
Tricyclic Derivative of Acyclovir and Its Esters in Relation to the Esters of Acyclovir Enzymatic Stability: Enzymatic Stability Study

 ,
,
Abstract
:1. Introduction
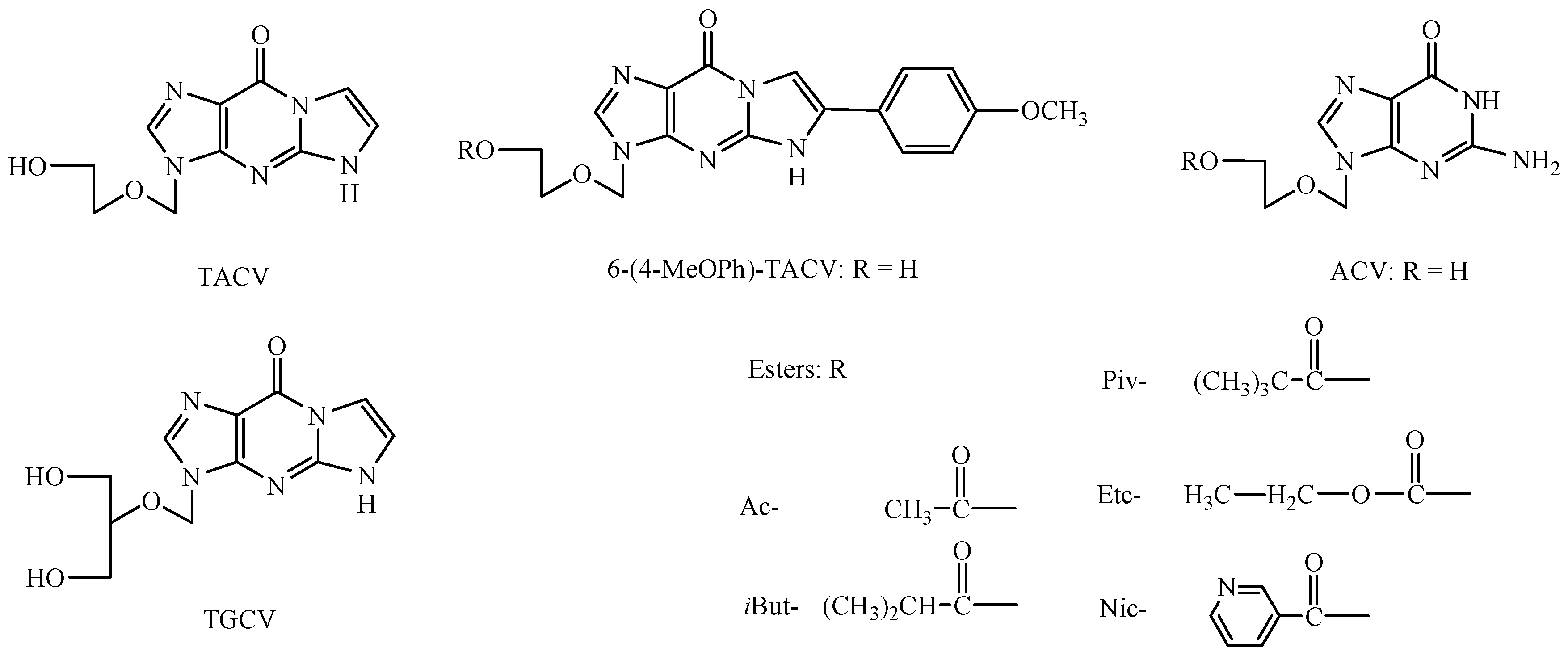
2. Results and Discussion
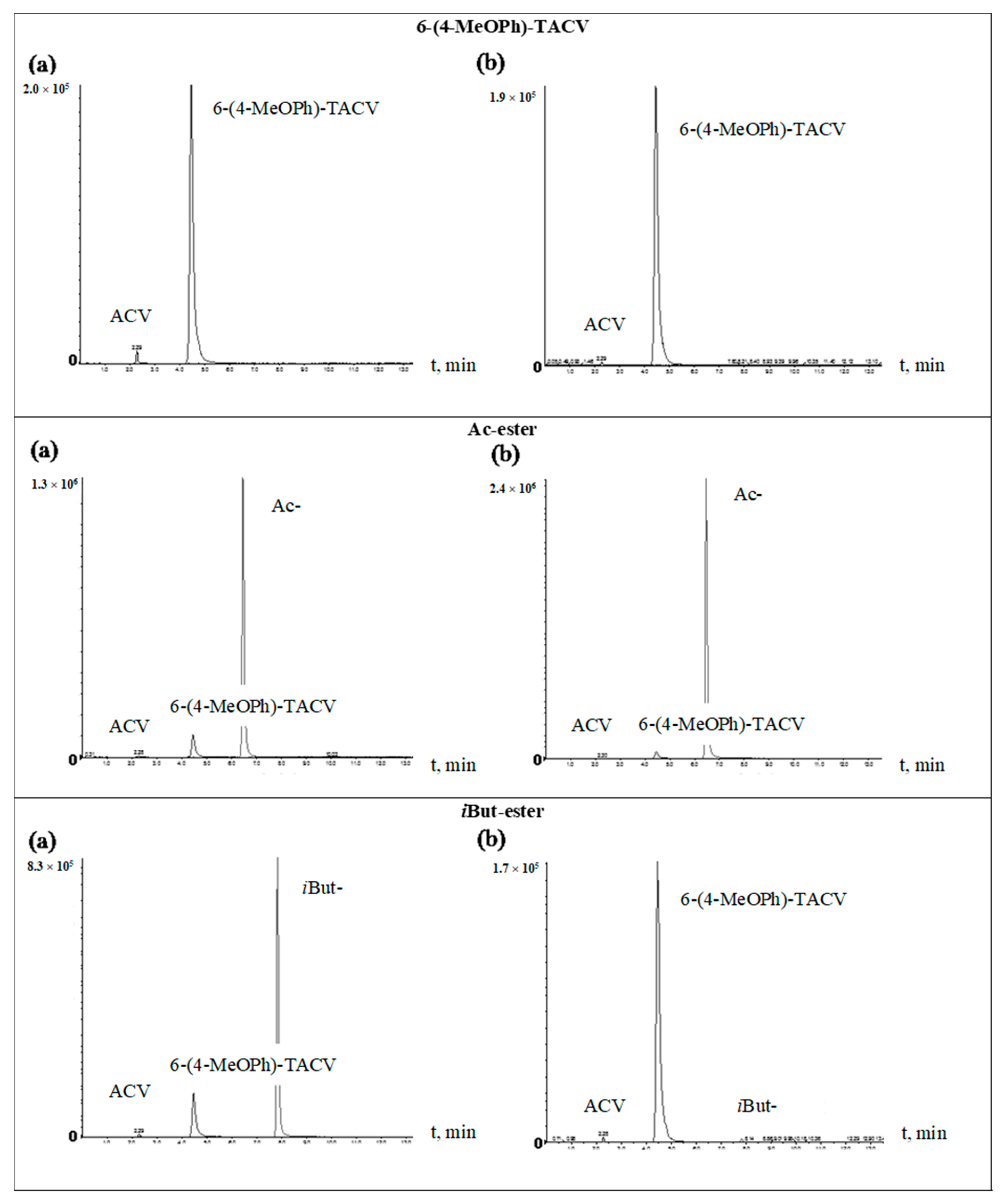
2.1. HPLC Method Validation
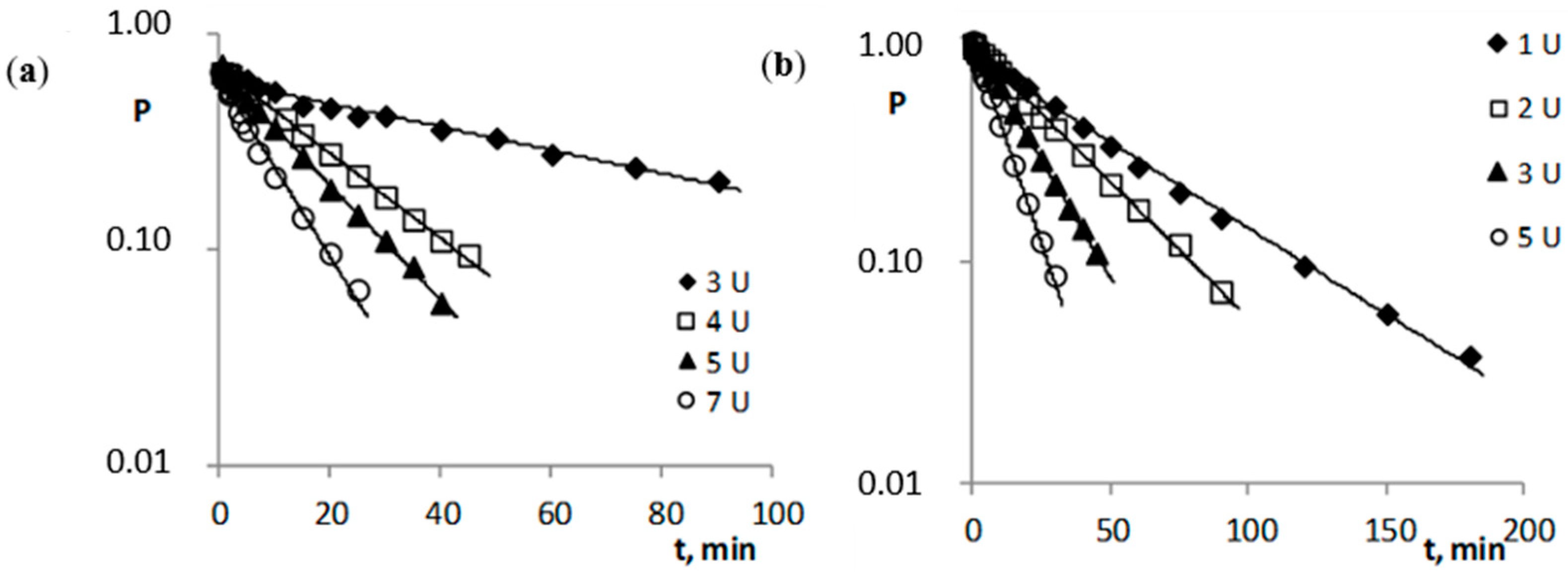
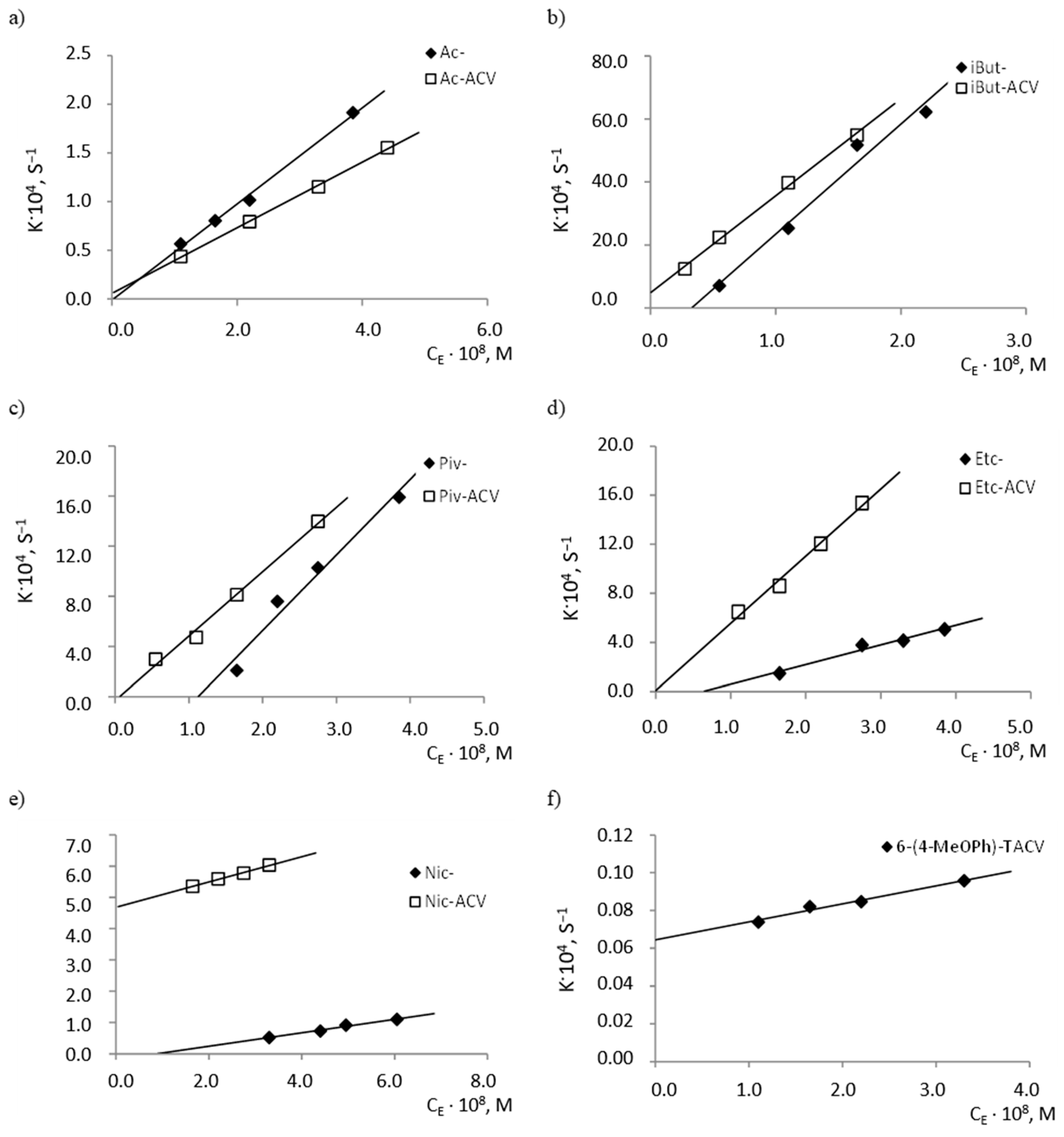
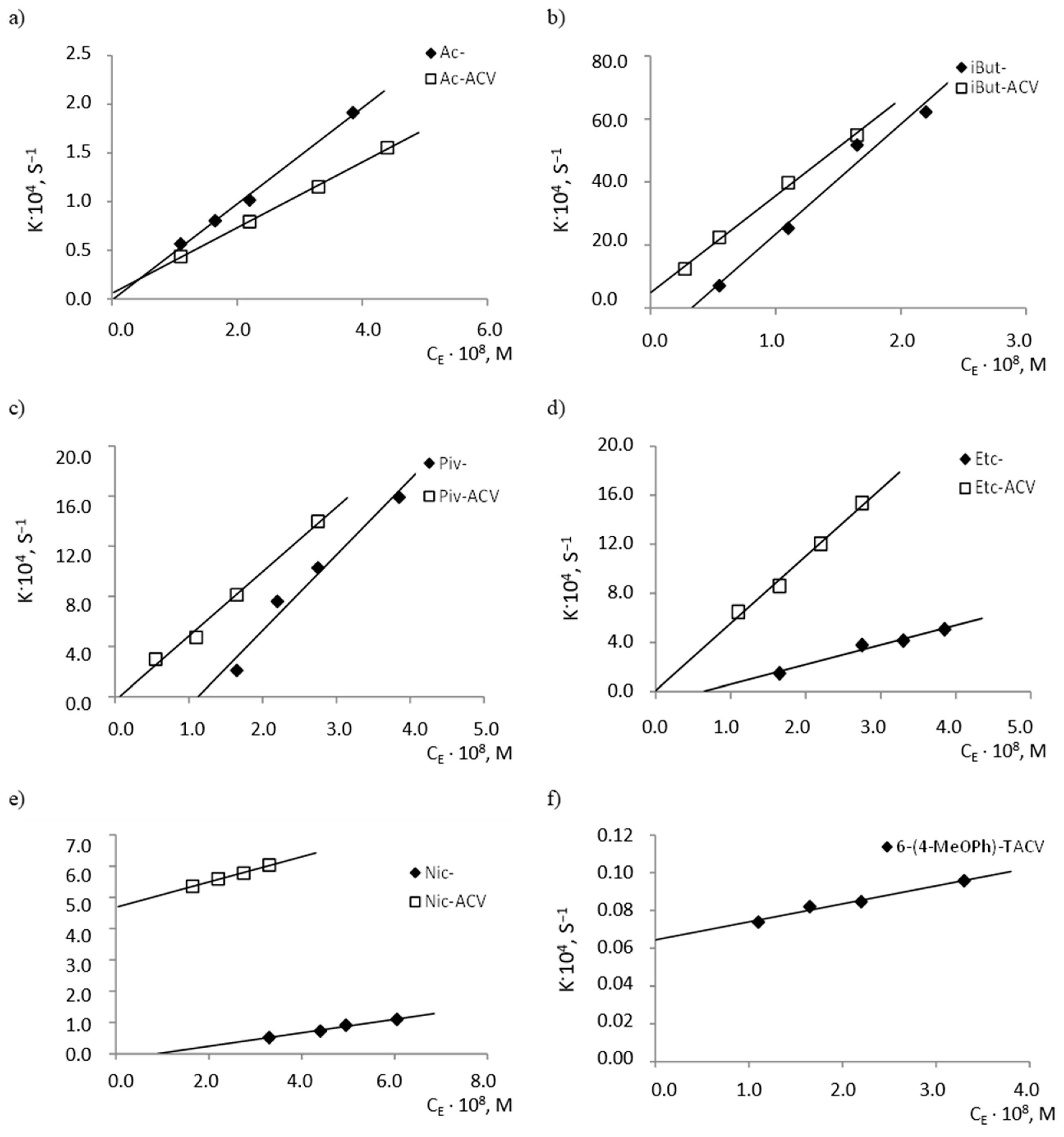
2.2. Enzymatic Stability
3. Experimental
3.1. Materials
3.2. Apparatus and Chromatographic Conditions
3.2.1. HPLC-UV
3.2.2. HPLC-MS/MS
3.2.3. Other Equipment
3.2.4. Phosphate Buffer PBS pH 7.4
3.3. HPLC Method Validation for Determination of the Tested Compounds in Plasma
3.3.1. Stock Solutions
3.3.2. Selectivity
3.3.3. Linearity
3.3.4. Precision and Accuracy
3.3.5. Stability of the Analyte
3.4. Enzymatic Stability of Tested Compounds
3.4.1. Stability Test in Plasma
3.4.2. Stability Test in Plasma with Esterase
3.4.3. HPLC-MS/MS Analysis of Enzymatic Hydrolysis Products
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Huttunen, K.M.; Raunio, H.; Rautio, J. Prodrugs—From Serendipity to Rational Design. Pharmacol. Rev. 2011, 63, 750–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liederer, B.M.; Borchardt, R.T. Enzymes involved in the bioconversion of ester-based prodrugs. J. Pharm. Sci. 2006, 95, 1177–1195. [Google Scholar] [CrossRef]
- Zając, M.; Pawełczyk, E.; Jelińska, A. Pro-leki, in: Chemia Leków dla Studentów Farmacji i Farmaceutów (Pro-drugs, in Chemistry of Medicines for Pharmacy Students and Pharmacists, in Polish); Akademia Medyczna im. K. Marcinkowskiego: Poznań, Poland, 2006; pp. 25–32. [Google Scholar]
- Miwa, M.; Ura, M.; Nishida, M.; Sawada, N.; Ishikawa, T.; Mori, K.; Shimma, N.; Umeda, I.; Ishitsuka, H. Design of a novel oral fluoropyrimidine carbamate, capecitabine, which generates 5-fluorouracil selectively in tumours by enzymes concentrated in human liver and cancer tissue. Eur. J. Cancer 1998, 34, 1274–1281. [Google Scholar] [CrossRef]
- Senter, P.D.; Marquardt, H.; Thomas, B.A.; Hammock, B.D.; Frank, I.S.; Svensson, H.P. The role of rat serum carboxylesterase in the activation of paclitaxel and camptothecin prodrugs. Cancer Res. 1996, 56, 1471–1474. [Google Scholar] [PubMed]
- Yoshigae, Y.; Imai, T.; Taketani, M.; Otagiri, M. Characterization of esterases involved in the stereoselective hydrolysis of ester-type prodrugs of propranolol in rat liver and plasma. Chirality 1999, 11, 10–13. [Google Scholar] [CrossRef]
- Quinn, D.M. Acetylcholinesterse: Enzyme structure, reaction dynamics, and virtual transition states. Chem. Rev. 1987, 87, 955–979. [Google Scholar] [CrossRef]
- Tak, R.V.; Pal, D.; Gao, H.; Dey, S.; Mitra, A.K. Transport of acyclovir ester prodrugs through rabbit cornea and SIRC-rabbit corneal epithelial cell line. J. Pharm. Sci. 2001, 90, 1505–1515. [Google Scholar] [CrossRef]
- Nakamura, M.; Shirasawa, E.; Hikida, M. Characterization of Esterases Involved in the Hydrolysis of Dipivefrin Hydrochloride. Ophthalmic Res. 1993, 25, 46–51. [Google Scholar] [CrossRef]
- Sun, H.; Pang, Y.-P.; Lockridge, O.; Brimijoin, S. Re-engineering butyrylcholinesterase as a cocaine hydrolase. Mol. Pharmacol. 2002, 62, 220–224. [Google Scholar] [CrossRef] [Green Version]
- Lockridge, O. Genetic variants of human serum cholinesterase influence metabolism of the muscle relaxant succinylcholine. Pharmacol. Ther. 1990, 47, 35–60. [Google Scholar] [CrossRef] [Green Version]
- Masson, E.; Froment, M.T.; Fortier, P.L.; Visicchio, J.E.; Bartels, C.F.; Lockridge, O. Butyrylcholineste-rase-catalysed hydrolysis of aspirin, a negatively charged ester, and aspirin related neutral esters. Biochim. Biophys. Acta 1998, 1387, 41–52. [Google Scholar] [CrossRef]
- Lockridge, O.; Mottershaw-Jackson, N.; Eckerson, H.W.; La Du, B.N. Hydrolysis of diacetylmorphine (heroin) by human serum cholinesterase. J. Pharmacol. Exp. Ther. 1980, 215, 1–8. [Google Scholar] [PubMed]
- Tunek, A.; Levin, E.; Svensson, L.-Å. Hydrolysis of 3H-bambuterol, a carbamate prodrug of terbutaline, in blood from humans and laboratory animals in vitro. Biochem. Pharmacol. 1988, 37, 3867–3876. [Google Scholar] [CrossRef]
- Meyers, C.; Lockridge, O.; La Du, B.N. Hydrolysis of methylprednisolone acetate by human serum cholinesterase. Drug Metab. Dispos. 1982, 10, 279–280. [Google Scholar]
- Gilmer, J.F.; Moriarty, L.M.; Lally, M.N.; Clancy, J.M. Isosorbide-based aspirin prodrugs. II. Hydrolysis kinetics of isosorbide diaspirinate. Eur. J. Pharm. Sci. 2002, 16, 297–304. [Google Scholar] [CrossRef]
- Laethem, R.M.; Blumenkopf, T.A.; Cory, M.; Elwell, L.; Moxham, C.P.; Ray, P.H.; Walton, L.M.; Smith, G.K. Expression and characterization of human pancreatic preprocarboxypeptidase A1 and preprocarboxypeptidase A2. Arch. Biochem. Biophys. 1996, 332, 8–18. [Google Scholar] [CrossRef]
- Sidhu, R.S.; Blair, A.H. Human liver aldehyde dehydrogenase. Esterase activity. J. Biol. Chem. 1975, 250, 7894–7898. [Google Scholar]
- Verpoorte, J.A.; Mehta, S.; Edsall, J.T. Esterase activities of human carbonic anhydrases B and C. J. Biol. Chem. 1967, 242, 4221–4229. [Google Scholar]
- Tsunematsu, H.; Yoshida, S.; Horie, K.; Yamamoto, M. Synthesis and the Stereoselective Enzymatic Hydrolysis of Flurbiprofen-Basic Amino Acid Ethyl Esters. J. Drug Target. 1995, 2, 517–525. [Google Scholar] [CrossRef]
- Tsujita, T.; Shirai, K.; Saito, Y.; Okuda, H. Relationship between lipase and esterase. Prog. Clin. Biol. Res. 1990, 344, 915–933. [Google Scholar]
- Field, H.J.; Hodge, R.A.V. Recent developments in anti-herpesvirus drugs. Br. Med. Bull. 2013, 106, 213–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beauchamp, L.M.; Dolmatch, B.L.; Schaeffer, H.J.; Collins, P.; Bauer, D.J.; Keller, P.M.; Fyfe, J.A. Modifications on the heterocyclic base of acyclovir: syntheses and antiviral properties. J. Med. Chem. 1985, 28, 982–987. [Google Scholar] [CrossRef] [PubMed]
- Robins, M.J.; Hatfield, P.W.; Balzarini, J.; De Clercq, E. Nucleic acid related compounds. 47. Synthesis and biological activities of pyrimidine and purine “acyclic” nucleoside analogues. J. Med. Chem. 1984, 27, 1486–1492. [Google Scholar] [CrossRef]
- Boryski, J.; Golankiewicz, B.; De Clercq, E. Synthesis and antiviral activity of novel N-substituted derivatives of acyclovir. J. Med. Chem. 1988, 31, 1351–1355. [Google Scholar] [CrossRef] [PubMed]
- Boryski, J.; Golankiewicz, B.; De Clercq, E. Synthesis and antiviral activity of 3-substituted derivatives of 3,9-dihydro-9-oxo-5H-imidazo[1,2-a]purines, tricyclic analogues of acyclovir and ganciclovir. J. Med. Chem. 1991, 34, 2380–2383. [Google Scholar] [CrossRef] [PubMed]
- Golankiewicz, B.; Ostrowski, T.; Boryski, J.; De Clercq, E. Synthesis of acyclowyosine and acyclo-3-methylguanosine, as probes for some chemical and biological properties resulting from the N-3 substitution of guanosine and its analogues. J. Chem. Soc. Perkin Trans. 1 1991, 589. [Google Scholar] [CrossRef]
- Sekiyama, T.; Hatsuya, S.; Tanaka, Y.; Uchiyama, M.; Ono, N.; Iwayama, S.; Oikawa, M.; Suzuki, K.; Okunishi, M.; Tsuji, T. Synthesis and Antiviral Activity of Novel Acyclic Nucleosides: Discovery of a Cyclopropyl Nucleoside with Potent Inhibitory Activity against Herpesviruses. J. Med. Chem. 1998, 41, 1284–1298. [Google Scholar] [CrossRef]
- Iwayama, S.; Ono, N.; Ohmura, Y.; Suzuki, K.; Aoki, M.; Nakazaw, H. Antiherpesvirus activities of (1′S,2′R)-9-{[1′,2′-bis(hydroxymethyl)cycloprop-1′-yl]methyl}guanine (A-5021) in cell culture. Antimicrob. Agents Chemother. 1998, 42, 1666–1670. [Google Scholar] [CrossRef] [Green Version]
- Balzarini, J.; Ostrowski, T.; Goslinski, T.; De Clercq, E.; Golankiewicz, B. Pronounced cytostatic activity and bystander effect of a novel series of fluorescent tricyclic acyclovir and ganciclovir derivatives in herpes simplex virus thymidine kinase gene-transduced tumor cell lines. Gene Ther. 2002, 9, 1173–1182. [Google Scholar] [CrossRef] [Green Version]
- Golankiewicz, B.; Ostrowski, T.; Goslinski, T.; Januszczyk, P.; Zeidler, J.; Baranowski, D.; De Clercq, E. Fluorescent Tricyclic Analogues of Acyclovir and Ganciclovir. A Structure−Antiviral Activity Study. J. Med. Chem. 2001, 44, 4284–4287. [Google Scholar] [CrossRef]
- Golankiewicz, B.; Ostrowski, T. Tricyclic nucleoside analogues as antiherpes agents. Antivir. Res. 2006, 71, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, T.; Zeidler, J.; Gośliński, T.; Golankiewicz, B. Substituent—Directed aralkylation and alkylation reactions of the tricyclic analogues of acyclovir and guanosine. Nucleos. Nucleot. Nucl. 2000, 19, 1911–1929. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, T.; Golankiewicz, B.; De Clercq, E.; Balzarini, J. Fluorosubstitution and 7-alkylation as prospective modifications of biologically active 6-aryl derivatives of tricyclic acyclovir and ganciclovir analogues. Bioorg. Med. Chem. 2005, 13, 2089–2096. [Google Scholar] [CrossRef] [PubMed]
- Goslinski, T.; Golankiewicz, B.; De Clercq, E.; Balzarini, J. Synthesis and Biological Activity of Strongly Fluorescent Tricyclic Analogues of Acyclovir and Ganciclovir. J. Med. Chem. 2002, 45, 5052–5057. [Google Scholar] [CrossRef] [PubMed]
- Goslinski, T.; Wenska, G.; Golankiewicz, B.; Balzarini, J.; De Clercq, E. Synthesis and Fluorescent Properties of 6-(4-Biphenylyl)-3,9-dihydro-9-oxo-5H-imidazo[1,2-A]purine Analogues of Acyclovir and Ganciclovir. Nucleosides Nucleotides Nucleic Acids 2003, 22, 911–914. [Google Scholar] [CrossRef] [PubMed]
- Gośliński, T.; Januszczyk, P.; Wenska, G.; Golankiewicz, B.; De Clercq, E.; Balzarini, J. Synthesis and fluorescent properties of the tricyclic analogues of acyclovir linked with nitrogen heterocyclic units. Nucleos. Nucleot. Nucl. 2005, 24, 571–575. [Google Scholar] [CrossRef] [PubMed]
- Lesniewska, M.A.; Ostrowski, T.; Zeidler, J.; Muszalska, I. Ester groups as carriers of antivirally active tricyclic analogue of acyclovir in prodrugs designing: Synthesis, lipophilicity—Comparative statistical study of the chromatografic and theoretical methods, validation of the HPLC method. Comb. Chem. High Throughput Scr. 2014, 17, 639–650. [Google Scholar] [CrossRef]
- Lesniewska, M.A.; Gdaniec, Z.; Muszalska, I. Calculation procedures and HPLC method for analysis of the lipophilicity of acyclovir esters. Drug Dev. Ind. Pharm. 2014, 41, 663–669. [Google Scholar] [CrossRef]
- Muszalska, I.; Lesniewska-Kowiel, M.A.; Ostrowski, T. Comparative analysis of stability of tricyclic analogues of acyclovir in an acidic environment. React. Kinet. Mech. Catal. 2019, 127, 283–299. [Google Scholar] [CrossRef] [Green Version]
- Lesniewska, M.A.; Gola, M.; Dutkiewicz, Z.; Muszalska, I. Comparative Analysis of Acyclovir Esters Stability in Solutions: The Influence of the Substituent Structure, Kinetics, and Steric Effects. Int. J. Chem. Kinet. 2015, 47, 724–733. [Google Scholar] [CrossRef]
- Bełtowska-Brzezinska, M. Podstawy Kinetyki Chemicznej—Skrypt do Wykładów (Basics of Chemical Kinetics—A Lecture Script, in Polish); Wydział Chemii UAM: Poznań, Poland, 2009; pp. 61–66. [Google Scholar]
- Molski, A. Wprowadzenie do Kinetyki Chemicznej (Introduction to Chemical Kinetics, in Polish); Wydawnictwo Naukowo-Techniczne: Warszawa, Poland, 2001; pp. 128–134. [Google Scholar]
- Katragadda, S.; Jain, R.; Kwatra, D.; Hariharan, S.; Mitra, A.K. Pharmacokinetics of amino acid ester prodrugs of acyclovir after oral administration: Interaction with the transporters on Caco-2 cells. Int. J. Pharm. 2008, 362, 93–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Sample Availability: Samples of the compounds are available from the authors. |

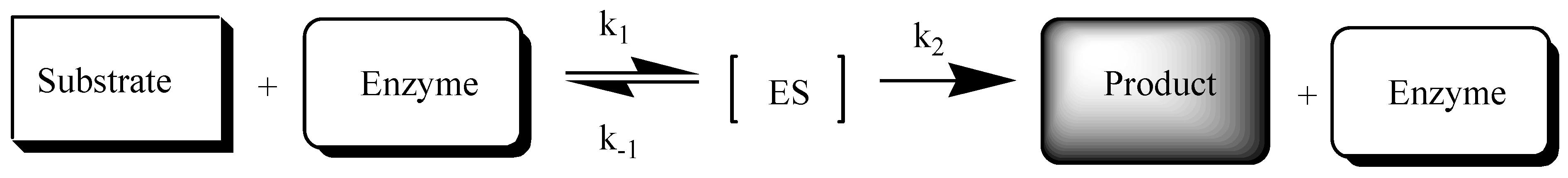


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Mobile Phase | Flow Rate, mL/min | Retention Time, min | Internal Standard—i.s. μg/mL | Retention Time of i.s., min |
|---|---|---|---|---|---|
| Acetonitrile—CH3COOH (2 mM), KCl (1 mM), v/v | |||||
| plasma residue 6-(4-MeOPh)-TACV Nic- Etc- iBut- | 35:65 | 1.0 | 2.01 4.29 8.16 10.83 13.49 | sulfadimethoxin (192) | 6.65 |
| plasma residue 6-(4-MeOPh)-TACV Ac- Piv- | 2.01 4.29 6.95 20.55 | nitrazepam (192) | 11.35 | ||
| plasma residue ACV Ac-ACV | 15:85 | 0.7 | 3.50 3.50 5.51 | sulfathiazole (96) | 9.72 |
| plasma residue ACV iBut-ACV Piv-ACV | 23:77 | 1.0 | 1.59 2.47 5.02 7.89 | sulfafurazol (112) | 1.59 |
| plasma residue ACV Etc-ACV | 20:80 | 1.0 | 2.41 3.33 4.84 | sulfafurazol (56) | 2.41 |
| plasma residue ACV Nic-ACV | 2.41 3.33 4.16 | sulfafurazol (112) |
| Compound | kobs. ± Δkobs., s−1 | t0.5, h | -r | n |
|---|---|---|---|---|
| 6-(4-MeOPh)-TACV | (2.81 ± 0.36)·10−6 | 68.40 | 0.9822 | 13 |
| Ac- | (9.36 ± 0.72)·10−6 | 20.57 | 0.9933 | 13 |
| iBut- | (1.67 ± 0.04)·10−5 | 11.52 | 0.9992 | 15 |
| Piv- | (6.84 ± 0.33)·10−6 | 28.13 | 0.9977 | 12 |
| Etc- | (9.74 ± 1.16)·10−6 | 19.77 | 0.9824 | 14 |
| Nic- | (1.15 ± 0.13)·10−5 | 16.78 | 0.9837 | 14 |
| Ac-ACV | (6.47 ± 0.49)·10−5 | 2.98 | 0.9912 | 16 |
| iBut-ACV | (4.16 ± 0.16)·10−4 | 0.46 | 0.9978 | 16 |
| Piv-ACV | (2.38 ± 0.17)·10−5 | 8.09 | 0.9932 | 15 |
| Etc-ACV | (7.99 ± 0.37)·10−5 | 2.41 | 0.9976 | 13 |
| Nic-ACV | (4.31 ± 0.23)·10−4 | 0.45 | 0.9959 | 15 |
| Compound | cE, U | cE, M | kobs. ± Δkobs., s−1 | t0.5 | -r | n |
|---|---|---|---|---|---|---|
| 6-(4-MeOPh)-TACV | 2.0 | 1.10·10−8 | (7.39 ± 0.49)·10−6 | 26.0 h | 0.9950 | 13 |
| 3.0 | 1.65·10−8 | (8.21 ± 0.58)·10−6 | 23.5 h | 0.9938 | 14 | |
| 4.0 | 2.20·10−8 | (8.37 ± 0.24)·10−6 | 22.7 h | 0.9988 | 16 | |
| 6.0 | 3.30·10−8 | (9.58 ± 0.39)·10−6 | 20.1 h | 0.9975 | 16 | |
| Ac- | 2.0 | 1.10·10−8 | (5.66 ± 0.36)·10−5 | 3.4 h | 0.9940 | 16 |
| 3.0 | 1.65·10−8 | (8.04 ± 0.50)·10−5 | 2.4 h | 0.9943 | 16 | |
| 4.0 | 2.20·10−8 | (1.02 ± 0.05)·10−4 | 1.9 h | 0.9966 | 16 | |
| 7.0 | 3.85·10−8 | (1.91 ± 0.10)·10−4 | 1.0 h | 0.9968 | 13 | |
| iBut- | 1.0 | 5.50·10−9 | (7.05 ± 0.39)·10−4 | 16.4 min | 0.9962 | 14 |
| 2.0 | 1.10·10−8 | (2.53 ± 0.14)·10−3 | 4.6 min | 0.9978 | 10 | |
| 3.0 | 1.65·10−8 | (5.17 ± 0.10)·10−3 | 2.2 min | 0.9997 | 11 | |
| 4.0 | 2.20·10−8 | (6.22 ± 0.45)·10−3 | 1.9 min | 0.9962 | 10 | |
| Piv- | 3.0 | 1.65·10−8 | (2.08 ± 0.19)·10−4 | 55.5 min | 0.9908 | 13 |
| 4.0 | 2.20·10−8 | (7.59 ± 0.18)·10−4 | 15.2 min | 0.9993 | 14 | |
| 5.0 | 2.75·10−8 | (1.03 ± 0.03)·10−3 | 11.2 min | 0.9992 | 13 | |
| 7.0 | 3.85·10−8 | (1.59 ± 0.11)·10−3 | 7.3 min | 0.9936 | 14 | |
| Etc- | 3.0 | 1.65·10−8 | (1.48 ± 0.07)·10−4 | 1.3 h | 0.9972 | 13 |
| 5.0 | 2.75·10−8 | (3.79 ± 0.27)·10−4 | 30.5 min | 0.9930 | 15 | |
| 6.0 | 3.30·10−8 | (4.15 ± 0.26)·10−4 | 27.8 min | 0.9964 | 11 | |
| 7.0 | 3.85·10−8 | (5.07 ± 0.43)·10−4 | 22.8 min | 0.9927 | 12 | |
| Nic- | 6.0 | 3.30·10−8 | (5.24 ± 0.38)·10−5 | 3.7 h | 0.9933 | 14 |
| 8.0 | 4.40·10−8 | (7.31 ± 0.49)·10−5 | 2.6 h | 0.9943 | 14 | |
| 9.0 | 4.95·10−8 | (9.18 ± 0.65)·10−5 | 2.1 h | 0.9937 | 14 | |
| 11.0 | 6.05·10−8 | (1.10 ± 0.17)·10−4 | 1.7 h | 0.9764 | 12 | |
| Ac-ACV | 2.0 | 1.10·10−8 | (4.37 ± 0.27)·10−5 | 4.4 h | 0.9946 | 15 |
| 4.0 | 2.20·10−8 | (7.94 ± 0.23)·10−5 | 2.4 h | 0.9988 | 15 | |
| 6.0 | 3.30·10−8 | (1.15 ± 0.02)·10−4 | 1.7 h | 0.9995 | 15 | |
| 8.0 | 4.40·10−8 | (1.55 ± 0.04)·10−4 | 1.2 h | 0.9992 | 15 | |
| iBut-ACV | 0.5 | 2.75·10−9 | (1.24 ± 0.12)·10−3 | 9.3 min | 0.9869 | 15 |
| 1.0 | 5.50·10−9 | (2.24 ± 0.09)·10−3 | 5.2 min | 0.9971 | 17 | |
| 2.0 | 1.10·10−8 | (3.98 ± 0.13)·10−3 | 2.9 min | 0.9985 | 16 | |
| 3.0 | 1.65·10−8 | (5.49 ± 0.20)·10−3 | 2.1 min | 0.9979 | 16 | |
| Piv-ACV | 1.0 | 5.50·10−9 | (2.97 ± 0.09)·10−4 | 38.9 min | 0.9988 | 16 |
| 2.0 | 1.10·10−8 | (4.71 ± 0.10)·10−4 | 24.5 min | 0.9995 | 16 | |
| 3.0 | 1.65·10−8 | (8.11 ± 0.15)·10−4 | 14.3 min | 0.9995 | 15 | |
| 5.0 | 2.75·10−8 | (1.40 ± 0.04)·10−3 | 8.3 min | 0.9989 | 15 | |
| Etc-ACV | 2.0 | 1.10·10−8 | (6.48 ± 0.35)·10−4 | 17.8 min | 0.9960 | 15 |
| 3.0 | 1.65·10−8 | (8.60 ± 0.34)·10−4 | 13.4 min | 0.9977 | 16 | |
| 4.0 | 2.20·10−8 | (1.20 ± 0.03)·10−3 | 9.6 min | 0.9991 | 15 | |
| 5.0 | 2.75·10−8 | (1.54 ± 0.07)·10−3 | 7.5 min | 0.9971 | 15 | |
| Nic-ACV | 3.0 | 1.65·10−8 | (5.35 ± 0.23)·10−4 | 21.6 min | 0.9971 | 16 |
| 4.0 | 2.20·10−8 | (5.59 ± 0.50)·10−4 | 20.7 min | 0.9892 | 15 | |
| 5.0 | 2.75·10−8 | (5.77 ± 0.32)·10−4 | 20.0 min | 0.9966 | 13 | |
| 6.0 | 3.30·10−8 | (6.03 ± 0.22)·10−4 | 19.2 min | 0.9981 | 15 |
| Compound | k2/KM, mol −1·L·s−1 | Compound | k2/KM, mol −1·L·s−1 |
|---|---|---|---|
| 6-(4-MeOPh)-TACV | (9.52 ± 4.26)·101 | ||
| Ac- | (4.89 ± 0.64)·103 | Ac-ACV | (3.54 ± 0.18)·103 |
| iBut- | (2.80 ± 1.00)·105 | iBut-ACV | (3.48 ± 0.30)·105 |
| Piv- | (3.66 ± 1.93)·104 | Piv-ACV | (4.97 ± 0.89)·104 |
| Etc- | (1.28 ± 0.51)·104 | Etc-ACV | (5.52 ± 1.02)·104 |
| Nic- | (1.77 ± 0.47)·103 | Nic-ACV | (4.04 ± 0.79)·103 |
| Time [min] | Phase A [%] | Phase B [%] |
|---|---|---|
| 0.0 | 65.0 | 35.0 |
| 1.0 | 65.0 | 35.0 |
| 8.0 | 15.0 | 85.0 |
| 10.0 | 15.0 | 85.0 |
| 11.0 | 65.0 | 35.0 |
| 17.0 | 65.0 | 35.0 |
| Compound | Molecular Weight, Da | Retention Time, min | Precursor Ion, m/z | Product Ions, m/z | DP, V | CE, V | CXP, V | DT, ms |
|---|---|---|---|---|---|---|---|---|
| ACV | 225.2 | 2.29 | 226.2 | 152.1 | 51 | 20 | 13 | 57 |
| 110.1 | 43 | 10 | ||||||
| 6-(4-MeOPh)-TACV | 355.35 | 4.46 | 356.3 | 294.2 | 50 | 24 | 16 | |
| 282.1 | 29 | 15 | ||||||
| Ac- | 397.39 | 6.46 | 398.2 | 294.3 | 85 | 27 | 17 | |
| 87.1 | 46 | 15 | ||||||
| iBut- | 425.45 | 7.82 | 426.2 | 294.3 | 100 | 28 | 17 | |
| 115.2 | 42 | 20 | ||||||
| Piv- | 439.47 | 8.45 | 440.5 | 294.3 | 112 | 27 | 24 | |
| 129.3 | 45 | 22 | ||||||
| Etc- | 427.42 | 7.36 | 428.2 | 194.3 | 110 | 27 | 17 | |
| 117.3 | 31 | 21 | ||||||
| Nic- | 460.45 | 6.68 | 461.2 | 294.3 | 100 | 30 | 17 | |
| 150.2 | 41 | 27 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muszalska-Kolos, I.; Lesniewska-Kowiel, M.A.; Plewa, S.; Klupczyńska, A. Tricyclic Derivative of Acyclovir and Its Esters in Relation to the Esters of Acyclovir Enzymatic Stability: Enzymatic Stability Study. Molecules 2020, 25, 2156. https://doi.org/10.3390/molecules25092156
Muszalska-Kolos I, Lesniewska-Kowiel MA, Plewa S, Klupczyńska A. Tricyclic Derivative of Acyclovir and Its Esters in Relation to the Esters of Acyclovir Enzymatic Stability: Enzymatic Stability Study. Molecules. 2020; 25(9):2156. https://doi.org/10.3390/molecules25092156
Chicago/Turabian StyleMuszalska-Kolos, Izabela, Monika A. Lesniewska-Kowiel, Szymon Plewa, and Agnieszka Klupczyńska. 2020. "Tricyclic Derivative of Acyclovir and Its Esters in Relation to the Esters of Acyclovir Enzymatic Stability: Enzymatic Stability Study" Molecules 25, no. 9: 2156. https://doi.org/10.3390/molecules25092156
APA StyleMuszalska-Kolos, I., Lesniewska-Kowiel, M. A., Plewa, S., & Klupczyńska, A. (2020). Tricyclic Derivative of Acyclovir and Its Esters in Relation to the Esters of Acyclovir Enzymatic Stability: Enzymatic Stability Study. Molecules, 25(9), 2156. https://doi.org/10.3390/molecules25092156