
Synthesis, Electrochemical and Spectroscopic Characterization of Selected Quinolinecarbaldehydes and Their Schiff Base Derivatives

 ,
,  , , , and
, , , and
Abstract
:1. Introduction
2. Results and Discussion
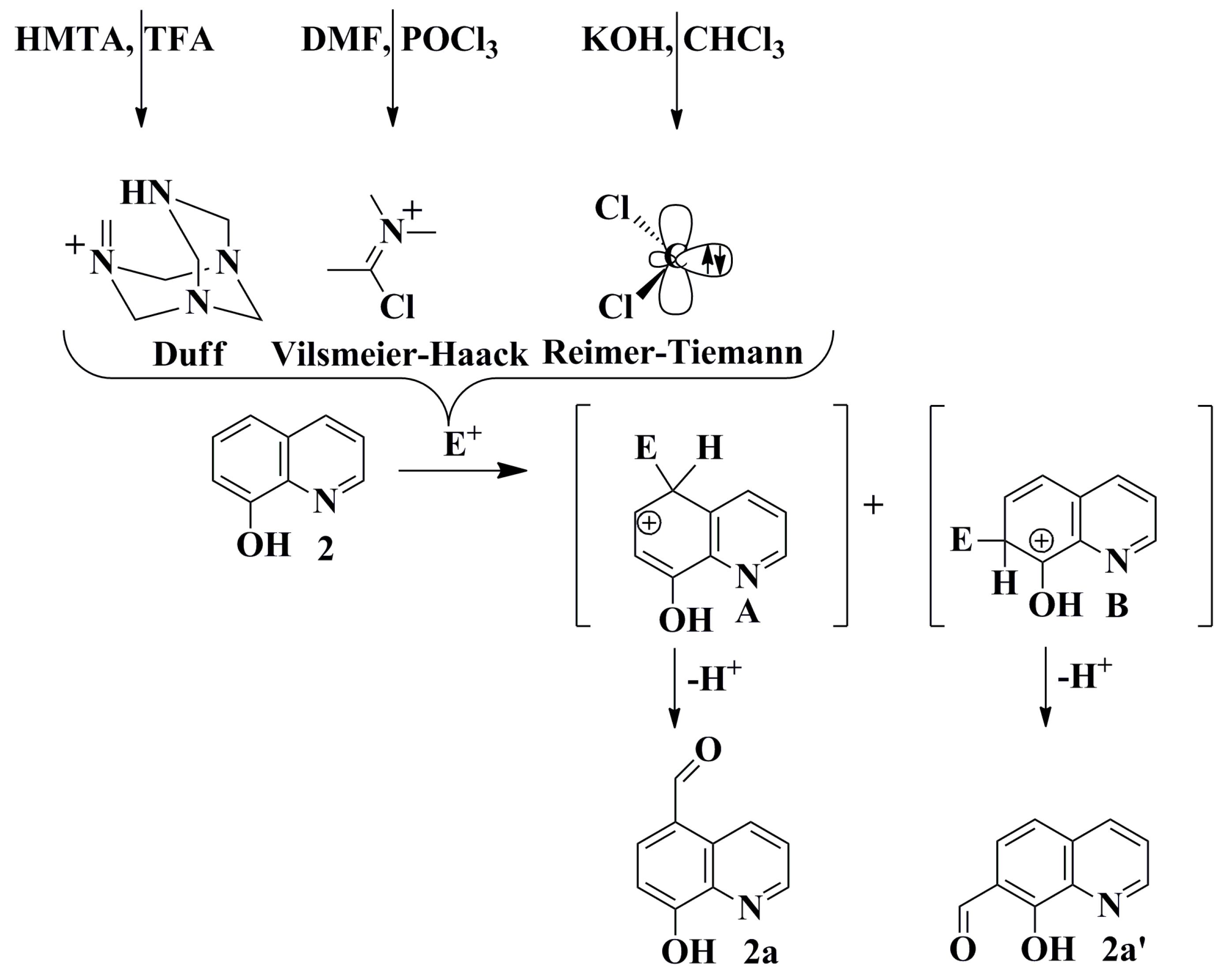
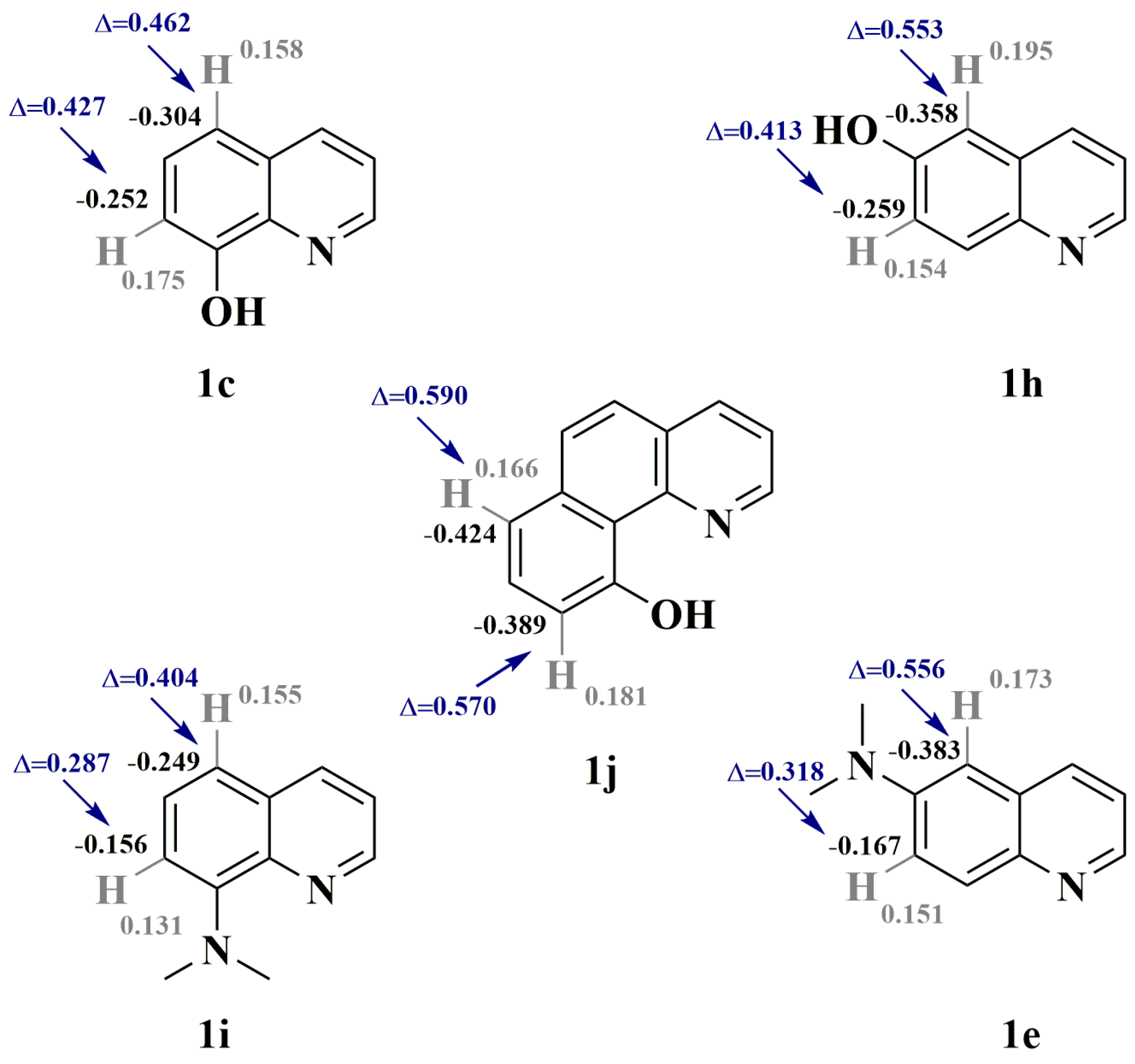
2.1. Synthesis and Structural Characterization
2.2. Crystal Structure Determination and Refinement
2.3. X-ray Studies
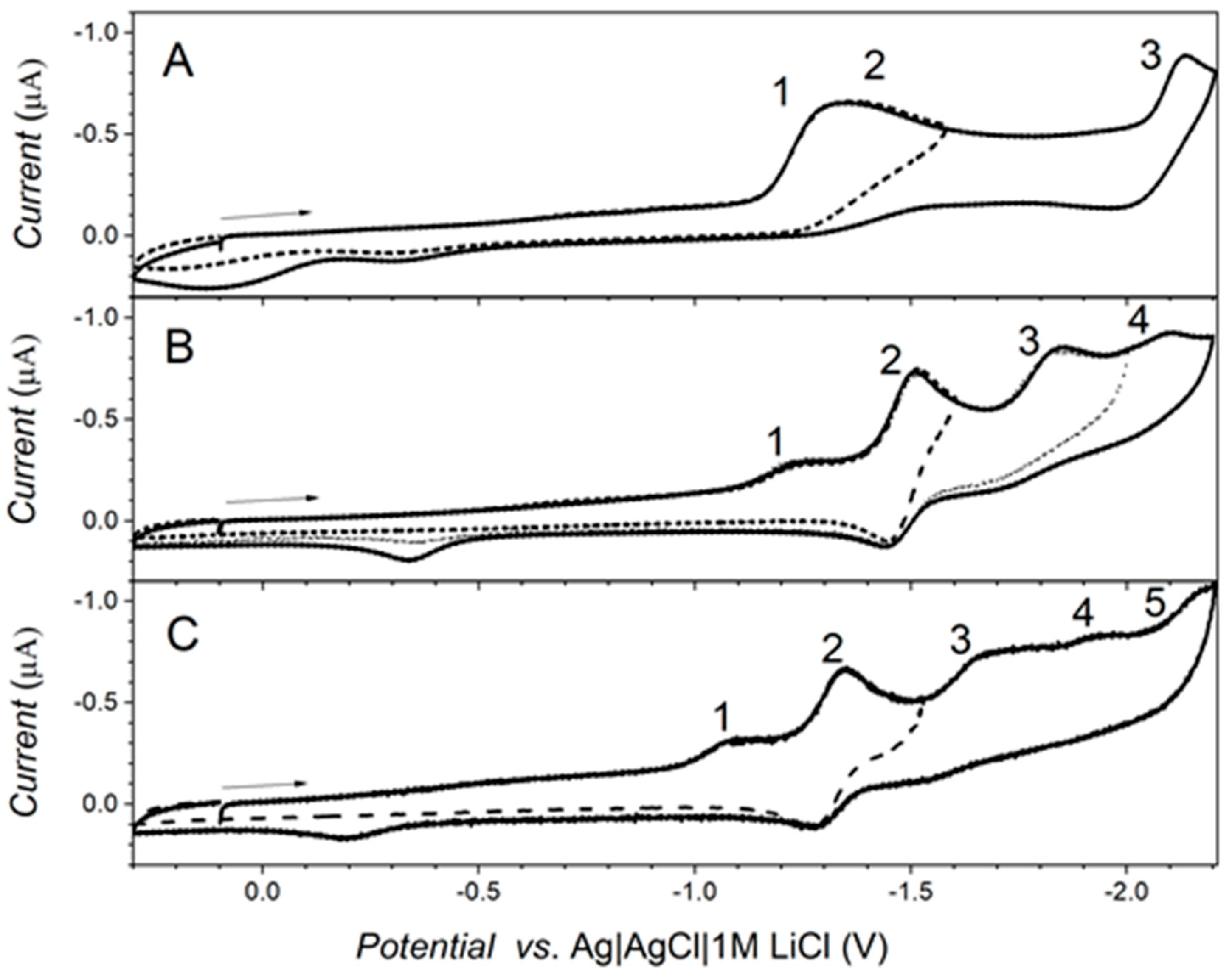
2.4. Electrochemical Measurements
3. Materials and Methods
3.1. Materials
3.2. Instrumentation
3.3. Electrochemical Measurements
3.4. Theoretical Calculations
3.5. General Procedures for Synthesis of Selected Quinolinecarbaldehydes Based on the Reimer-Tiemann Protocol
3.6. Synthesis of 7-Bromo-8-hydroxy-2-methylquinoline-5-carbaldehyde through a Carbene Insertion Reaction
3.7. General Procedures for Synthesis of Selected Quinoline-5-carbaldehydes Based on Vilsmeier-Haack Protocol
3.8. General Procedures for the Synthesis of Selected Quinolinecarbaldehydes Based on the Duff Protocol
3.9. General Procedure the for Synthesis of Selected Schiff Base Derivatives of 2,6-Diisopropylbenzenamine
3.10. Crystallization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wynberg, H. The Reimer-Tiemann reaction. Chem. Rev. 1960, 169–184. [Google Scholar] [CrossRef]
- González-Vera, J.A.; Luković, E.; Imperiali, B. Synthesis of Red-Shifted 8-Hydroxyquinoline Derivatives Using Click Chemistry and Their Incorporation into Phosphorylation Chemosensors. J. Org. Chem. 2009, 74, 7309–7314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Wang, H.; Wan, L.; Bian, Y.; Jiang, J. 8-Hydroxyquinoline-Substituted Boron-Dipyrromethene Compounds: Synthesis, Structure, and OFF-ON-OFF Type of pH-Sensing Properties. J. Org. Chem. 2011, 76, 3774–3781. [Google Scholar] [CrossRef]
- Ding, X.; Zhang, F.; Bai, Y.; Zhao, J.; Chen, X.; Ge, M.; Sun, W. Quinoline-based highly selective and sensitive fluorescent probe specific for Cd2+ detection in mixed aqueous media. Tetrahedron Lett. 2017, 58, 3868–3874. [Google Scholar] [CrossRef]
- Nycz, J.E.; Malecki, G.J. Synthesis, spectroscopy and computational studies of selected hydroxyquinoline carboxylic acids and their selected fluoro-, thio-, and dithioanalogues. J. Mol. Struct. 2013, 1032, 159–168. [Google Scholar] [CrossRef]
- Szala, M.; Nycz, J.E.; Malecki, G.; Sokolova, R.; Ramesova, S.; Switlicka-Olszewska, A.; Strzelczyk, R.; Podsiadly, R.; Machura, B. Synthesis of 5-azo-8-hydroxy-2-methylquinoline dyes and relevant spectroscopic, electrochemical and computational studies. Dyes Pigm. 2017, 142, 277–292. [Google Scholar] [CrossRef]
- Ogata, Y.; Kawasaki, A.; Suguria, F. Kinetics and mechanism of the Duff reaction. Tetrahedron 1968, 24, 5001–5010. [Google Scholar] [CrossRef]
- Lu, Z.-N.; Wang, L.; Zhang, X.; Zhu, Z.-J. A selective fluorescent chemosensor for Cd2+ based on 8-hydroxylquinoline-benzothiazole conjugate and imaging application. Spectrochim. Acta A 2019, 213, 57–63. [Google Scholar] [CrossRef]
- Mahajan, S.S.; Scian, M.; Sripathy, S.; Posakony, J.; Lao, U.; Loe, T.K.; Leko, V.; Thalhofer, A.; Schuler, A.D.; Bedalov, A.; et al. Development of Pyrazolone and Isoxazol-5-one Cambinol Analogues as Sirtuin Inhibitors. J. Med. Chem. 2014, 57, 3283–3294. [Google Scholar] [CrossRef]
- Olah, G.A. Mechanism of Electrophilic Aromatic Substitutions1. Accounts Chem. Res. 1971, 4, 240–248. [Google Scholar] [CrossRef]
- DeAngelis, F.; Inesi, A.; Feroci, M.; Nicoletti, R. Reaction of electrogenerated dichlorocarbene with methylindoles. J. Org. Chem. 1995, 60, 445–447. [Google Scholar] [CrossRef]
- Nandhakumar, R.; Suresh, T.; Calistus Jude, A.L.; Kannan, V.R.; Mohan, P.S. Synthesis, antimicrobial activities and cytogenetic studies of newer diazepino quinoline derivatives via Vilsmeier-Haack reaction. Eur. J. Med. Chem. 2007, 42, 1128–1136. [Google Scholar] [CrossRef] [PubMed]
- Bollyn, M. Thermal Hazards of the Vilsmeier-Haack Reaction on N,N-Dimethylaniline. Org. Process Res. Dev. 2005, 9, 982–996. [Google Scholar] [CrossRef]
- Hitchcock, P.B.; Lappert, M.F.; Nycz, J.E. Synthesis, structure and reductive dechlorination of the C-centred phosphorus(III) b-diketiminate PCl(Ph)L [L = C{C(Me)NC6H3Pr2i-2,6}{C(Me)NHC(6)H(3)Pr(i)2-2,6}]. Chem. Commun. 2003, 10, 1142–1143. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [Google Scholar]
- Lund, H.; Hammerich, O. Organic Electrochemistry, 4th ed.; Marcel Dekker, Inc.: New York, NY, USA, 2001. [Google Scholar]
- Sokolova, R.; Hromadova, M.; Ludvik, J.; Pospisil, L.; Giannarelli, S. The Autoprotonation in Reduction Mechanism of Pesticide Ioxynil. Electrochim. Acta 2010, 55, 8336–8340. [Google Scholar] [CrossRef]
- Sokolova, R.; Gal, M.; Valasek, M. New Proton Donors in Electrochemical Mechanistic Studies in Non-Aqueous Solution Dimethylsulfoxide: Chlorinated Hydroxybenzonitriles. J. Electroanal. Chem. 2012, 685, 33–36. [Google Scholar] [CrossRef]
- Smith, P.J.; Mann, C.K. Electrochemical dealkylation of aliphatic amines. J. Org. Chem. 1969, 34, 1821–1826. [Google Scholar] [CrossRef]
- Nycz, J.E.; Szala, M.; Malecki, G.J.; Nowak, M.; Kusz, J. Synthesis, spectroscopy and computational studies of selected hydroxyquinolines and their analogues. Spectrochim Acta A 2014, 117, 351–359. [Google Scholar] [CrossRef]
- Petit, M.; Tran, C.; Roger, T.; Gallavardin, T.; Dhimane, H.; Palma-Cerda, F.; Blanchard-Desce, M.; Acher, F.C.; Ogden, D.; Dalko, P.I. Substitution Effect on the One- and Two-photon Sensitivity of DMAQ “Caging” Groups. Org. Lett. 2012, 14, 6366–6369. [Google Scholar] [CrossRef]
- Wang, D.; Kuang, D.; Zhang, F.; Yang, C.; Zhu, X. Room-Temperature Copper-Catalyzed Arylation of Dimethylamine and Methylamine in Neat Water. Adv. Synth. Catal. 2015, 357, 714–718. [Google Scholar] [CrossRef]
- Whiteoak, C.J.; Planas, O.; Company, A.; Ribas, X. A First Example of Cobalt-Catalyzed Remote C–H Functionalization of 8-Aminoquinolines Operating through a Single Electron Transfer Mechanism. Adv. Synth. Catal. 2016, 358, 1679–1688. [Google Scholar] [CrossRef]
- Gąsiorowska, M.; Typek, J.; Soroka, J.A.; Sawicka, M.J.; Wróblewska, E.K.; Guskos, N.; Żołnierkiewicz, G. Spectroscopic and magnetic properties of solvatochromic complex of Cu2+ and novel 3H-indolium derivative. Spectrochim. Acta A 2014, 124, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Fiedler, H. Synthesis of methyl-8-hydroxyquinoline aldehydes. Arch. Pharm. 1960, 293, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Voloshin, N.A.; Chernyshev, A.V.; Bezuglyi, S.O.; Metelitsa, A.V.; Voloshina, E.N.; Minkin, V.I. Spiropyrans and spirooxazines 4. Synthesis and spectral properties of 6 ‘-halo-substituted spiro[indoline-2,2’-2H-pyrano [3,2-h]quinolines]. Russ. Chem. Bull. Int. Ed. 2008, 57, 151–158. [Google Scholar] [CrossRef]
- Skonieczny, K.; Charalambidis, G.; Tasior, M.; Krzeszewski, M.; Kalkan-Burat, A.; Coutsolelos, A.G.; Gryko, D.T. General and Efficient Protocol for Formylation of Aromatic and Heterocyclic Phenols. Synthesis-Stuttgart 2012, 44, 3683–3687. [Google Scholar]
- Hansen, P.E.; Kamounah, F.S.; Gryko, D.T. Deuterium Isotope Effects on C-13-NMR Chemical Shifts of 10-Hydroxybenzo[h]quinolines. Molecules 2013, 18, 4544–4560. [Google Scholar] [CrossRef]
- Bobranski, B. The oxy-6-quinolin-aldehyde-5 and several resultant represented 5,6-substituted quinoline derivatives. J. Prakt. Chem. 1932, 134, 141–152. [Google Scholar]
- Kisin-Finfer, E.; Redy-Keisar, O.; Roth, M.; Ben-Eliyahu, R.; Shabat, D. Molecular Insight into Long-Wavelength Fluorogenic Dye Design: Hydrogen Bond Induces Activation of a Dormant Acceptor. Chem. Eur. J. 2015, 21, 18566–18570. [Google Scholar] [CrossRef]
- Morimura, S.; Horiuchi, H.; Murayama, K. Vilsmeier Reaction of Phenols. I. Synthesis of Aryl Formates. Bull. Chem. Soc. Jpn. 1977, 8, 2189–2190. [Google Scholar] [CrossRef] [Green Version]
Sample Availability: Samples of the compounds 2e, 2f, 2j, 2h, 2k, 3a, 3b, 3c, 3d are available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2h | 3a | 3b | 3c | 3d | |
|---|---|---|---|---|---|
| Empirical formula | C12H9NO3,CHCl3 | 3(C22H24N2O)·CH3CN | C25H31N3 | C24H29N3 | C23H26N2O |
| Temperature (K) | 100(1) | 100(1) | 100(1) | 293(1) | 80(1) |
| Wavelength (Å) | 0.71073 (Mo Kα) | ||||
| Crystal system | Orthorhombic | Trigonal | Orthorhombic | Monoclinic | Monoclinic |
| Space group | Pnma | P31c | Pbca | P21/c | P21/c |
| Unit cell dimensions | |||||
| a (Å) | 26.1040(14) | 29.4970(4) | 18.8459(8) | 9.0651(3) | 24.0519(9) |
| b (Å) | 6.4668(4) | 29.4970(4) | 11.0427(3) | 11.2850(3) | 16.2882(7) |
| c (Å) | 8.2056(4) | 12.0258(3) | 20.8067(6) | 20.8767(7) | 9.7079(3) |
| α (o) | 90 | 90 | 90 | 90 | 90 |
| β (o) | 90 | 90 | 90 | 93.873(3) | 99.189(3) |
| γ (o) | 90 | 120 | 90 | 90 | 90 |
| Volume (Å3) | 1385.18(13) | 9061.5(3) | 4330.1(3) | 2130.80(12) | 3754.4(2) |
| Z | 4 | 6 | 8 | 4 | 8 |
| Calculated density (Mg/m3) | 1.604 | 1.142 | 1.146 | 1.121 | 1.226 |
| Absorption coefficient (mm−1) | 0.666 | 0.070 | 0.068 | 0.066 | 0.075 |
| F (000) | 680 | 3336 | 1616 | 776 | 1488 |
| Crystal dimensions (mm) | 0.12 × 0.10 × 0.02 | 0.18 × 0.13 × 0.03 | 0.25 × 0.02 × 0.02 | 0.56 × 0.22 × 0.09 | 0.30 × 0.05 × 0.02 |
| θ range for data collection (o) | 2.932 to 26.371 | 2.875 to 24.710 | 3.452 to 26.370 | 3.401 to 26.367 | 3.033 to 26.371 |
| Index ranges | −32 ≤ h ≤ 32; −6 ≤ k ≤ 8; −10 ≤ l ≤ 10 | −34 ≤ h ≤ 34; −34 ≤ k ≤ 31; −14 ≤ l ≤ 14 | −21 ≤ h ≤ 23; −13 ≤ k ≤ 9; −26 ≤ l ≤ 26 | −11 ≤ h ≤ 11; −14 ≤ k ≤ 9; −26 ≤ l ≤ 26 | −30 ≤ h ≤ 30; −20 ≤k ≤ 20; −9 ≤ l ≤ 12 |
| Reflections collected | 13863 | 69751 | 25986 | 25693 | 30149 |
| Independent reflections | 1543 [R(int) = 0.0162] | 10284 [R(int) = 0.0713] | 4420 [R(int) = 0.0680] | 4343 [R(int) = 0.0509] | 7669 [R(int) = 0.0599] |
| Data/restraints/parameters | 1543/0/120 | 10284/1/721 | 4420/0/260 | 4343/0/251 | 7669/0/493 |
| Goodness-of-fit on F2 | 1.223 | 1.070 | 1.072 | 1.026 | 1.032 |
| Final R indices (I > 2σ(I)) | R1 = 0.0547; wR2 = 0.1379 | R1 = 0.0581; wR2 = 0.1287 | R1 = 0.0488; wR2 = 0.1082 | R1 = 0.0494; wR2 = 0.1180 | R1 = 0.0521; wR2 = 0.1228 |
| R indices (all data) | R1 = 0.0556; wR2 = 0.1385 | R1 = 0.0811; wR2 = 0.1436 | R1 = 0.0776; wR2 = 0.1262 | R1 = 0.0763; wR2 = 0.1329 | R1 = 0.0884; wR2 = 0.1499 |
| Largest diff. Peak and hole | 0.696 and −0.377 | 0.159 and −0.257 | 0.203 and −0.191 | 0.158 and −0.154 | 0.325 and −0.297 |
| CCDC-Number | 1890715 | 1501807 | 1501808 | 1829344 | 1829345 |
| Bond Lengths (Å) | |||||
|---|---|---|---|---|---|
| 2h | 3a | 3b | 3c | 3d | |
| C(10)–N(2) | - | 1.269(6) | 1.2741 | 1.265(2) | 1.2816 |
| C(33)–N(4) | - | 1.421(6) | 1.417(2) | 1.426(2) | 1.4263 |
| C(5)–C(10) | - | 1.459(7) | 1.465(2) | 1.4631 | 1.4488 |
| C(5)–C(11) | 1.448 | - | - | - | - |
| C(7)–C(12) | 1.452 | - | - | - | - |
| C(8)–O(1) | 1.243 | 1.350(5) | - | - | 1.341 |
| C(6)–N(3) | - | - | 1.415(2) | 1.403(2) | - |
| Angles (o) | |||||
| C(10)–N(2)–C(11) | 116.8(4) | 120.5(1) | 117.5(1) | 121.18 | |
| C(5)–C(10)–N(2) | 127.2(5) | 124.0(1) | 126.1(1) | 121.52 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wantulok, J.; Szala, M.; Quinto, A.; Nycz, J.E.; Giannarelli, S.; Sokolová, R.; Książek, M.; Kusz, J. Synthesis, Electrochemical and Spectroscopic Characterization of Selected Quinolinecarbaldehydes and Their Schiff Base Derivatives. Molecules 2020, 25, 2053. https://doi.org/10.3390/molecules25092053
Wantulok J, Szala M, Quinto A, Nycz JE, Giannarelli S, Sokolová R, Książek M, Kusz J. Synthesis, Electrochemical and Spectroscopic Characterization of Selected Quinolinecarbaldehydes and Their Schiff Base Derivatives. Molecules. 2020; 25(9):2053. https://doi.org/10.3390/molecules25092053
Chicago/Turabian StyleWantulok, Jakub, Marcin Szala, Andrea Quinto, Jacek E. Nycz, Stefania Giannarelli, Romana Sokolová, Maria Książek, and Joachim Kusz. 2020. "Synthesis, Electrochemical and Spectroscopic Characterization of Selected Quinolinecarbaldehydes and Their Schiff Base Derivatives" Molecules 25, no. 9: 2053. https://doi.org/10.3390/molecules25092053