Studies of Coumarin Derivatives for Constitutive Androstane Receptor (CAR) Activation

 ,
,  , , , ,
, , , ,
Abstract
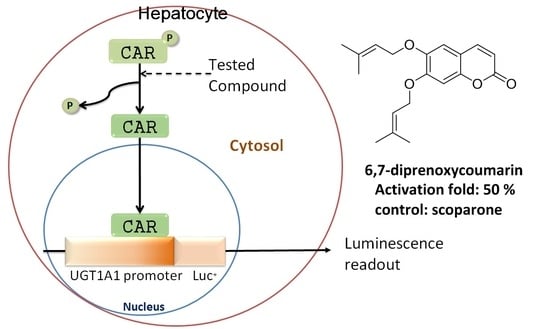
:
1. Introduction
2. Results and Discussion
Chemistry
3. Establishment of In Vitro CAR Activation Screening Assay
4. Structure-Activity Relationships
5. Mechanistic Studies of 6,7-Diprenoxycoumarin (50)
6. Further In Vivo Hypoglycemic Effect and Pharmacokinetic Studies
7. Conclusions
8. Materials and Methods
8.1. General Information
8.2. Chemistry
8.3. General Procedure for the Synthesis of Various Alkoxyl Courmarin Derivatives
8.4. Cell Lines
8.5. Cell Culture
8.6. Cell Cytotoxicity Assay of Coumarin Derivatives
8.7. Reverse Transcription Polymerase Chain Reaction (RT-PCR)
8.8. Primer Sequences
| Primer | Sequence |
| UGT1A1 | Forward: 5′-TGCAAAGCGCATGGAGACTA-3′ |
| Reverse: 5′-GAGGCGCATGATGTTCTCCT-3′ | |
| MRP2 | Forward: 5′-AGGTGAGGATTGACACCAACCA-3′ |
| Reverse: 5′-AGGCAGTTTGTGAGGGATGACT-3′ | |
| CAR | Forward: 5′-GTGCTGCCTCTGGTCACACACT-3′ |
| Reverse: 5′-GAGGCCCGCAGAGGAAGTTT-3′ | |
| GAPDH | Forward: 5′-GTCTCCTCTGACTTCAACAGCG-3′ |
| Reverse: 5′-ACCACCCTGTTGCTGTAGCCAA-3′ | |
| gtPBREM | Forward: 5′-GTTTCCGCTAGCTACACTAGTAAAGGTCACTC-3′ |
| Reverse: 5′-GTTTAACTCGAGCCCTCTAGCCATTCTGGATC-3′ |
8.9. Cloning and Transfection
8.10. Luciferase Assays
8.11. Nuclear and Cytoplasmic Extraction
8.12. Protein Extraction
8.13. Western Blots
8.14. Immunofluorescence
8.15. Animals
8.16. Oral Glucose Tolerance Test (OGTT)
8.17. Determination of Blood Insulin and Fructosamine
8.18. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Banerjee, M.; Robbins, D.; Chen, T. Targeting xenobiotic receptors PXR and CAR in human diseases. Drug Discov. Today 2015, 20, 618–628. [Google Scholar] [CrossRef] [Green Version]
- Trauner, M.; Wagner, M.; Fickert, P.; Zollner, G. Molecular regulation of hepatobiliary transport systems: Clinical implications for understanding and treating cholestasis. J. Clin. Gastroenterol. 2005, 39, S111–S124. [Google Scholar] [CrossRef]
- Kachaylo, E.M.; Pustylnyak, V.O.; Lyakhovich, V.V.; Gulyaeva, L.F. Constitutive androstane receptor (CAR) is a xenosensor and target for therapy. Biochemistry 2011, 76, 1087–1097. [Google Scholar] [CrossRef]
- Goodwin, B.; Hodgson, E.; D’Costa, D.J.; Robertson, G.R.; Liddle, C. Transcriptional regulation of the human CYP3A4 gene by the constitutive androstane receptor. Mol. Pharmacol. 2002, 62, 359–365. [Google Scholar] [CrossRef] [Green Version]
- Echchgadda, I.; Song, C.S.; Oh, T.; Ahmed, M.; De La Cruz, I.J.; Chatterjee, B. The xenobiotic-sensing nuclear receptors pregnane X receptor, constitutive androstane receptor, and orphan nuclear receptor hepatocyte nuclear factor 4alpha in the regulation of human steroid-/bile acid-sulfotransferase. Mol. Endocrinol. 2007, 21, 2099–2111. [Google Scholar] [CrossRef] [Green Version]
- Wagner, M.; Halilbasic, E.; Marschall, H.U.; Zollner, G.; Fickert, P.; Langner, C.; Zatloukal, K.; Denk, H.; Trauner, M. CAR and PXR agonists stimulate hepatic bile acid and bilirubin detoxification and elimination pathways in mice. Hepatology 2005, 42, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Huang, W.; Qatanani, M.; Evans, R.M.; Moore, D.D. The constitutive androstane receptor and pregnane X receptor function coordinately to prevent bile acid-induced hepatotoxicity. J. Biol. Chem. 2004, 279, 49517–49522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; He, J.; Zhai, Y.; Wada, T.; Xie, W. The constitutive androstane receptor is an anti-obesity nuclear receptor that improves insulin sensitivity. J. Biol. Chem. 2009, 284, 25984–25992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, B.; Saha, P.K.; Huang, W.; Chen, W.; Abu-Elheiga, L.A.; Wakil, S.J.; Stevens, R.D.; Ilkayeva, O.; Newgard, C.B.; Chan, L.; et al. Activation of nuclear receptor CAR ameliorates diabetes and fatty liver disease. Proc. Natl. Acad. Sci. USA 2009, 106, 18831–18836. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Coslo, D.M.; Chen, T.; Zhang, L.; Tian, Y.; Smith, P.B.; Patterson, A.D.; Omiecinski, C.J. Metabolomic Approaches Reveal the Role of CAR in Energy Metabolism. J. Proteome Res. 2019, 18, 239–251. [Google Scholar] [CrossRef] [Green Version]
- Tzameli, I.; Pissios, P.; Schuetz, E.G.; Moore, D.D. The xenobiotic compound 1,4-bis [2-(3,5-dichloropyridyloxy)] benzene is an agonist ligand for the nuclear receptor CAR. Mol. Cell. Biol. 2000, 20, 2951–2958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, W.; Zhang, J.; Moore, D.D. A traditional herbal medicine enhances bilirubin clearance by activating the nuclear receptor CAR. J. Clin. Investig. 2004, 113, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Elferink, R.O. Yin Zhi Huang and other plant-derived preparations: Where herbal and molecular medicine meet. J. Hepatol. 2004, 41, 691–693. [Google Scholar] [CrossRef]
- Chen, Z.; Ma, X.; Zhao, Y.; Wang, J.; Zhang, Y.; Li, J.; Wang, R.; Zhu, Y.; Wang, L.; Xiao, X. Yinchenhao decoction in the treatment of cholestasis: A systematic review and meta-analysis. J. Ethnopharm. 2015, 168, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Zhang, Q.; Sun, R.; Wu, J.; Li, X.; Liu, R. Recent progress in the study of Artemisiae Scopariae Herba (Yin Chen), a promising medicinal herb for liver diseases. Biomed. Pharmacother. 2020, 130, 11053. [Google Scholar] [CrossRef]
- Yang, D.; Yang, J.; Shi, D.; Deng, R.; Yan, B. Scoparone potentiates transactivation of the bile salt export pump gene and this effect is enhanced by cytochrome P450 metabolism but abolished by a PKC inhibitor. Br. J. Pharmacol. 2011, 164, 154–157. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.S.; Tsang, Z.J.; Wu, P.L.; Lin, F.W.; Li, C.Y.; Teng, C.M.; Lee, K.H. New constituents and antiplatelet aggregation and anti-HIV principles of Artemisia capillaris. Bioorg. Med. Chem. 2001, 9, 77–83. [Google Scholar] [CrossRef]
- Perkin, W.H. On the artificial production of coumarin and formation of its homologues. J. Chem. Soc. 1868, 21, 53–63. [Google Scholar] [CrossRef] [Green Version]
- Bogdal, D. Coumarins: Fast synthesis by Knoevenagel condensation under microwave irradiation. J. Chem. Res. 1998, 8, 468–469. [Google Scholar] [CrossRef]
- Joule, J.A.; Mills, K. Heterocyclic Chemistry; Wiley: Hoboken, NJ, USA, 2013. [Google Scholar]
- Desai, V.G.; Shet, J.B.; Tilve, S.G.; Mali, R.S. Intramolecular Wittig reactions. A new synthesis of coumarins and 2-quinolones. J. Chem. Res. 2003, 2003, 628–629. [Google Scholar] [CrossRef]
- Boeck, F.; Blazejak, M.; Anneser, M.R.; Hintermann, L. Cyclization of ortho-hydroxycinnamates to coumarins under mild conditions: A nucleophilic organocatalysis approach. Beilstein J. Org. Chem. 2012, 8, 1630–1636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, H.S.P.; Sivakumar, S. Condensation of α-Aroylketene dithioacetals and 2-hydroxyarylaldehydes results in facile synthesis of a combinatorial library of 3-aroylcoumarins. J. Org. Chem. 2006, 71, 8715–8723. [Google Scholar] [CrossRef] [PubMed]
- Oyamada, J.; Kitamura, T. Synthesis of coumarins by Pt-catalyzed hydroarylation of propiolic acids with phenols. Tetrahedron 2006, 62, 6918–6925. [Google Scholar] [CrossRef]
- Patnam, R.; Chang, F.-R.; Chen, C.-Y.; Kuo, R.-Y.; Lee, Y.-H.; Wu, Y.-C. Hydrachine A, a novel alkaloid from the roots of Hydrangea chinensis. J. Nat. Prod. 2001, 64, 948–949. [Google Scholar] [CrossRef]
- Borba, I.C.G.; Bridi, H.; Soares, K.D.; Apel, M.A.; Poser, G.L.; Ritter, M.R. New natural coumarins from Trichocline macrocephala (Asteraceae). Phytochem. Lett. 2019, 32, 129–133. [Google Scholar] [CrossRef]
- Bratulescu, G. A Quick and advantageous synthesis of 2H-1-benzopyran-2-ones unsubstituted on the pyranic nucleus. Synthesis 2008, 2008, 2871–2873. [Google Scholar] [CrossRef]
- Kaighen, M.; Williams, R.T. The metabolism of [3-14C] coumarin. J. Med. Pharm. Chem. 1961, 3, 25–43. [Google Scholar] [CrossRef]
- Takaishi, K.; Izumi, M.; Baba, N.; Kawazu, K.; Nakajima, S. Synthesis and biological evaluation of alkoxycoumarins as novel nematicidal constituents. Bioorg. Med. Chem. Lett. 2008, 18, 5614–5617. [Google Scholar] [CrossRef]
- Iranshahi, M.; Jabbari, A.; Orafaie, A.; Mehri, R.; Zeraatkar, S.; Ahmadi, T.; Alimardani, M.; Sadeghian, H. Synthesis and SAR studies of mono O-prenylated coumarins as potent 15-lipoxygenase inhibitors. Eur. J. Med. Chem. 2012, 57, 134–142. [Google Scholar] [CrossRef]
- Farooq, S.; Shakeel-u-Rehman Dangroo, N.A.; Priya, D.; Banday, J.A.; Sangwan, P.L.; Qurishi, M.A.; Koul, S.; Saxena, A.K. Isolation, cytotoxicity evaluation and HPLC-quantification of the chemical constituents from Prangos pabularia. PLoS ONE 2014, 9, e108713. [Google Scholar] [CrossRef]
- Ding, C.; Zhang, W.; Li, J.; Lei, J.; Yu, J. Cytotoxic constituents of ethyl acetate fraction from Dianthus superbus. Nat. Prod. Res. 2013, 27, 1691–1694. [Google Scholar] [CrossRef] [PubMed]
- Garro, H.A.; Reta, G.F.; Donadel, O.J.; Pungitore, C.R. Cytotoxic and antitumor activity of some coumarin derivatives. Nat. Prod. Commun. 2016, 11, 1289–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chattopadhyay, S.K.; Mondal, P.; Ghosh, D. A new route to pyranocoumarins and their benzannulated derivatives. Synthesis 2014, 46, 3331–3340. [Google Scholar] [CrossRef]
- Adfa, M.; Itoh, T.; Hattori, Y.; Koketsu, M. Inhibitory effects of 6-alkoxycoumarin and 7-alkoxycoumarin derivatives on lipopolysaccharide/interferon γ-stimulated nitric oxide production in RAW264 cells. Biol. Pharm. Bull. 2012, 35, 963–966. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Recillas, A.; Navarrete-Vázquez, G.; Hidalgo-Figueroa, S.; Rios, M.Y.; Ibarra-Barajas, M.; Estrada-Soto, S. Semisynthesis, ex vivo evaluation, and SAR studies of coumarin derivatives as potential antiasthmatic drugs. Eur. J. Med. Chem. 2014, 77, 400–408. [Google Scholar] [CrossRef]
- Liu, G.-L.; Hao, B.; Liu, S.-P.; Wang, G.-X. Synthesis and anthelmintic activity of osthol analogs against Dactylogyrus intermedius in goldfish. Eur. J. Med. Chem. 2012, 54, 582–590. [Google Scholar] [CrossRef]
- Huang, W.-J.; Wang, Y.-C.; Chao, S.-W.; Yang, C.-Y.; Chen, L.-C.; Lin, M.-H.; Hou, W.-C.; Chen, M.-Y.; Lee, T.-L.; Yang, P.; et al. Synthesis and biological evaluation of ortho-aryl N-hydroxycinnamides as potent histone deacetylase (HDAC) 8 isoform-selective inhibitors. Chem. Med. Chem. 2012, 7, 1815–1824. [Google Scholar] [CrossRef]
- Riveiro, M.E.; Maes, D.; Vazquez, R.; Vermeulen, M.; Mangelinckx, S.; Jacobs, J.; Debenedetti, S.; Shayo, C.; De Kimpe, N.; Davio, C. Toward establishing structure-activity relationships for oxygenated coumarins as differentiation inducers of promonocytic leukemic cells. Bioorg. Med. Chem. 2009, 17, 6547–6559. [Google Scholar] [CrossRef]
- Zhu, L.J.; Hou, Y.L.; Shen, X.Y.; Pan, X.D.; Zhang, X.; Yao, X.S. Monoterpene pyridine alkaloids and phenolics from Scrophularia ningpoensis and their cardioprotective effect. Fitoterapia 2013, 88, 44–49. [Google Scholar] [CrossRef]
- Nagarajan, G.R.; Rani, U.; Parmar, V.S. Coumarins from Fraxinus floribunda leaves. Phytochemistry 1980, 19, 2494–2495. [Google Scholar] [CrossRef]
- Litinas, K.E.; Mangos, A.; Nikkou, T.E.; Hadjipavlou-Litina, D.J. Synthesis and biological evaluation of fused oxepinocoumarins as free radicals scavengers. J. Enzyme Inhib. Med. Chem. 2011, 26, 805–812. [Google Scholar] [CrossRef] [Green Version]
- Sellers, E.M.; Tyndale, R.F. NICOGEN INC., CYP2a Enzymes and Their Use in Therapeutic and Diagnostic Methods. WO1999027919A2, 6 October 1999. [Google Scholar]
- Garg, S.S.; Gupta, J.; Sharma, S.; Sahu, D. An insight into the therapeutic applications of coumarin compounds and their mechanisms of action. Eur. J. Pharm. Sci. 2020, 152, 105424. [Google Scholar] [CrossRef] [PubMed]
- Maqua, M.P.; Vines, A.C.G.; Caballero, E.; Grande, M.C.; Medarde, M.; Bellido, I.S. Components from Santolina rosmarinifolia, subspecies rosmarinifolia and canescens. Phytochemistry 1988, 27, 3664–3667. [Google Scholar] [CrossRef]
- Bohlmann, F.; Suwita, A.; King, R.M.; Robinson, H. Neue ent-labdan-derivate aus Austroeupatorium chaparense. Phytochemistry 1980, 19, 111–114. [Google Scholar] [CrossRef]
- Guillaumet, G.; Hretani, M.; Coudert, G.; Averbeck, D.; Averbeck, S. Synthesis and photoinduced biological properties of linear dioxinocoumarins. Eur. J. Med. Chem. 1990, 25, 45–51. [Google Scholar] [CrossRef]
- Ballantyne, M.M.; McCabe, P.H.; Murray, R.D.H. Claisen rearrangements—II: Synthesis of six natural coumarins. Tetrahedron 1971, 27, 871–877. [Google Scholar] [CrossRef]
- Genovese, S.; Epifano, F.; Medina, P.; Caron, N.; Rives, A.; Poirot, M.; Silvente-Poirot, S.; Fiorito, S. Natural and semisynthetic oxyprenylated aromatic compounds as stimulators or inhibitors of melanogenesis. Bioorg. Chem. 2019, 87, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Orhan, I.E.; Deniz, F.S.S.; Salmas, R.E.; Durdagi, S.; Epifano, F.; Genovese, S.; Fiorito, S. Combined molecular modeling and cholinesterase inhibition studies on some natural and semisynthetic O-alkylcoumarin derivatives. Bioorg. Chem. 2019, 84, 355–362. [Google Scholar] [CrossRef]
- Blagbrough, I.S.; Bayoumi, S.A.; Rowan, M.G.; Beeching, J.R. Cassava: An appraisal of its phytochemistry and its biotechnological prospects. Phytochemistry 2010, 71, 1940–1951. [Google Scholar] [CrossRef]
- Medeiros-Neves, B.; Teixeira, H.F.; Poser, G.L. The genus Pterocaulon (Asteraceae)—A review on traditional medicinal uses, chemical constituents and biological properties. J. Ethnopharm. 2018, 224, 451–464. [Google Scholar] [CrossRef]
- Kim, K.S.; Lee, S.; Shin, J.S.; Shim, S.H.; Kim, B. Arteminin, a new coumarin from Artemisia apiacea. Fitoterapia 2002, 73, 266–268. [Google Scholar] [CrossRef]
- Maglich, J.M.; Parks, D.J.; Moore, L.B.; Collins, J.L.; Goodwin, B.; Billin, A.N.; Stoltz, C.A.; Kliewer, S.A.; Lambert, M.H.; Willson, T.M.; et al. Identification of a novel human constitutive androstane receptor (CAR) agonist and its use in the identification of CAR target genes. J. Biol. Chem. 2003, 278, 17277–17283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugatani, J. Function, genetic polymorphism, and transcriptional regulation of human UDP-glucuronosyltransferase (UGT) 1A1. Drug Metab. Pharmacokinet. 2013, 28, 83–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutoh, S.; Sobhany, M.; Moore, R.; Perera, L.; Pedersen, L.; Sueyoshi, T.; Negishi, M. Phenobarbital indirectly activates the constitutive active androstane receptor (CAR) by inhibition of epidermal growth factor receptor signaling. Sci. Signal. 2013, 6, ra31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, M.B.; Nielsen, S.E.; Berg, K. Re-examination and further development of a precise and rapid dye method for measuring cell growth/cell kill. J. Immunol. Methods 1989, 119, 203–210. [Google Scholar] [CrossRef]
- Institute of Laboratory Animal Resources Commission on Life Sciences. National Research Council. In Guide for the Care and Use of Laboratory Animals, 8th ed.; National Academy Press: Washington, DC, USA, 2011. [Google Scholar]
- Wallace, T.M.; Levy, J.C.; Matthews, D.R. Use and abuse of HOMA modeling. Diabetes Care 2004, 27, 1487–1495. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
| Compound | R1 | R2 | R3 | R4 | CAR Activation Fold a | Ref |
| 1 5-hydroxycoumarin | OH | H | H | H | 6% ± 22% | [25] |
| 2 5-methoxycoumarin | O-methyl | H | H | H | −20% ± 6% | [26,27] |
| 3 | O-acetyl | H | H | H | −32% ± 12% | [28] |
| 4 | O-allyl | H | H | H | −15% ± 6% | [29] |
| 5 | O-n-butyl | H | H | H | −9% ± 14% | [29] |
| 6 | O-prenyl | H | H | H | −5% ± 9% | [30] |
| 7 | O-geranyl | H | H | H | −18% ± 11% | [30] |
| 8 | O-farnasyl | H | H | H | 2% ± 14% | [30] |
| 9 6-hydroxycoumarin | H | OH | H | H | −8% ± 11 % | [31] |
| 10 6-methoxycoumarin | H | O-methyl | H | H | 32% ± 5% | [32] |
| 11 | H | O-trifluoromethyl | H | H | −34% ± 1% | New |
| 12 | H | O-acetyl | H | H | −22% ± 3% | [33] |
| 13 | H | O-allyl | H | H | −17% ± 6% | [34] |
| 14 | H | O-n-butyl | H | H | 37% ± 6% | [35] |
| 15 | H | O-prenyl | H | H | 22% ± 2% | [30] |
| 16 | H | O-geranyl | H | H | 18% ± 7% | [30] |
| 17 | H | O-farnasyl | H | H | −41% ± 9% | [30] |
| 18 umbelliferone | H | H | OH | H | −1% ± 18% | [31,36] |
| 19 7-methoxycoumarin | H | H | O-methyl | H | −9% ± 6% | [25,27,36] |
| 20 | H | H | O-trifluoromethyl | H | 18% ± 2% | New |
| 21 | H | H | O-acetyl | H | −37% ± 12% | [37] |
| 22 | H | H | O-allyl | H | −46% ± 11% | [37] |
| 23 | H | H | O-n-butyl | H | −51% ± 7% | [38] |
| 24 | H | H | O-prenyl | H | −35% ± 13% | [30,39] |
| 25 aurapten | H | H | O-geranyl | H | −13% ± 4% | [30] |
| 26 | H | H | O-farnasyl | H | −29% ± 10% | [30] |
| 27 8-hydroxycoumarin | H | H | H | OH | 40% ± 38% | [40] |
| 28 8-methoxycoumarin | H | H | H | O-methyl | −8% ± 10% | [41] |
| 29 | H | H | H | O-acetyl | −33% ± 13% | [28] |
| 30 | H | H | H | O-allyl | −34% ± 12% | [42] |
| 31 | H | H | H | O-n-butyl | 4% ± 9% | [43] |
| 32 | H | H | H | O-prenyl | 2% ± 23% | [30] |
| 33 | H | H | H | O-geranyl | −30% ± 4% | [30] |
| 34 | H | H | H | O-farnasyl | 19% ± 25% | [30] |
 | ||||
| Compound | R1 | R2 | CAR Activation Fold a | Ref |
| 35 esculetin | OH | OH | −22% ± 12% | [36,44] |
| 36 | methyl | methyl | −17% ± 5% | [24] |
| 37 isoscopoletin | OH | O-methyl | −10% ± 6% | [45] |
| 38 scopoletin | O-methyl | OH | −1% ± 5% | [44] |
| 39 scoparone | O-methyl | O-methyl | - | [15,17] |
| 40 ayapin | -OCH2O- | 7% ± 2% | [46] | |
| 41 | -OCH2CH2O- | −50% ± 9% | [47] | |
| 42 | O-ethyl | O-ethyl | −26% ± 8% | [37] |
| 43 | O-acetyl | O-acetyl | −11% ± 2% | [37] |
| 44 | O-allyl | O-allyl | −18% ± 15% | New |
| 45 | O-propyl | O-propyl | −37% ± 5% | [37] |
| 46 | O-n-butyl | O-n-butyl | −39% ± 13% | New |
| 47 | O-pentyl | O-pentyl | −10% ± 18% | New |
| 48 | O-isopentyl | O-isopentyl | −61% ± 2% | New |
| 49 | O-hexyl | O-hexyl | −5% ± 9% | New |
| 50 | O-prenyl | O-prenyl | 50% ± 1% | [48,49,50] |
| 51 | O-geranyl | O-geranyl | −32% ± 11% | New |
| 52 isoscopolin | O-Glc | O-methyl | 35% ± 5% | [17,51] |
| 53 scopolin | O-methyl | O-Glc | 17% ± 26% | [17] |
 | ||||||
| Compound | R1 | R2 | R3 | R4 | CAR Activation Fold a | Ref |
| 54 | -OCH3 | -OCH3 | -OCH3 | H | −19% ± 3% | [39,44] |
| 55 fraxinol | -OCH3 | -OH | -OCH3 | H | −17% ± 2% | [39] |
| 56 | -OCH3 | H | -OCH3 | -OCH3 | −14% ± 2% | [39] |
| 57 leptodactylone | -OCH3 | H | -OCH3 | -OH | 2% ± 4% | [39] |
| 58 | H | -OCH3 | -OCH3 | -OCH3 | 0% ± 2% | [52] |
| 59 | H | -OCH3 | -OCH2O- | 36% ± 23% | [17,53] | |
| 60 | -OCH3 | -OCH3 | -OCH2O- | 12% ± 1% | [17,53] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Juang, S.-H.; Hsieh, M.-T.; Hsu, P.-L.; Chen, J.-L.; Liu, H.-K.; Liang, F.-P.; Kuo, S.-C.; Chiu, C.-Y.; Liu, S.-H.; Chou, C.-H.; et al. Studies of Coumarin Derivatives for Constitutive Androstane Receptor (CAR) Activation. Molecules 2021, 26, 164. https://doi.org/10.3390/molecules26010164
Juang S-H, Hsieh M-T, Hsu P-L, Chen J-L, Liu H-K, Liang F-P, Kuo S-C, Chiu C-Y, Liu S-H, Chou C-H, et al. Studies of Coumarin Derivatives for Constitutive Androstane Receptor (CAR) Activation. Molecules. 2021; 26(1):164. https://doi.org/10.3390/molecules26010164
Chicago/Turabian StyleJuang, Shin-Hun, Min-Tsang Hsieh, Pei-Ling Hsu, Ju-Ling Chen, Hui-Kang Liu, Fong-Pin Liang, Sheng-Chu Kuo, Chen-Yuan Chiu, Shing-Hwa Liu, Chen-Hsi Chou, and et al. 2021. "Studies of Coumarin Derivatives for Constitutive Androstane Receptor (CAR) Activation" Molecules 26, no. 1: 164. https://doi.org/10.3390/molecules26010164
APA StyleJuang, S.-H., Hsieh, M.-T., Hsu, P.-L., Chen, J.-L., Liu, H.-K., Liang, F.-P., Kuo, S.-C., Chiu, C.-Y., Liu, S.-H., Chou, C.-H., Wu, T.-S., & Hung, H.-Y. (2021). Studies of Coumarin Derivatives for Constitutive Androstane Receptor (CAR) Activation. Molecules, 26(1), 164. https://doi.org/10.3390/molecules26010164