
A One-Pot Divergent Sequence to Pyrazole and Quinoline Derivatives



Abstract
:
1. Introduction
2. Results and Discussion
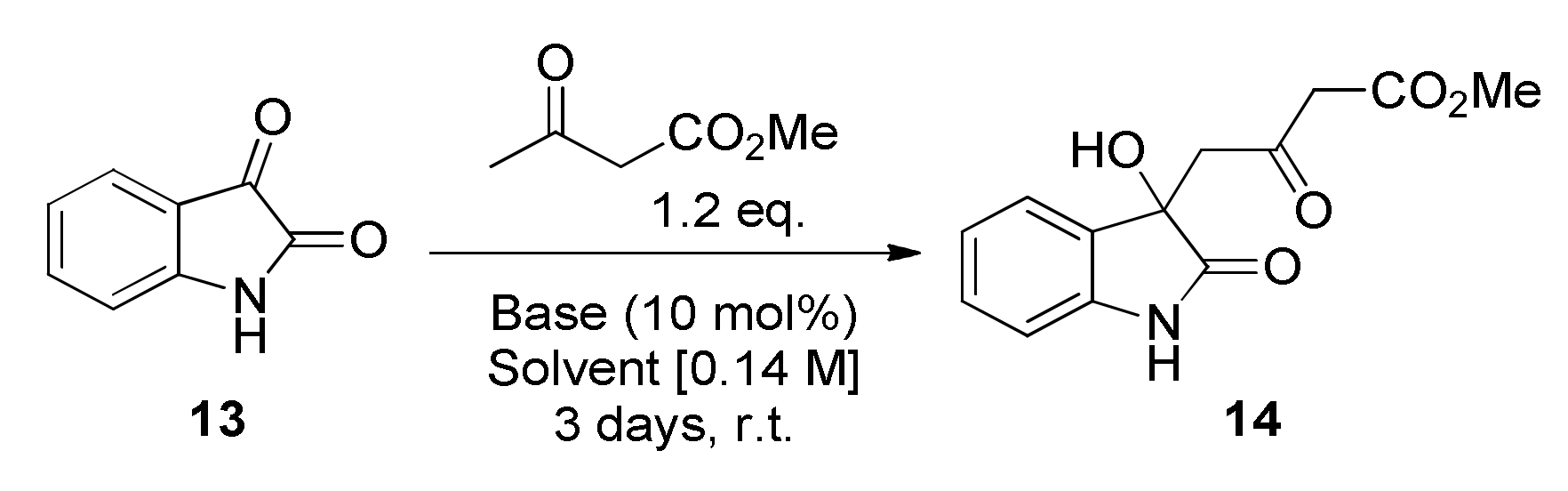
2.1. Optimisation of the Synthesis Method
2.2. Observations of the Spectroscopic Data Obtained Illustrated for Compound 15
3. Materials and Methods
3.1. General Procedure for the Synthesis of 14
3.2. General Procedure for the Synthesis of 15a–j
3.3. Characterisation of the Compounds 14 and 15a-j and 16 Derivatives
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Karrouchi, K.; Radi, S.; Ramli, Y.; Taoufik, J.; Mabkhot, Y.N.; Al-Aizari, F.A.; Ansar, M. Synthesis and Pharmacological Activities of Pyrazole Derivatives: A Review. Molecules 2018, 23, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Zhan, P.; De Clercq, E.; Liu, X. Strategies for the Design of HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors: Lessons from the Development of Seven Representative Paradigms. J. Med. Chem. 2012, 55, 3595–3613. [Google Scholar] [CrossRef] [PubMed]
- Ohsumi, K.; Matsueda, H.; Hatanaka, T.; Hirama, R.; Umemura, T.; Oonuki, A.; Ishida, N.; Kageyama, Y.; Maezono, K.; Kondo, N. Pyrazole-O-glucosides as novel Na(+)-glucose cotransporter (SGLT) inhibitors. Bioorg. Med. Chem. Lett. 2003, 13, 2269–2272. [Google Scholar] [CrossRef]
- Pinnetti, S.; Streicher, R.; Thomas, L.; Dugi, K. Pharmaceutical Composition Comprising a Pyrazole-O-Glucoside Derivative. U.S. Patent 12/521,644, 16 December 2008. [Google Scholar]
- Fushimi, N.; Fujikura, H.; Shiohara, H.; Teranishi, H.; Shimizu, K.; Yonekubo, S.; Ohno, K.; Miyagi, T.; Itoh, F.; Shibazaki, T.; et al. Structure–activity relationship studies of 4-benzyl-1H-pyrazol-3-yl β-d-glucopyranoside derivatives as potent and selective sodium glucose co-transporter 1 (SGLT1) inhibitors with therapeutic activity on postprandial hyperglycemia. Bioorg. Med. Chem. 2012, 20, 6598–6612. [Google Scholar] [CrossRef] [PubMed]
- Foley, K.P.; Du, Z. Method for Treating Proliferative Disorders Associated with Mutations in C-Met. U.S. Patent 9,108,933, 18 August 2015. [Google Scholar]
- Pfefferkorn, J.A.; Choi, C.; Winters, T.; Kennedy, R.; Chi, L.; Perrin, L.A.; Lu, G.; Ping, Y.-W.; McClanahan, T.; Schroeder, R.; et al. P2Y1 receptor antagonists as novel antithrombotic agents. Bioorg. Med. Chem. Lett. 2008, 18, 3338–3343. [Google Scholar] [CrossRef] [PubMed]
- Song, M.-X.; Zheng, C.-J.; Deng, X.-Q.; Sun, L.-P.; Wu, Y.; Hong, L.; Li, Y.-J.; Liu, Y.; Wei, Z.-Y.; Jin, M.-J.; et al. Synthesis and antibacterial evaluation of rhodanine-based 5-aryloxy pyrazoles against selected methicillin resistant and quinolone-resistant Staphylococcus aureus (MRSA and QRSA). Eur. J. Med. Chem. 2013, 60, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Jardosh, H.; Sangani, C.B.; Patel, M.P.; Patel, R.G. One step synthesis of pyrido[1,2-a]benzimidazole derivatives of aryloxypyrazole and their antimicrobial evaluation. Chin. Chem. Lett. 2013, 24, 123–126. [Google Scholar] [CrossRef]
- Tang, Y.-Q.; Sattler, I.; Thiericke, R.; Grabley, S.; Feng, X.-Z. ChemInform Abstract: Maremycins C (I) and D (II), New Diketopiperazines, and Maremycins E (III) and F (IV), Novel Polycyclic spiro-Indole Metabolites Isolated from Streptomyces sp. Eur. J. Org. Chem. 2010, 33, 261–267. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, C.; Lin, H.; Liu, Y.; Li, Y.; Zhao, Y.; Li, P.; Liu, J. Discovery of the Potential Biomarkers for Discrimination between Hedyotis diffusa and Hedyotis corymbosa by UPLC-QTOF/MS Metabolome Analysis. Molecules 2018, 23, 1525. [Google Scholar] [CrossRef] [Green Version]
- Qian, P.; Zhang, Y.-B.; Yang, Y.-F.; Xu, W.; Yang, X. Pharmacokinetics Studies of 12 Alkaloids in Rat Plasma after Oral Administration of Zuojin and Fan-Zuojin Formulas. Molecules 2017, 22, 214. [Google Scholar] [CrossRef] [Green Version]
- Koguchi, Y.; Kohno, J.; Nishio, M.; Takahashi, K.; Okuda, T.; Ohnuki, T.; Komatsubara, S. TMC-95A, B, C, and D, novel proteasome inhibitors produced by Apiospora montagnei Sacc. TC 1093. Taxonomy, production, isolation, and biological activities. J. Antibiot. 2000, 53, 105–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.; Yang, Z.-Q.; Kwok, B.H.B.; Koldobskiy, M.; Crews, C.M.; Danishefsky, S.J. Total Synthesis of TMC-95A and -B via a New Reaction Leading toZ-Enamides. Some Preliminary Findings as to SAR. J. Am. Chem. Soc. 2004, 126, 6347–6355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantouris, G.; Loudon-Griffiths, J.; Mowat, C.G. Insights into the mechanism of inhibition of tryptophan 2,3-dioxygenase by isatin derivatives. J. Enzym. Inhib. Med. Chem. 2016, 31, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raj, M.; Veerasamy, N.; Singh, V.K. Highly enantioselective synthesis of 3-cycloalkanone-3-hydroxy-2-oxindoles, potential anticonvulsants. Tetrahedron Lett. 2010, 51, 2157–2159. [Google Scholar] [CrossRef]
- Prathima, P.S.; Rajesh, P.; Rao, V.J.; Kailash, U.S.; Sridhar, B.; Rao, M.M. “On water” expedient synthesis of 3-indolyl-3-hydroxy oxindole derivatives and their anticancer activity in vitro. Eur. J. Med. Chem. 2014, 84, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, T.; Hume, W.E.; Umezome, T.; Okazaki, K.; Ueki, Y.; Kumagai, K.; Hourai, S.; Nagamine, J.; Seki, H.; Taiji, M.; et al. Oxindole derivatives as orally active potent growth hormone secretagogues. J. Med. Chem. 2001, 44, 4641–4649. [Google Scholar] [CrossRef] [PubMed]
- Tomita, D.; Yamatsugu, K.; Kanai, M.; Shibasaki, M. Enantioselective Synthesis of SM-130686 Based on the Development of Asymmetric Cu(I)F Catalysis to Access 2-Oxindoles Containing a Tetrasubstituted Carbon. J. Am. Chem. Soc. 2009, 131, 6946–6948. [Google Scholar] [CrossRef]
- Bergman, J. Oxindoles. Advances in Heterocyclic Chemistry 2015, 117, 1–81. [Google Scholar] [CrossRef]
- Medvedev, A.E.; Buneeva, O.; Glover, V. Biological targets for isatin and its analogues: Implications for therapy. Boil. Targets Ther. 2007, 1, 151–162. [Google Scholar]
- Liu, H.; Wu, H.-Y.; Luo, Z.; Shen, J.; Kang, G.; Liu, B.; Wan, Z.; Jiang, J. Regioselectivity-Reversed Asymmetric Aldol Reaction of 1,3-Dicarbonyl Compounds. Chem. A Eur. J. 2012, 18, 11899–11903. [Google Scholar] [CrossRef]
- Zhang, D.; Chen, Y.; Cai, H.; Yin, L.; Zhong, J.; Man, J.; Zhang, Q.-F.; Bethi, V.; Tanaka, F. Direct Catalytic Asymmetric Synthesis of Oxindole-Derived δ-Hydroxy-β-ketoesters by Aldol Reactions. Org. Lett. 2019, 22, 6–10. [Google Scholar] [CrossRef]
- Nagaraju, S.; Satyanarayana, N.; Paplal, B.; Vasu, A.K.; Kanvah, S.; Kashinath, D. Synthesis of functionalized isoxazole–oxindole hybrids via on water, catalyst free vinylogous Henry and 1,6-Michael addition reactions. RSC Adv. 2015, 5, 81768–81773. [Google Scholar] [CrossRef]
- Shvekhgeimer, M.G.-A. The Pfitzinger Reaction. Eur. J. Org. Chem. 2004, 35, 257–294. [Google Scholar] [CrossRef]
- Ramann, G.A.; Cowen, B. Recent Advances in Metal-Free Quinoline Synthesis. Molecules 2016, 21, 986. [Google Scholar] [CrossRef] [PubMed]
- Health and Safety Executive. EH40/2005 Workplace Exposure Limits Limits for Use with the Control of Substances, 4th ed.; The Stationery Office: London, UK, 2020. [Google Scholar]
- Ayoub, M.T.; Shandala, M.Y.; Bashi, G.M.G.; Pelter, A. The conversion of 5,6-dihydro-4-methoxy-2-pyrones into 3-alkyl-5-hydroxypyrazoles. J. Chem. Soc. Perkin Trans. 1 1981, 697. [Google Scholar] [CrossRef]
- Dolomanov, O.; Bourhis, L.J.; Gildea, R.; Howard, J.A.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT - integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry a | Base (10 mol%) | Solvent | Conversion (%) b | Yield (%) c |
|---|---|---|---|---|
| 1 | Triethylamine | MeOH | 38 | 36 |
| 2 | DBU | MeOH | 13 | 0 |
| 3d | DBU | THF | 88 | 46 |
| 4 | Pyrrolidine | MeOH | 92 | 84 |
| 5 | K2CO3 | MeOH | 77 | 26 |
| 6 | Morpholine | MeOH | 36 | 36 |
| 7 | N,N-diethylethylendiamine | MeOH | 55 | 33 |
| 8 | Piperidine | MeOH | 93 | 87 |
| 9e | Piperidine | MeOH | - | - |
| 10 | Piperidine | iPrOH | 96 | 84 |
| 11 | Piperidine | H2O | 53 | 15 |
| 12 | Piperidine | MeCN | 87 | 59 |
| 13 | Piperidine | THF | 90 | 67 |
| 14 | Piperidine | Toluene | 61 | 27 |
| 15 | Piperidine | AcOEt | 84 | 81 |

| Entry a | Reagent b | Conv. 15 (%) c | Conv. 16 (%) c |
|---|---|---|---|
| 1 | NH2NH2 ∙H2O | 21 | 36 |
| 2 | NH2NH2 ∙HCl | 20 | 20 |
| 3d | NH2NH2 | 53 | 42 |

| Entry a | Reaction Time (h) | Hydrazine Addition | Conv. 15 (%) b | Conv. 16 (%) b |
|---|---|---|---|---|
| 1 | 96 | fast | 21 | 36 |
| 2 | 2 | fast | 28 | 17 |
| 3 | 2 | dropwise c | 50 (38) | 16 (15) |
| 4d | 2 | dropwise | 43 | 11 |
| 5e | 2 | dropwise | 53 | 10 |

| Entry a | X | Name | Conv. 15 (%) b | Conv. 16 (%) b |
|---|---|---|---|---|
| 1 | 4-F | a | 13 (10) | 9 (- c) |
| 2 | 4-Cl | b | 92 (81) | 0 |
| 3 | 4-Br | c | 93 (91) | 0 |
| 4 | 5-Cl | d | 46 (40) | 22 (20) |
| 5 | 5-MeO | e | 45 (41) | - c (11) |
| 6 | 5-F | f | 46 (44) | 30 (25) |
| 7 | 6-Cl | g | 57 (55) | 10 (5) |
| 8 | 7-Cl | h | 44 (38) | 12 (10) |
| 9 | 7-CF3 | i | 54 (53) | 5 (- c) |
| 10 | 7-F | j | 32 (31) | - c (3) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gambacorta, G.; Apperley, D.C.; Baxendale, I.R. A One-Pot Divergent Sequence to Pyrazole and Quinoline Derivatives. Molecules 2020, 25, 2160. https://doi.org/10.3390/molecules25092160
Gambacorta G, Apperley DC, Baxendale IR. A One-Pot Divergent Sequence to Pyrazole and Quinoline Derivatives. Molecules. 2020; 25(9):2160. https://doi.org/10.3390/molecules25092160
Chicago/Turabian StyleGambacorta, Guido, David C. Apperley, and Ian R. Baxendale. 2020. "A One-Pot Divergent Sequence to Pyrazole and Quinoline Derivatives" Molecules 25, no. 9: 2160. https://doi.org/10.3390/molecules25092160
APA StyleGambacorta, G., Apperley, D. C., & Baxendale, I. R. (2020). A One-Pot Divergent Sequence to Pyrazole and Quinoline Derivatives. Molecules, 25(9), 2160. https://doi.org/10.3390/molecules25092160