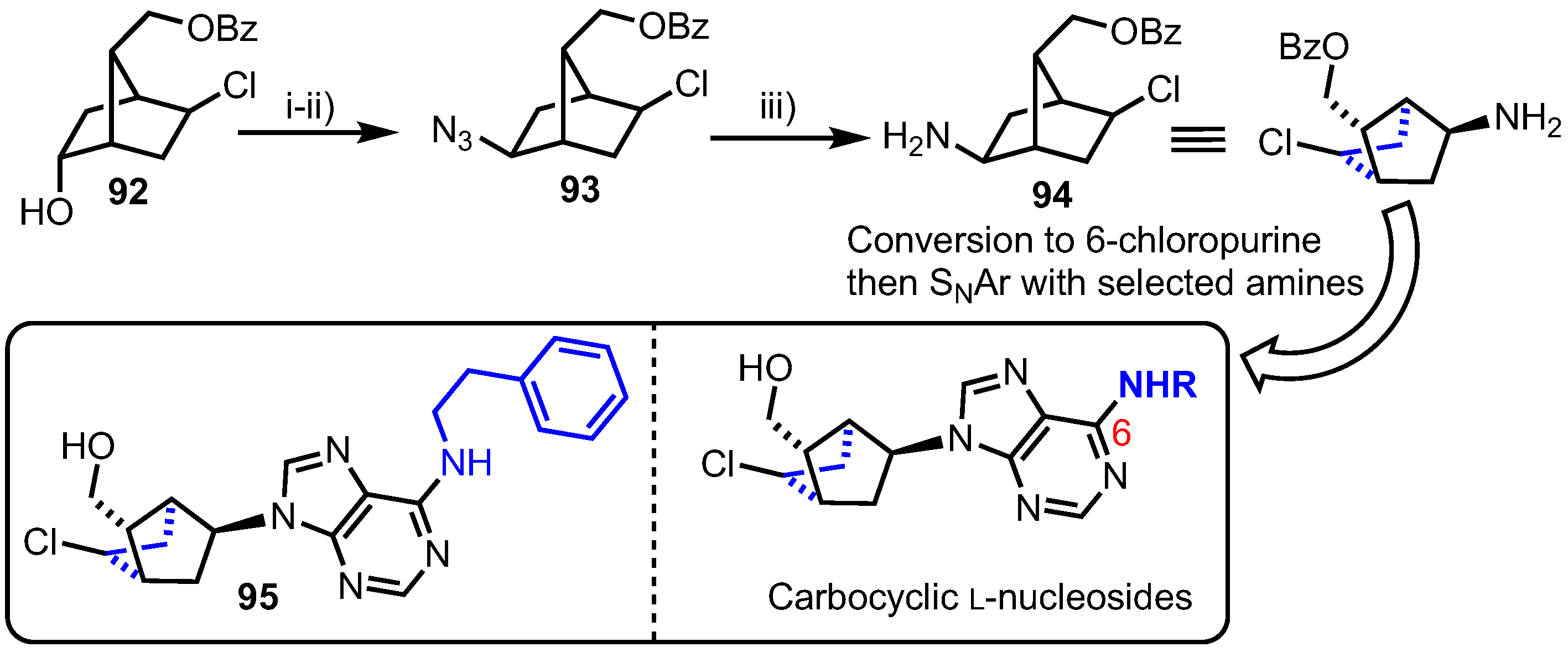
2.4.3. C3’,C5’-Bridged Carbocyclic L-Nucleosides
3. 2’-, 3’- and 5’-Furanose Ring Modifications
3.1. 2’-Furanose Modifications
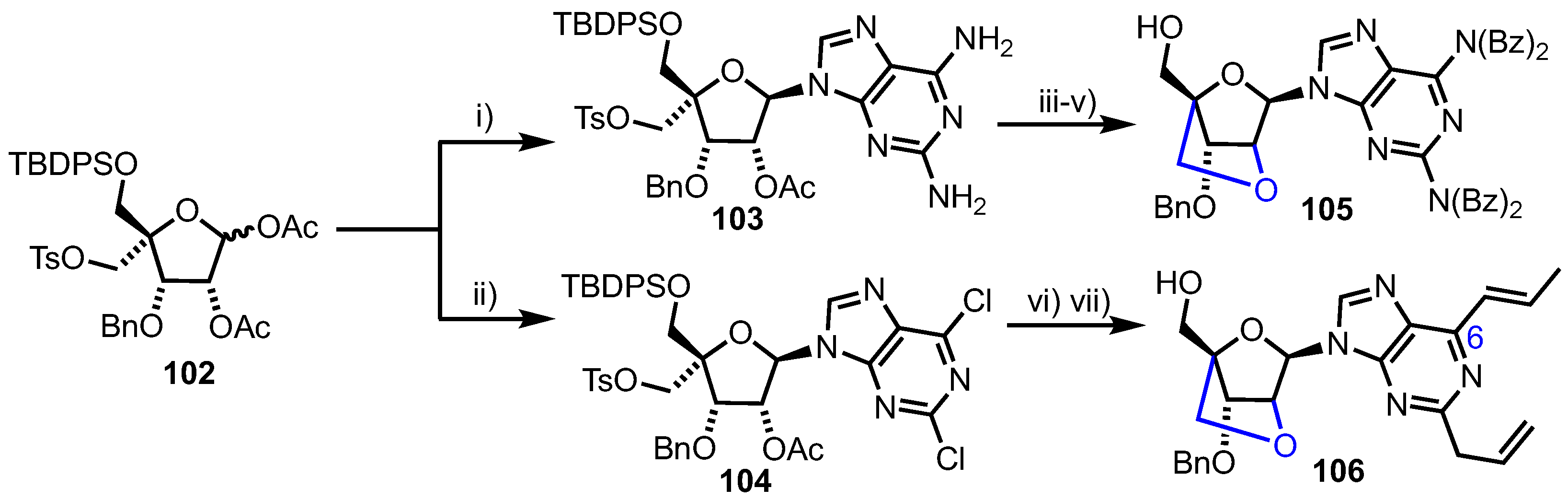
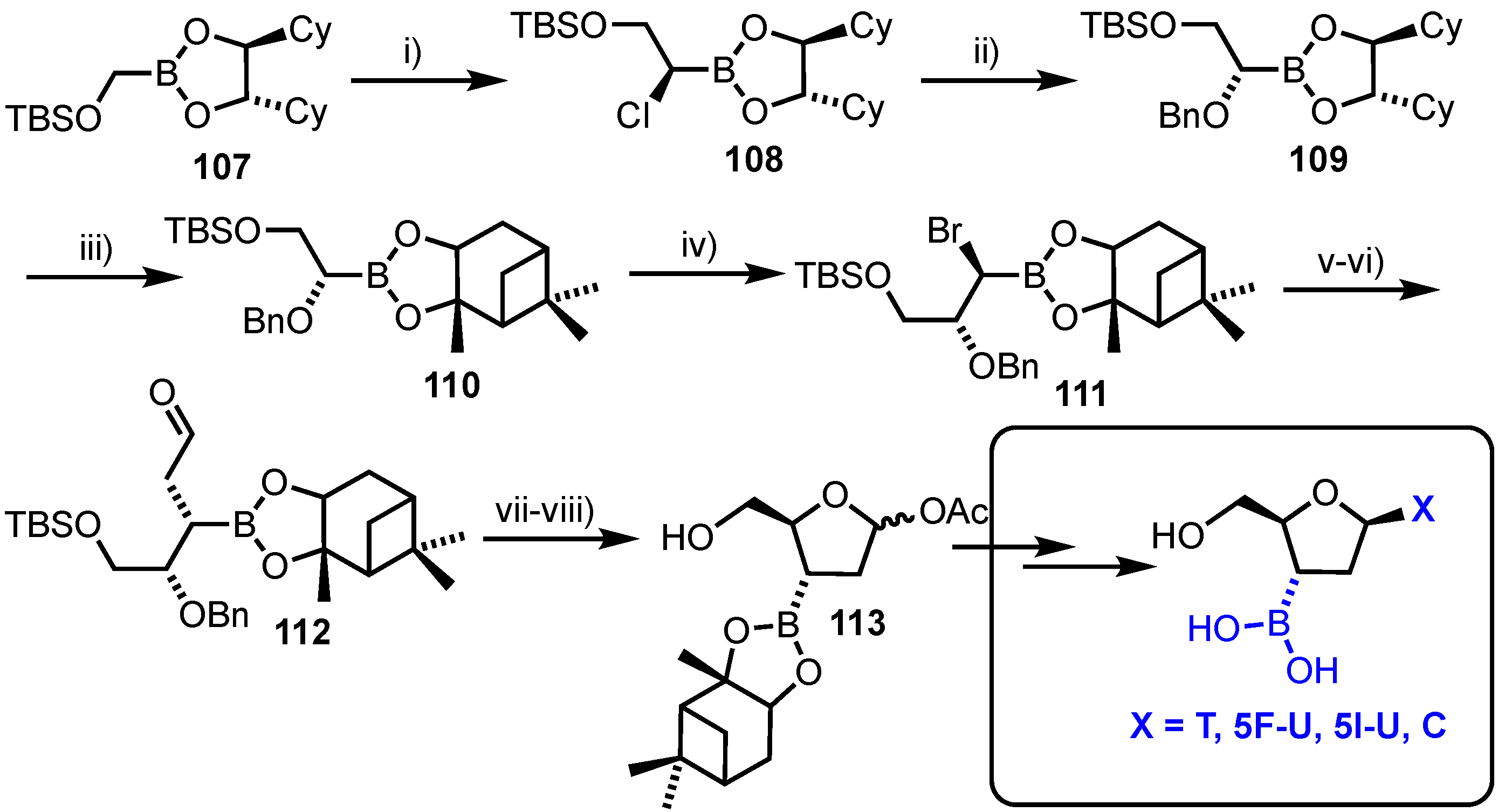
3.2. 2’-O,4-C’-Bridged Nucleosides
3.3. 3’-Modified Nucleosides
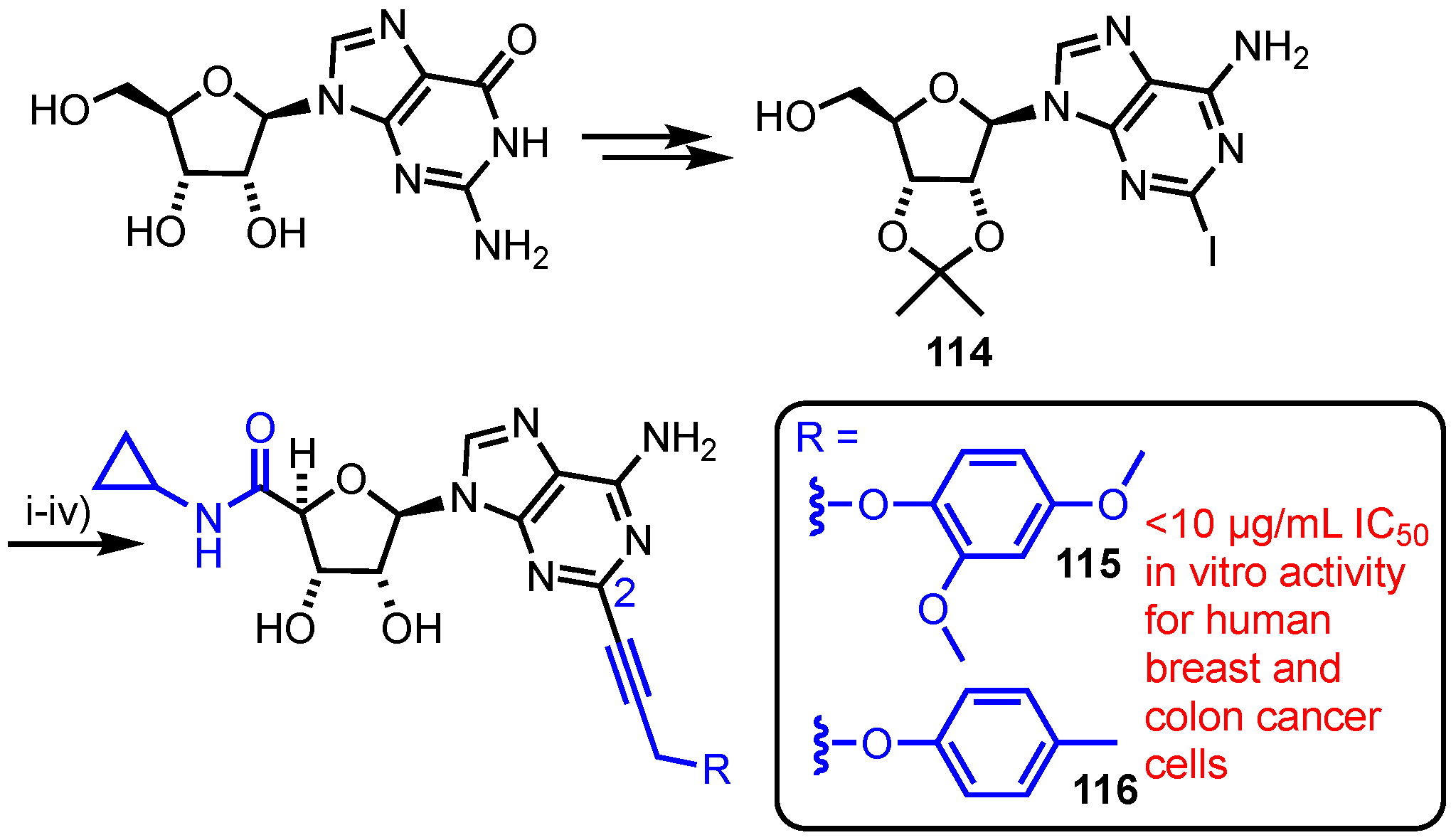
3.4. C5′-N-Cyclopropylcarboxamido-C6-amino-C2-alkynylated Analogues
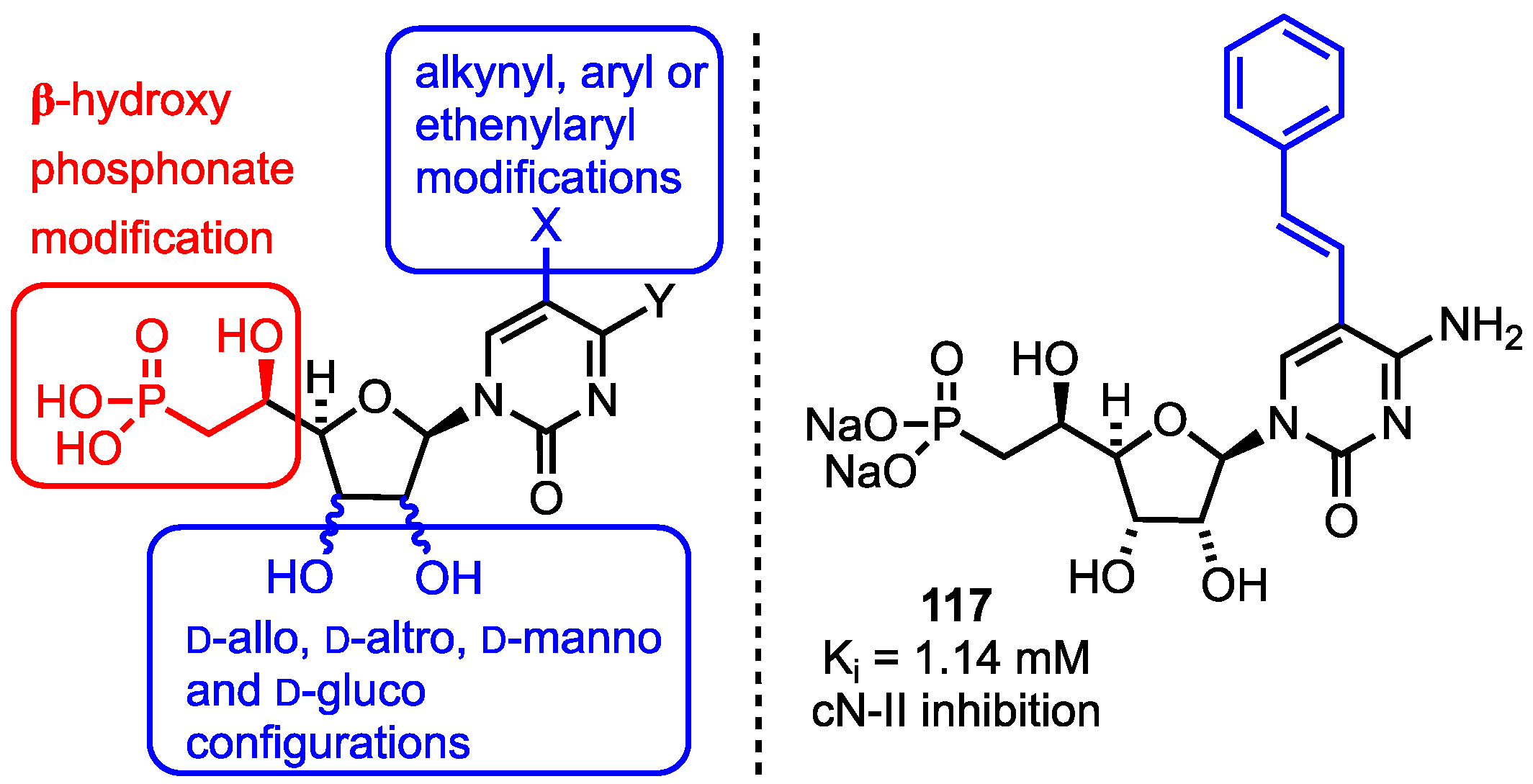
3.5. 5′-β-Hydroxyphosphonate Analogues
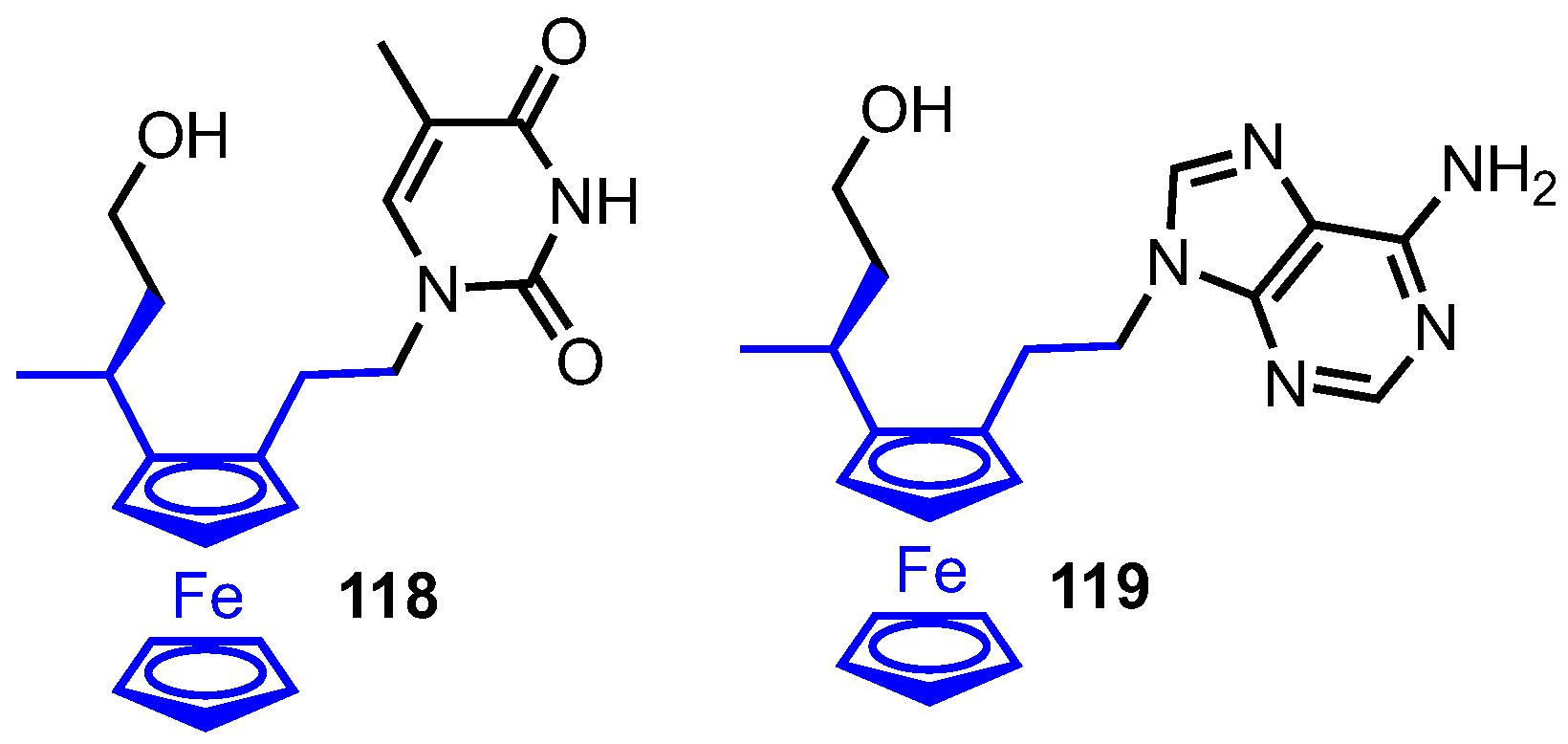
3.6. Ferronucleosides
4. Nucleoside Analogue Prodrugs
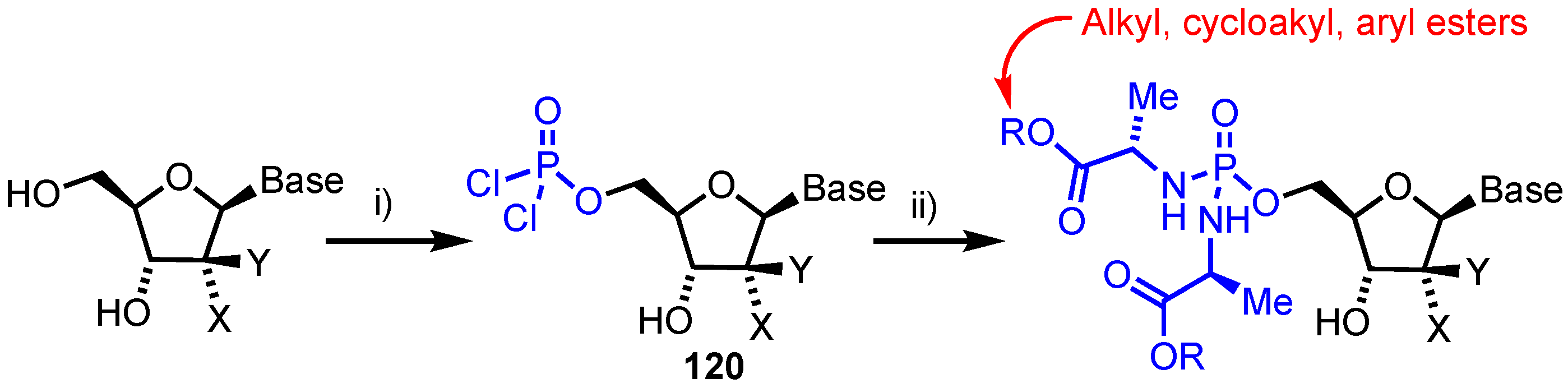
4.1. Phosphorodiamidate Prodrugs
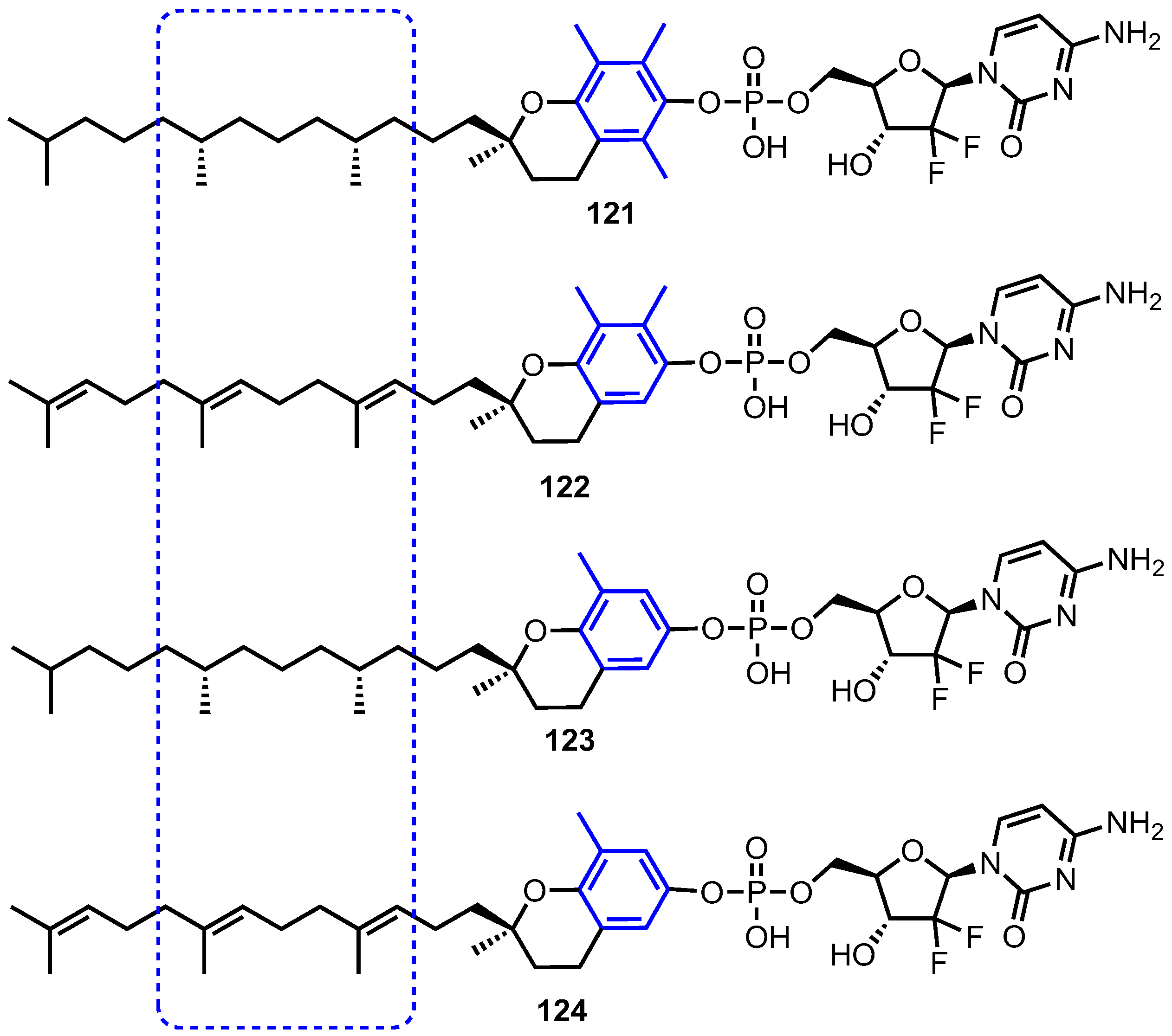
4.2. Vitamin E Phosphate Prodrugs
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Jordheim, L.P.; Durantel, D.; Zoulim, F.; Dumontet, C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug. Discov. 2013, 12, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Clercq, E.D.; Neyts, J. Handbook of Experimental Pharmacology. Handb. Exp. Pharmacol. 2009, 53–84. [Google Scholar]
- Chapman, T.R.; Kinsella, T.J. Ribonucleotide reductase inhibitors: A new look at an old target for radiosensitization. Front. Oncol. 2012, 1, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Flotho, C.; Claus, R.; Batz, C.; Schneider, M.; Sandrock, I.; Ihde, S.; Plass, C.; Niemeyer, C.M.; Lübbert, M. The DNA methyltransferase inhibitors azacitidine, decitabine and zebularine exert differential effects on cancer gene expression in acute myeloid leukemia cells. Leukemia 2009, 23, 1019–1028. [Google Scholar] [CrossRef] [PubMed]
- Galmarini, C.; Mackey, J.; Dumontet, C. Nucleoside analogues: Mechanisms of drug resistance and reversal strategies. Leukemia 2001, 15, 875–890. [Google Scholar] [CrossRef] [PubMed]
- Jordheim, L.; Dumontet, C. Review of recent studies on resistance to cytotoxic deoxynucleoside analogues. Biochim. Biophys. Acta 2007, 1776, 138–159. [Google Scholar] [CrossRef]
- Yates, M.K.; Seley-Radtke, K.L. The evolution of antiviral nucleoside analogues: A review for chemists and non-chemists. Part II: Complex modifications to the nucleoside scaffold. Antivir. Res. 2018, 162, 5–21. [Google Scholar] [CrossRef]
- Seley-Radtke, K.L.; Yates, M.K. The evolution of nucleoside analogue antivirals: A review for chemists and non-chemists. Part 1: Early structural modifications to the nucleoside scaffold. Antivir. Res. 2018, 154, 66–86. [Google Scholar] [CrossRef]
- Siegel, D.; Hui, H.C.; Doerffler, E.; Clarke, M.O.; Chun, K.; Zhang, L.; Neville, S.; Carra, E.; Lew, W.; Ross, B.; et al. Discovery and Synthesis of a Phosphoramidate Prodrug of a Pyrrolo[2,1-f][triazin-4-amino] Adenine C-Nucleoside (GS-5734) for the Treatment of Ebola and Emerging Viruses. J. Med. Chem. 2017, 60, 1648–1661. [Google Scholar] [CrossRef]
- Warren, T.K.; Jordan, R.; Lo, M.K.; Ray, A.S.; Mackman, R.L.; Soloveva, V.; Siegel, D.; Perron, M.; Bannister, R.; Hui, H.C.; et al. Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature 2016, 531, 381–385. [Google Scholar] [CrossRef]
- Sheahan, T.P.; Sims, A.C.; Leist, S.R.; Schäfer, A.; Won, J.; Brown, A.J.; Montgomery, S.A.; Hogg, A.; Babusis, D.; Clarke, M.O.; et al. Comparative therapeutic efficacy of remdesivir and combination lopinavir, ritonavir, and interferon beta against MERS-CoV. Nat. Commun. 2020, 11, 222. [Google Scholar] [CrossRef] [PubMed]
- Cho, A.; Saunders, O.L.; Butler, T.; Zhang, L.; Xu, J.; Vela, J.E.; Feng, J.Y.; Ray, A.S.; Kim, C.U. Synthesis and antiviral activity of a series of 1′-substituted 4-aza-7,9-dideazaadenosine C-nucleosides. Bioorg. Med. Chem. Lett. 2012, 22, 2705–2707. [Google Scholar] [CrossRef] [PubMed]
- Serpi, M.; Ferrari, V.; Pertusati, F. Nucleoside Derived Antibiotics to Fight Microbial Drug Resistance: New Utilities for an Established Class of Drugs? J. Med. Chem. 2016, 59, 10343–10382. [Google Scholar] [CrossRef] [PubMed]
- Romeo, G.; Chiacchio, U.; Corsaro, A.; Merino, P. Chemical Synthesis of Heterocyclic−Sugar Nucleoside Analogues. Chem. Rev. 2010, 110, 3337–3370. [Google Scholar] [CrossRef] [PubMed]
- Roy, B.; Depaix, A.; Peérigaud, C.; Peyrottes, S. Recent Trends in Nucleotide Synthesis. Chem. Rev. 2016, 116, 7854–7897. [Google Scholar] [CrossRef]
- Kicsák, M.; Mándi, A.; Varga, S.; Herczeg, M.; Batta, G.; Bényei, A.; Borbás, A.; Herczegh, P. Tricyclanos: Conformationally constrained nucleoside analogues with a new heterotricycle obtained from a d-ribofuranose unit. Org. Biomol. Chem. 2018, 16, 393–401. [Google Scholar] [CrossRef]
- Hernández, D.; Boto, A. Nucleoside Analogues: Synthesis and Biological Properties of Azanucleoside Derivatives. Eur. J. Org. Chem. 2014, 2014, 2201–2220. [Google Scholar] [CrossRef]
- Quintás-Cardama, A.; Santos, F.P.S.; Garcia-Manero, G. Therapy with azanucleosides for myelodysplastic syndromes. Nat. Rev. Clin. Oncol. 2010, 7, 433–444. [Google Scholar] [CrossRef]
- Bouton, J.; Hecke, K.V.; Calenbergh, S.V. Efficient diastereoselective synthesis of a new class of azanucleosides: 2′-homoazanucleosides. Tetrahedron 2017, 73, 4307–4316. [Google Scholar] [CrossRef]
- Bantia, S.; Miller, P.J.; Parker, C.D.; Ananth, S.L.; Horn, L.L.; Kilpatrick, J.M.; Morris, P.E.; Hutchison, T.L.; Montgomery, J.A.; Sandhu, J.S. Purine nucleoside phosphorylase inhibitor BCX-1777 (Immucillin-H)—A novel potent and orally active immunosuppressive agent. Int. Immunopharmacol. 2001, 1, 1199–1210. [Google Scholar] [CrossRef]
- Miles, R.W.; Tyler, P.C.; Furneaux, R.H.; Bagdassarian, C.K.; Schramm, V.L. One-third-the-sites transition-state inhibitors for purine nucleoside phosphorylase. Biochemistry 1998, 37, 8615–8621. [Google Scholar] [CrossRef] [PubMed]
- Makita, S.; Maeshima, A.M.; Maruyama, D.; Izutsu, K.; Tobinai, K. Forodesine in the treatment of relapsed/refractory peripheral T-cell lymphoma: An evidence-based review. Oncotargets Ther. 2018, 11, 2287–2293. [Google Scholar] [CrossRef] [PubMed]
- Al-Kali, A.; Gandhi, V.; Ayoubi, M.; Keating, M.; Ravindi, F. Forodesine: Review of preclinical and clinical data. Future Oncol. 2010, 6, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Warren, T.K.; Wells, J.; Panchal, R.G.; Stuthman, K.S.; Garza, N.L.; Tongeren, S.A.V.; Dong, L.; Retterer, C.J.; Eaton, B.P.; Pegoraro, G.; et al. Protection against filovirus diseases by a novel broad-spectrum nucleoside analogue BCX4430. Nature 2014, 508, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Evans, G.B.; Furneaux, R.H.; Gainsford, G.J.; Schramm, V.L.; Tyler, P.C. Synthesis of Transition State Analogue Inhibitors for Purine Nucleoside Phosphorylase and N-Riboside Hydrolases. Tetrahedron 2000, 56, 3053–3062. [Google Scholar] [CrossRef]
- Fleet, G.W.J.; Son, J.C. Polyhydroxylated pyrrolidines from sugar lactomes: Synthesis of 1,4-dideoxy-1,4-imino-d-glucitol from d-galactonolactone and syntheses of 1,4-dideoxy-1,4-imino-d-allitol, 1,4-dideoxy-1,4-imino-d-ribitol, and (2s,3r,4s)-3,4-dihydroxyproline from d-gulonolactone. Tetrahedron 1988, 44, 2637–2647. [Google Scholar]
- Kezar, H.S.; Kilpatrick, J.M.; Phillips, D.; Kellogg, D.; Zhang, J.; Morris, P.E. Synthesis and Pharmacokinetic and Pharmacodynamic Evaluation of the Forodesine HCl Analog BCX-3040. Nucleosides Nucleotides Nucleic Acids 2005, 24, 1817–1830. [Google Scholar] [CrossRef]
- Wortmann, R.L.; Andres, C.; Kaminska, J.; Mejias, E.; Gelf, E.; Arnold, W.; Rich, K.; Fox, I.H. Purine Nucleoside Phosphorylase Deficiency. Arthritis Rheum. 1979, 22, 524–531. [Google Scholar] [CrossRef]
- Witczak, Z.J.; Culhane, J.M. Thiosugars: New perspectives regarding availability and potential biochemical and medicinal applications. Appl. Microbiol. Biotechnol. 2005, 69, 237–244. [Google Scholar] [CrossRef]
- Secrist, J.A.; Tiwari, K.N.; Riordan, J.M.; Montgomery, J.A. Synthesis and biological activity of 2’-deoxy-4’-thio pyrimidine nucleosides. J. Med. Chem. 1991, 34, 2361–2366. [Google Scholar] [CrossRef]
- Pejanović, V.; Stokić, Z.; Stojanović, B.; Piperski, V.; Popsavin, M.; Popsavin, V. Synthesis and biological evaluation of some novel 4′-Thio-L-ribonucleosides with modified nucleobase moieties. Bioorg. Med. Chem. Lett. 2003, 13, 1849–1852. [Google Scholar] [CrossRef]
- Zheng, F.; Zhang, X.-H.; Qiu, X.-L.; Zhang, X.; Qing, F.-L. Synthesis of l-β-3‘-Deoxy-3‘,3‘-difluoro-4‘-thionucleosides. Org. Lett. 2006, 8, 6083–6086. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, Y.; Kitano, K.; Yamada, K.; Satoh, H.; Watanabe, M.; Miura, S.; Sakata, S.; Sasaki, T.; Matsuda, A. A Novel Synthesis of 2′-Modified 2′-Deoxy-4′-thiocytidines from D-Glucose. J. Org. Chem. 1997, 62, 3140–3152. [Google Scholar] [CrossRef]
- Yoshimura, Y.; Kitano, K.; Satoh, H.; Watanabe, M.; Miura, S.; Sakata, S.; Sasaki, T.; Matsuda, A. A Novel Synthesis of New Antineoplastic 2‘-Deoxy-2‘-substituted-4‘-thiocytidines. J. Org. Chem. 1996, 61, 822–823. [Google Scholar] [CrossRef]
- Yoshimura, Y.; Saito, Y.; Natori, Y.; Wakamatsu, H. Synthesis of 4′-Thionucleosides as Antitumor and Antiviral Agents. Chem. Pharm. Bull. 2018, 66, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, Y.; Kano, F.; Miyazaki, S.; Ashida, N.; Sakata, S.; Haraguchi, K.; Itoh, Y.; Tanaka, H.; Miyasaka, T. Synthesis and biological evaluation of 1′-C-cyano-pyrimidine nucleosides. Nucleosides Nucleotides 1996, 15, 305–324. [Google Scholar] [CrossRef]
- Thottassery, J.V.; Sambandam, V.; Allan, P.W.; Maddry, J.A.; Maxuitenko, Y.Y.; Tiwari, K.; Hollingshead, M.; Parker, W.B. Novel DNA methyltransferase-1 (DNMT1) depleting anticancer nucleosides, 4′-thio-2′-deoxycytidine and 5-aza-4′-thio-2′-deoxycytidine. Cancer Chemother. Pharmacol. 2014, 74, 291–302. [Google Scholar] [CrossRef]
- Haraguchi, K.; Kumamoto, H.; Konno, K.; Yagi, H.; Tatano, Y.; Odanaka, Y.; Matsubayashi, S.S.; Snoeck, R.; Andrei, G. Synthesis of 4′-substituted 2′-deoxy-4′-thiocytidines and its evaluation for antineoplastic and antiviral activities. Tetrahedron 2019, 75, 4542–4555. [Google Scholar] [CrossRef]
- Haraguchi, K.; Takahashi, H.; Shiina, N.; Horii, C.; Yoshimura, Y.; Nishikawa, A.; Sasakura, E.; Nakamura, K.T.; Tanaka, H. Stereoselective Synthesis of the β-Anomer of 4‘-Thionucleosides Based on Electrophilic Glycosidation to 4-Thiofuranoid Glycals. J. Org. Chem. 2002, 67, 5919–5927. [Google Scholar] [CrossRef]
- Jayakanthan, K.; Johnston, B.D.; Pinto, B.M. Stereoselective synthesis of 4′-selenonucleosides using the Pummerer glycosylation reaction. Carbohydr. Res. 2008, 343, 1790–1800. [Google Scholar] [CrossRef]
- Haraguchi, K.; Shimada, H.; Kimura, K.; Akutsu, G.; Tanaka, H.; Abe, H.; Hamasaki, T.; Baba, M.; Gullen, E.A.; Dutschman, G.E.; et al. Synthesis of 4′-Ethynyl-2′-deoxy-4′-thioribonucleosides and Discovery of a Highly Potent and Less Toxic NRTI. ACS. Med. Chem. Lett. 2011, 2, 692–697. [Google Scholar] [CrossRef] [PubMed]
- Jeong, L.S.; Tosh, D.K.; Kim, H.O.; Wang, T.; Hou, X.; Yun, H.S.; Kwon, Y.; Lee, S.K.; Choi, J.; Zhao, L.X. First Synthesis of 4‘-Selenonucleosides Showing Unusual Southern Conformation. Org. Lett. 2008, 10, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Varela, O.; Zunszain, P.A. First synthesis of aldopentono-1,4-thiolactones. J. Org. Chem. 1993, 58, 7860–7864. [Google Scholar] [CrossRef]
- Jeong, L.S.; Tosh, D.K.; Choi, W.J.; Lee, S.K.; Kang, Y.-J.; Choi, S.; Lee, J.H.; Lee, H.; Lee, H.W.; Kim, H.O. Discovery of a New Template for Anticancer Agents: 2′-deoxy-2′-fluoro-4′-selenoarabinofuranosyl-cytosine (2′-F-4′-Seleno-ara-C). J. Med. Chem. 2009, 52, 5303–5306. [Google Scholar] [CrossRef]
- Kim, J.-H.; Yu, J.; Alexander, V.; Choi, J.H.; Song, J.; Lee, H.W.; Kim, H.O.; Choi, J.; Lee, S.K.; Jeong, L.S. Structure–activity relationships of 2′-modified-4′-selenoarabinofuranosyl-pyrimidines as anticancer agents. Eur. J. Med. Chem. 2014, 83, 208–225. [Google Scholar] [CrossRef] [PubMed]
- Alexander, V.; Song, J.; Yu, J.; Choi, J.H.; Kim, J.-H.; Lee, S.K.; Choi, W.J.; Jeong, L.S. Synthesis and biological evaluation of 2’-substituted-4’-selenoribofuranosyl pyrimidines as antitumor agents. Arch. Pharm. Sci. Res. 2014, 38, 966–972. [Google Scholar] [CrossRef]
- Marquez, V.E. Advances in Antiviral Drug Design. In Advances in Antiviral Drug Design; Clercq, E.D., Ed.; Elsevier: Amsterdam, The Netherlands, 1996; pp. 89–146. [Google Scholar]
- Marquez, V.E.; Lim, M.I.; Treanor, S.P.; Plowman, J.; Priest, M.A.; Markovac, A.; Khan, M.S.; Kaskar, B.; Driscoll, J.S. Cyclopentenylcytosine. A carbocyclic nucleoside with antitumor and antiviral properties. J. Med. Chem. 1988, 31, 1687–1694. [Google Scholar] [CrossRef]
- Cavaliere, A.; Probst, K.C.; Westwell, A.D.; Slusarczyk, M. Fluorinated nucleosides as an important class of anticancer and antiviral agents. Future Med. Chem. 2017, 9, 1809–1833. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, X.; Wang, Q.; Zhang, Y.; Jiang, J.; Guo, X.; Fan, Q.; Zheng, L.; Yu, X.; Wang, N.; et al. FNC, a novel nucleoside analogue inhibits cell proliferation and tumor growth in a variety of human cancer cells. Biochem. Pharmacol. 2011, 81, 848–855. [Google Scholar] [CrossRef]
- Jeong, L.S.; Zhao, L.X.; Choi, W.J.; Pal, S.; Park, Y.H.; Lee, S.K.; Chun, M.W.; Lee, Y.B.; Ahn, C.H.; Moon, H.R. Synthesis and Antitumor Activity of Fluorocyclopentenyl-Pyrimidines. Nucleosides Nucleotides Nucleic Acids 2007, 26, 713–716. [Google Scholar] [CrossRef]
- Moon, H.R.; Choi, W.J.; Kim, H.O.; Jeong, L.S. Improved and alternative synthesis of d- and l-cyclopentenone derivatives, the versatile intermediates for the synthesis of carbocyclic nucleosides. Tet. Asymm. 2002, 13, 1189–1193. [Google Scholar] [CrossRef]
- Choi, W.J.; Moon, H.R.; Kim, H.O.; Yoo, B.N.; Lee, J.A.; Shin, D.H.; Jeong, L.S. Preparative and Stereoselective Synthesis of the Versatile Intermediate for Carbocyclic Nucleosides: Effects of the Bulky Protecting Groups to Enforce Facial Selectivity. J. Org. Chem. 2004, 69, 2634–2636. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.J.; Chung, H.-J.; Chandra, G.; Alexander, V.; Zhao, L.X.; Lee, H.W.; Nayak, A.; Majik, M.S.; Kim, H.O.; Kim, J.-H.; et al. Fluorocyclopentenyl-cytosine with Broad Spectrum and Potent Antitumor Activity. J. Med. Chem. 2012, 55, 4521–4525. [Google Scholar] [CrossRef] [PubMed]
- Balboni, B.; Hassouni, B.E.; Honeywell, R.J.; Sarkisjan, D.; Giovannetti, E.; Poore, J.; Heaton, C.; Peterson, C.; Benaim, E.; Lee, Y.B.; et al. RX-3117 (fluorocyclopentenyl cytosine): A novel specific antimetabolite for selective cancer treatment. Expert. Opin. Inv. Drug. 2019, 28, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Peters, G.J.; Smid, K.; Vecchi, L.; Kathmann, I.; Sarkisjan, D.; Honeywell, R.J.; Losekoot, N.; Ohne, O.; Orbach, A.; Blaugrund, E.; et al. Metabolism, mechanism of action and sensitivity profile of fluorocyclopentenylcytosine (RX-3117; TV-1360). Investig. New Drugs 2013, 31, 1444–1457. [Google Scholar] [CrossRef]
- Yang, M.Y.; Lee, Y.B.; Ahn, C.-H.; Kaye, J.; Fine, T.; Kashi, R.; Ohne, O.; Smid, K.; Peters, G.J.; Kim, D.J. A novel cytidine analog, RX-3117, shows potent efficacy in xenograft models, even in tumors that are resistant to gemcitabine. Anticancer Res. 2014, 34, 6951–6959. [Google Scholar]
- Udvaros, I.; Rethy, A.; Lang, I.; Hitre, E.; Peterson, C. A phase 1 exploratory study of RX-3117 to determine oral bioavailability in cancer subjects with solid tumors. J. Clin. Oncol. 2015, 33, e13545. [Google Scholar] [CrossRef]
- Dejmek, M.; Šála, M.; Hřebabecký, H.; Dračínský, M.; Procházková, E.; Chalupská, D.; Klíma, M.; Plačková, P.; Hájek, M.; Andrei, G.; et al. Norbornane-based nucleoside and nucleotide analogues locked in North conformation. Bioorg. Med. Chem. 2015, 23, 184–191. [Google Scholar] [CrossRef]
- Marquez, V.E.; Siddiqui, M.A.; Ezzitouni, A.; Russ, P.; Wang, J.; Wagner, R.W.; Matteucci, M.D. Nucleosides with a Twist. Can Fixed Forms of Sugar Ring Pucker Influence Biological Activity in Nucleosides and Oligonucleotides? J. Med. Chem. 1996, 39, 3739–3747. [Google Scholar] [CrossRef]
- Tănase, C.I.; Drăghici, C.; Căproiu, M.T.; Shova, S.; Mathe, C.; Cocu, F.G.; Enache, C.; Maganu, M. New carbocyclic nucleoside analogues with a bicyclo[2.2.1]heptane fragment as sugar moiety; Synthesis, X-ray crystallography and anticancer activity. Bioorg. Med. Chem. 2014, 22, 513–522. [Google Scholar] [CrossRef]
- Tănase, C.I.; Drăghici, C.; Cojocaru, A.; Galochkina, A.V.; Orshanskaya, J.R.; Zarubaev, V.V.; Shova, S.; Enache, C.; Maganu, M. New carbocyclic N6-substituted adenine and pyrimidine nucleoside analogues with a bicyclo[2.2.1]heptane fragment as sugar moiety; synthesis, antiviral, anticancer activity and X-ray crystallography. Bioorg. Med. Chem. 2015, 23, 6346–6354. [Google Scholar] [CrossRef] [PubMed]
- Waud, W.R.; Schmid, S.M.; Montgomery, J.A.; Secrist, J.A. Preclinical Antitumor Activity of 2-Chloro-9-(2-deoxy-2-fluoro-β-D-arabinofuranosyl)adenine (C1-F-Ara-A). Nucleosides Nucleotides Nucleic Acids 2000, 19, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Albertioni, F.; Lindemalm, S.; Reichelova, V.; Pettersson, B.; Eriksson, S.; Juliusson, G.; Liliemark, J. Pharmacokinetics of cladribine in plasma and its 5’-monophosphate and 5’-triphosphate in leukemic cells of patients with chronic lymphocytic leukemia. Clin. Cancer Res. 1998, 4, 653–658. [Google Scholar] [PubMed]
- Pui, C.H.; Jeda, S.; Kirkpatrick, P. Clofarabine. Nat. Rev. Drug. Discov. 2005, 4, 369–370. [Google Scholar] [CrossRef] [PubMed]
- Cen, Y.; Sauve, A.A. Efficient syntheses of clofarabine and gemcitabine from 2-deoxyribonolactone. Nucleosides Nucleotides Nucleic Acids 2010, 29, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Ellery, S.P.; Rivas, F.; Saye, K.; Rogers, E.; Workinger, T.J.; Schallenberger, M.; Tawatao, R.; Montero, A.; Hessell, A.; et al. Synthesis and biological evaluation of 2′,4′- and 3′,4′-bridged nucleoside analogues. Bioorg. Med. Chem. 2011, 19, 5648–5669. [Google Scholar] [CrossRef][Green Version]
- Obika, S.; Morio, K.; Nanbu, D.; Hari, Y.; Itoh, H.; Imanishi, T. Synthesis and conformation of 3′,4′-BNA monomers, 3′-O,4′-C-methyleneribonucleosides. Tetrahedron 2002, 58, 3039–3049. [Google Scholar] [CrossRef]
- Youssefyeh, R.D.; Verheyden, J.P.H.; Moffatt, J.G. 4′-Substituted nucleosides. 4. Synthesis of some 4′-hydroxymethyl nucleosides. J. Org. Chem. 1979, 44, 1301–1309. [Google Scholar] [CrossRef]
- Kim, B.J.; Zhang, J.; Tan, S.; Matteson, D.S.; Prusoff, W.H.; Cheng, Y.-C. Synthesis and properties of 1-(3′-dihydroxyboryl-2′,3′-dideoxyribosyl)pyrimidines. Org. Biomol. Chem. 2012, 10, 9349. [Google Scholar] [CrossRef]
- Bege, M.; Kiss, A.; Kicsák, M.; Bereczki, I.; Baksa, V.; Király, G.; Szemán-Nagy, G.; Szigeti, M.Z.; Herczegh, P.; Borbás, A. Synthesis and Cytostatic Effect of 3’-deoxy-3’-C-Sulfanylmethyl Nucleoside Derivatives with d-xylo Configuration. Molecules 2019, 24, 2173. [Google Scholar] [CrossRef]
- Mohan, A.A.; Sharma, G.V.R.; Vidavalur, S. Synthesis, characterization and biological evaluation of C5’-N-cyclopropylcarboxamido-C6-amino-C2-alkynylated purine nucleoside analogues. Nucleosides Nucleotides Nucleic Acids 2017, 36, 1–15. [Google Scholar]
- Meurillon, M.; Marton, Z.; Hospital, A.; Jordheim, L.P.; Béjaud, J.; Lionne, C.; Dumontet, C.; Périgaud, C.; Chaloin, L.; Peyrottes, S. Structure–activity relationships of β-hydroxyphosphonate nucleoside analogues as cytosolic 5′-nucleotidase II potential inhibitors: Synthesis, in vitro evaluation and molecular modeling studies. Eur. J. Med. Chem. 2014, 77, 18–37. [Google Scholar] [CrossRef] [PubMed]
- Gallier, F.; Lallemand, P.; Meurillon, M.; Jordheim, L.P.; Dumontet, C.; Périgaud, C.; Lionne, C.; Peyrottes, S.; Chaloin, L. Structural Insights into the Inhibition of Cytosolic 5′-Nucleotidase II (cN-II) by Ribonucleoside 5′-Monophosphate Analogues. PLoS Comput. Biol. 2011, 7, e1002295. [Google Scholar] [CrossRef] [PubMed]
- Galmarini, C.M.; Thomas, X.; Graham, K.; Jafaari, A.E.; Cros, E.; Jordheim, L.; Mackey, J.R.; Dumontet, C. Deoxycytidine kinase and cN-II nucleotidase expression in blast cells predict survival in acute myeloid leukaemia patients treated with cytarabine. Br. J. Haematol. 2003, 122, 53–60. [Google Scholar] [CrossRef]
- Nguyen, H.V.; Sallustrau, A.; Balzarini, J.; Bedford, M.R.; Eden, J.C.; Georgousi, N.; Hodges, N.J.; Kedge, J.; Mehellou, Y.; Tselepis, C.; et al. Organometallic nucleoside analogues with ferrocenyl linker groups: Synthesis and cancer cell line studies. J. Med. Chem. 2014, 57, 5817–5822. [Google Scholar] [CrossRef]
- Slusarczyk, M.; Ferrari, V.; Serpi, M.; Gönczy, B.; Balzarini, J.; McGuigan, C. Symmetrical Diamidates as a Class of Phosphate Prodrugs to Deliver the 5′-Monophosphate Forms of Anticancer Nucleoside Analogues. ChemMedChem 2018, 13, 2305–2316. [Google Scholar] [CrossRef]
- Cahard, D.; McGuigan, C.; Balzarini, J. Aryloxy phosphoramidate triesters as pro-tides. Mini-Rev. Med. Chem. 2004, 4, 371–381. [Google Scholar] [CrossRef]
- Liu, W.; Zhang, L.; Zhou, H.; Yang, C.; Miao, Z.; Zhao, Y. Synthesis of Novel Nucleoside Analogue Phosphorothioamidate Prodrugs and in vitro Anticancer Evaluation Against RKO Human Colon Carcinoma Cells. Nucleosides Nucleotides Nucleic Acids 2013, 32, 161–173. [Google Scholar] [CrossRef]
- Sofia, M.J.; Bao, D.; Chang, W.; Du, J.; Nagarathnam, D.; Rachakonda, S.; Reddy, P.G.; Ross, B.S.; Wang, P.; Zhang, H.-R.; et al. Discovery of a β- d -2′-Deoxy-2′-α-fluoro-2′-β- C -methyluridine Nucleotide Prodrug (PSI-7977) for the Treatment of Hepatitis C Virus. J. Med. Chem. 2010, 53, 7202–7218. [Google Scholar] [CrossRef]
- Mehellou, Y.; Valente, R.; Mottram, H.; Walsby, E.; Mills, K.I.; Balzarini, J.; McGuigan, C. Phosphoramidates of 2’-beta-D-arabinouridine (AraU) as phosphate prodrugs; design, synthesis, in vitro activity and metabolism. Bioorg. Med. Chem. 2010, 18, 2439–2446. [Google Scholar] [CrossRef]
- Jordheim, L.P.; Galmarini, C.M.; Dumontet, C. Gemcitabine resistance due to deoxycytidine kinase deficiency can be reverted by fruitfly deoxynucleoside kinase, DmdNK, in human uterine sarcoma cells. Cancer Chemother. Pharmacol. 2006, 58, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Damaraju, V.L.; Damaraju, S.; Young, J.D.; Baldwin, S.A.; Mackey, J.; Sawyer, M.B.; Cass, C.E. Nucleoside anticancer drugs: The role of nucleoside transporters in resistance to cancer chemotherapy. Oncogene 2003, 22, 7524–7536. [Google Scholar] [CrossRef] [PubMed]
- Daifuku, R.; Koratich, M.; Stackhouse, M. Vitamin E Phosphate Nucleoside Prodrugs: A Platform for Intracellular Delivery of Monophosphorylated Nucleosides. Pharmaceuticals 2018, 11, 16. [Google Scholar] [CrossRef] [PubMed]
- Shipley, L.A.; Brown, T.J.; Cornpropst, J.D.; Hamilton, M.; Daniels, W.D.; Culp, H.W. Metabolism and disposition of gemcitabine, an oncolytic deoxycytidine analog, in mice, rats, and dogs. Drug Metab. Dispos. 1992, 20, 849–855. [Google Scholar] [PubMed]





























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antineoplastic Activities IC50 (µg/mL) | |||
|---|---|---|---|
| Compound (Anomer) | 2′-Substituent | CCRF-HSB-2 a | KB Cells b |
| 28α | CH2 | >10 | ND c |
| 28β | CH2 | 0.01 | 0.12 |
| 33α | F (arabino) | >10 | ND |
| 33β | F (arabino) | 0.05 | 0.02 |
| 37α | F2 | >10 | ND |
| 37β | F2 | 1.5 | 17 |
| Ara-C 3 | 0.05 | 0.26 | |
| DMDC | 0.02 | 0.44 | |
| IC50 (μM) | ||||||
|---|---|---|---|---|---|---|
| Compound | HCT116 a | A549 b | SNU638 c | T47D d | PC-3 e | K562 f |
| 65 | 1.1 | 0.47 | 0.14 | 0.79 | 0.58 | 0.63 |
| 66 | 7.13 | 8.83 | 4.72 | ND | ND | 86.6 |
| 67 | >100 | >100 | >100 | >100 | >100 | >100 |
| 68 | >100 | >100 | >100 | >100 | >100 | >100 |
| 3 | 5.30 | 1.90 | 0.15 | 2.70 | 55.9 | 0.05 |
| 1 | 0.01 | 0.09 | ND | ND | 0.04 | ND |
| Cancer Cell Line | |||||||
|---|---|---|---|---|---|---|---|
| HTC-116 a | MDA-MB-231 b | PANC-1 c | MCF-7 d | A549 e | MKN45 f | U251 g | |
| IC50 (μM) | 0.39 | 0.18 | 0.62 | 0.34 | 0.34 | 0.50 | 0.83 |
| Dose (mg) | Tmax (h) | Cmax (ng/mL) | t1/2 (h) | Oral Bioavailability (%) |
|---|---|---|---|---|
| 20 * | 0.3 | 1144 | - | - |
| 50 | 2.2 | 303 | 14 | 56 |
| 100 | 2.5 | 311 | 21 | 33 |
| Cancer Cell Line | ||||||
|---|---|---|---|---|---|---|
| Breast MDA-MB-231 (μM) | Non-Small Cell Lung NCI-H460 (μM) | Colon HCT-116 (μM) | ||||
| Compound | DP (−) | DP (20 μM) | DP (−) | DP (20 μM) | DP (−) | DP (20 μM) |
| 1 | 3.08 | 56.8 | 0.02 | 0.82 | 0.03 | 2.39 |
| 122 | 30.3 | 27.8 | 7.16 | 16.0 | 5.55 | 12.6 |
| 123 | 17.2 | 23.3 | 2.14 | 1.47 | 3.07 | 6.74 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guinan, M.; Benckendorff, C.; Smith, M.; Miller, G.J. Recent Advances in the Chemical Synthesis and Evaluation of Anticancer Nucleoside Analogues. Molecules 2020, 25, 2050. https://doi.org/10.3390/molecules25092050
Guinan M, Benckendorff C, Smith M, Miller GJ. Recent Advances in the Chemical Synthesis and Evaluation of Anticancer Nucleoside Analogues. Molecules. 2020; 25(9):2050. https://doi.org/10.3390/molecules25092050
Chicago/Turabian StyleGuinan, Mieke, Caecilie Benckendorff, Mark Smith, and Gavin J. Miller. 2020. "Recent Advances in the Chemical Synthesis and Evaluation of Anticancer Nucleoside Analogues" Molecules 25, no. 9: 2050. https://doi.org/10.3390/molecules25092050
APA StyleGuinan, M., Benckendorff, C., Smith, M., & Miller, G. J. (2020). Recent Advances in the Chemical Synthesis and Evaluation of Anticancer Nucleoside Analogues. Molecules, 25(9), 2050. https://doi.org/10.3390/molecules25092050