
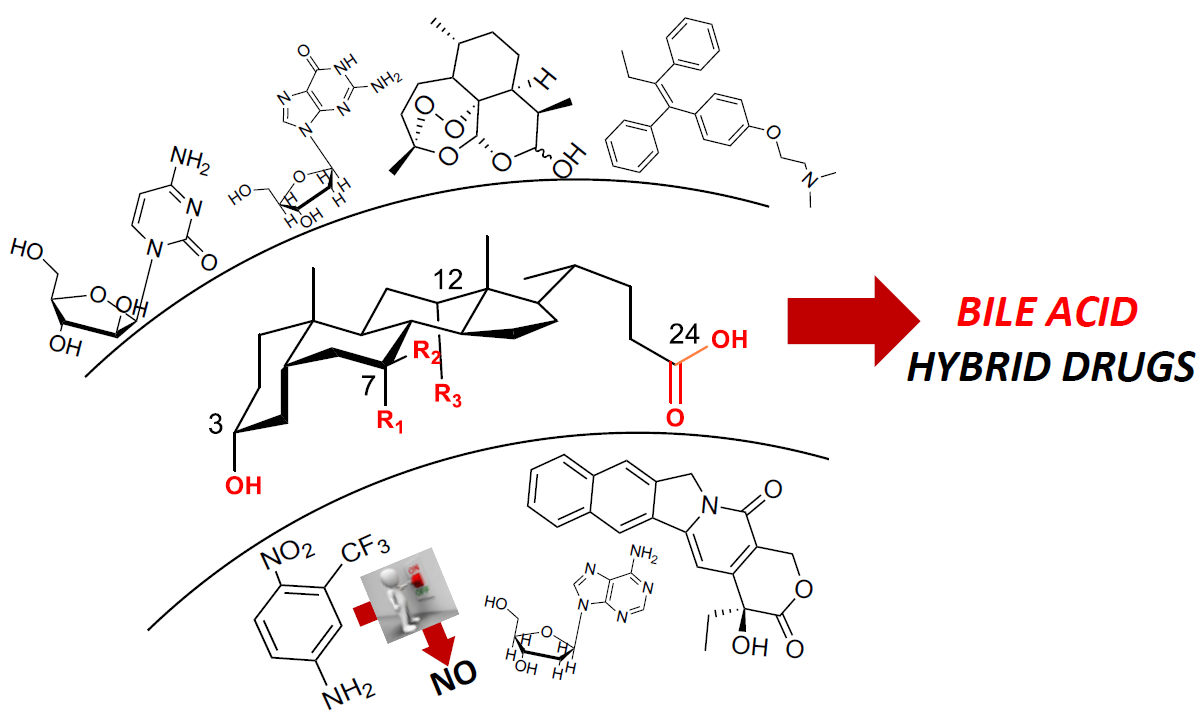
Bile Acid Conjugates with Anticancer Activity: Most Recent Research




Abstract
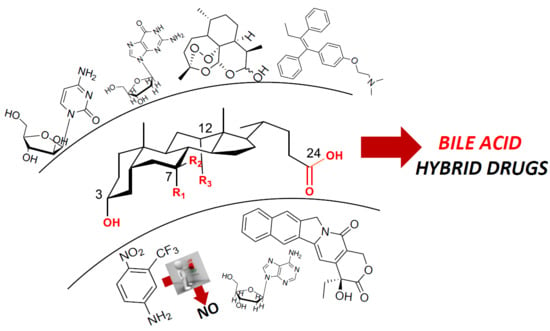
:
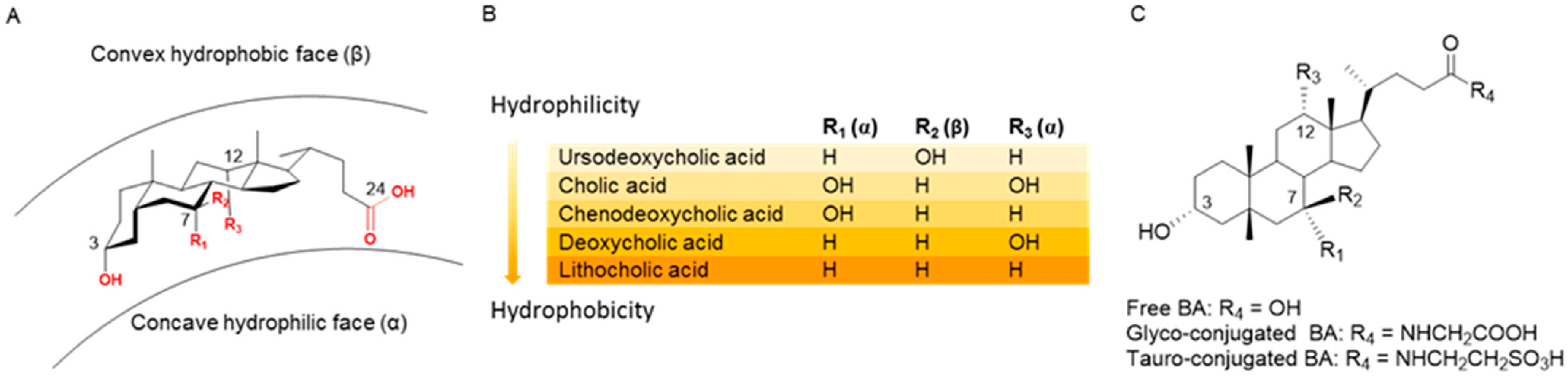
1. Introduction
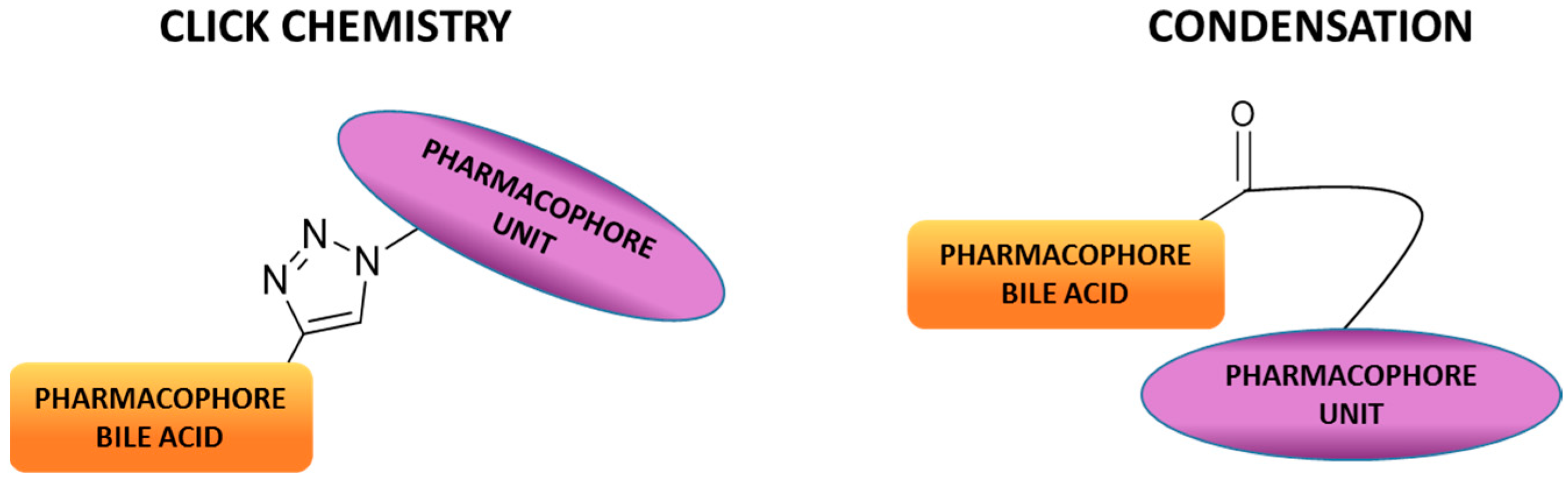
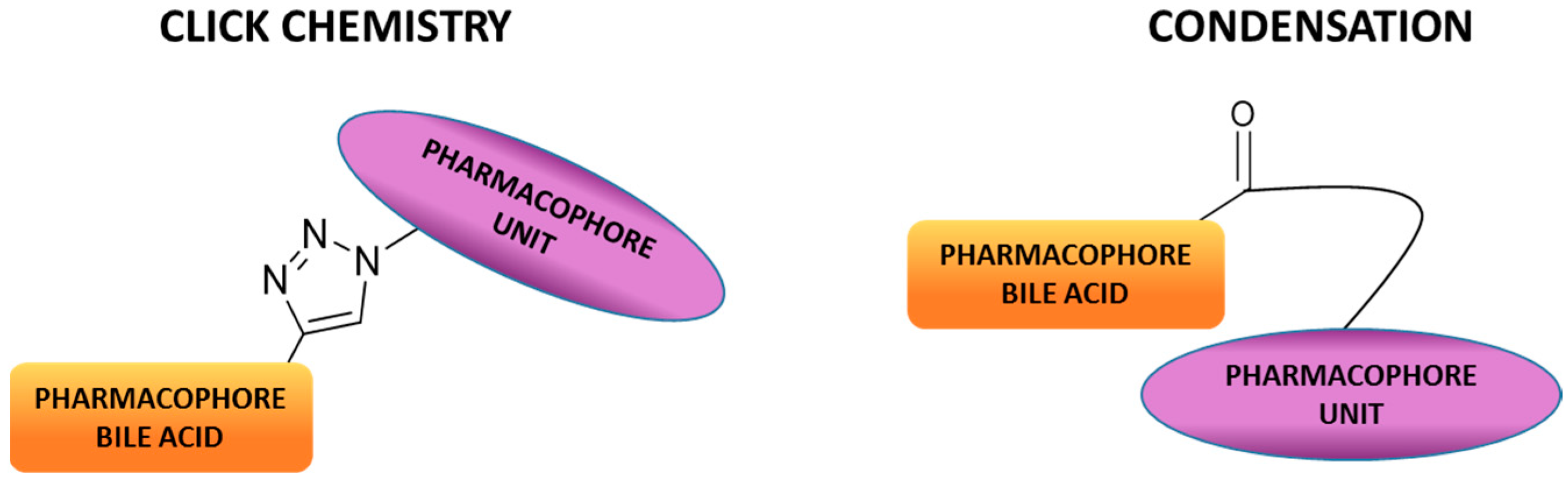
2. Synthetic Pathways to Bile Acid Hybrid Molecules
3. Bile Acid Hybrid Molecules Based on Natural Bioactive Molecules
4. Bile Acid Hybrid Molecules Based on Synthetic Bioactive Molecules
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Croteau, R.; Ketchum, R.E.B.; Long, R.M.; Kaspera, R.; Wildung, M.R. Taxol biosynthesis and molecular genetics. Phytochem. Rev. 2006, 5, 75–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moudi, M.; Go, R.; Yien, C.Y.; Nazre, M. Vinca alkaloids. Int. J. Prev. Med. 2013, 11, 1231–1235. [Google Scholar]
- Lutz, F.; Tietze, L.F.; Bell, H.P.; Chandrasekhar, S. Natural product hybrids as new leads for drug discovery. Angew. Chem. Int. Ed. 2003, 42, 3996–4028. [Google Scholar]
- Zimmermann, G.R.; Lehar, J.; Keith, C.T. Multi-target therapeutics: When the whole is greater than the sum of the parts. Drug Discov. Today 2007, 12, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Ridlon, J.M.; Kang, D.J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006, 47, 241–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, A.F.; Hagey, L.R. Key discovery in bile acid chemistry and biology and their clinical applications: History of the last eight decades. J. Lipid Res. 2014, 55, 1553–1595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorucci, S.; Distrutti, E. The pharmacology of bile acids and their receptors. In Handbook of Experimental Pharmacology; Springer: Cham, Switzerland, 2019; Volume 256, pp. 3–18. [Google Scholar]
- Hegyi, P.; Maléth, J.; Walters, J.R.; Hofmann, A.F.; Keely, S.J. Guts and gall: Bile acids in regulation of intestinal epithelial function in health and disease. Physiol. Rev. 2018, 98, 1983–2023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Chiang, J.Y.L. Bile acid signaling in metabolic disease and drug therapy. Pharmacol. Rev. 2014, 66, 948–983. [Google Scholar] [CrossRef] [Green Version]
- Fiorucci, S.; Baldoni, M.; Ricci, P.; Zampella, A.; Distrutti, E.; Biagioli, M. Bile acid-activated receptors and the regulation of macrophages function in metabolic disorders. Curr. Opin. Pharmacol. 2020, 53, 45–54. [Google Scholar] [CrossRef]
- Zhou, H.; Hylemon, P.B. Bile acids are nutrient signaling hormones. Steroids 2014, 86, 62–68. [Google Scholar] [CrossRef] [Green Version]
- Chavez-Talavera, O.; Tailleux, A.; Lefebvre, P.; Staels, B. bile acid control of metabolism and inflammation in obesity, type 2 diabetes, dyslipidemia, and nonalcoholic fatty liver disease. Gastroenterology 2017, 152, 1679–1694. [Google Scholar] [CrossRef] [PubMed]
- Vitek, L.; Haluzik, M. The role of bile acids in metabolic regulation. J. Endocrinol. 2016, 228, R85–R96. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-García, A.; Sahebkar, A.; Simental-Mendía, M.; Simental-Mendía, L.E. Effect of ursodeoxycholic acid on glycemic markers: A systematic review and metanalysis of clinical trials. Pharmacol. Res. 2018, 135, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, H.; Bernstein, C.; Payne, C.M.; Dvorak, K. Bile acids as endogenous etiologic agents in gastrointestinal cancer. World J. Gastroenterol. 2009, 15, 3329–3340. [Google Scholar] [CrossRef] [PubMed]
- di Ciaula, A.; Wang, Q.-H.; Molina-Molina, E.; Baccetto, R.L.; Calamita, G.; Palmieri, V.O.; Portincasa, P. Bile acid and cancer: Direct and environmental-dependent effects. Ann. Hepatol. 2017, 16, s87–s105. [Google Scholar] [CrossRef] [PubMed]
- Goossens, J.-F.; Bailly, C. Ursodeoxycholic acid and cancer: From chemoprevention to chemotherapy. Pharmacol. Ther. 2019, 203, 107396. [Google Scholar] [CrossRef] [PubMed]
- Dalpiaz, A.; Paganetto, G.; Pavan, B.; Fogagnolo, M.; Medici, A.; Beggiato, S.; Perrone, D. Zidovudine and ursodeoxycholic acid conjugation: Design of a new prodrug potentially able to bypass the active efflux transport systems of the central nervous system. Mol. Pharm. 2012, 9, 957–968. [Google Scholar] [CrossRef]
- Faustino, C.; Serafim, C.; Rijo, P.; Reis, C.P. Bile acids and bile acid derivatives: Use in drug delivery systems and as therapeutic agents. Expert Opin. Drug Deliv. 2016, 13, 1133–1148. [Google Scholar] [CrossRef]
- Sievänen, E. Exploitation of bile acid transport systems in prodrug design. Molecules 2007, 12, 1859–1889. [Google Scholar] [CrossRef] [Green Version]
- Obaidat, A.; Roth, M.; Hagenbuch, B. The expression and function of organic anion transporting polypeptides in normal tissues and in cancer. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 135–151. [Google Scholar] [CrossRef] [Green Version]
- Komori, Y.; Arisawa, S.; Takai, M.; Yokoyama, K.; Honda, M.; Hayashi, K.; Ishigami, M.; Katano, Y.; Goto, H.; Ueyama, J.; et al. Ursodeoxycholic acid inhibits overexpression of P-glycoprotein induced by doxorubicin in HepG2 cells. Eur. J. Pharmacol. 2014, 724, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise huisgen cycloaddition process: Copper(I)-catalyzed regioselective “Ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Xu, Z.; Zhao, S.-J.; Liu, Y. 1,2,3-Triazole-containing hybrids as potential anticancer agents: Current developments, action mechanisms and structure-activity relationships. Eur. J. Med. Chem. 2019, 183, 111700. [Google Scholar] [CrossRef] [PubMed]
- Bonandi, E.; Christodoulou, M.S.; Fumagalli, G.; Perdicchia, D.; Rastelli, G.; Passarella, D. The 1,2,3-triazole ring as a bioisostere in medicinal chemistry. Drug Discov. Today 2017, 22, 1572–1581. [Google Scholar] [CrossRef] [PubMed]
- Kharb, R.; Sharma, P.C.; Yar, M.S. Pharmacological significance of triazole scaffold. J. Enzym. Inhib. Med. Chem. 2011, 26, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Sztanke, K.; Tuzimski, T.; Rzymowska, J.; Pasternak, K.; Szerszen, M.K. Synthesis, structure elucidation and identification of antitumoural properties of novel fused 1,2,4-triazine aryl derivatives. Eur. J. Med. Chem. 2008, 43, 1085–1094. [Google Scholar] [CrossRef] [PubMed]
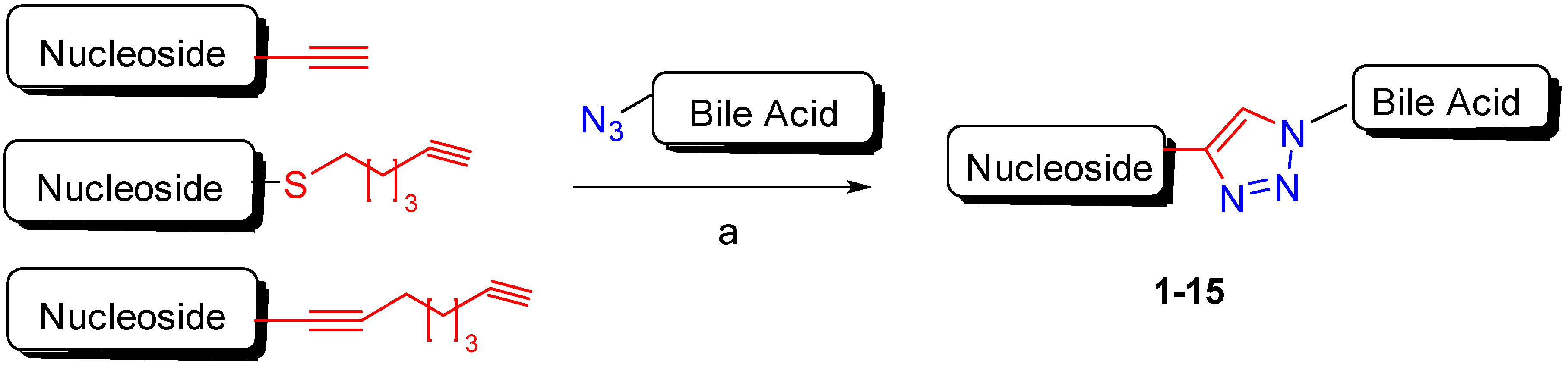
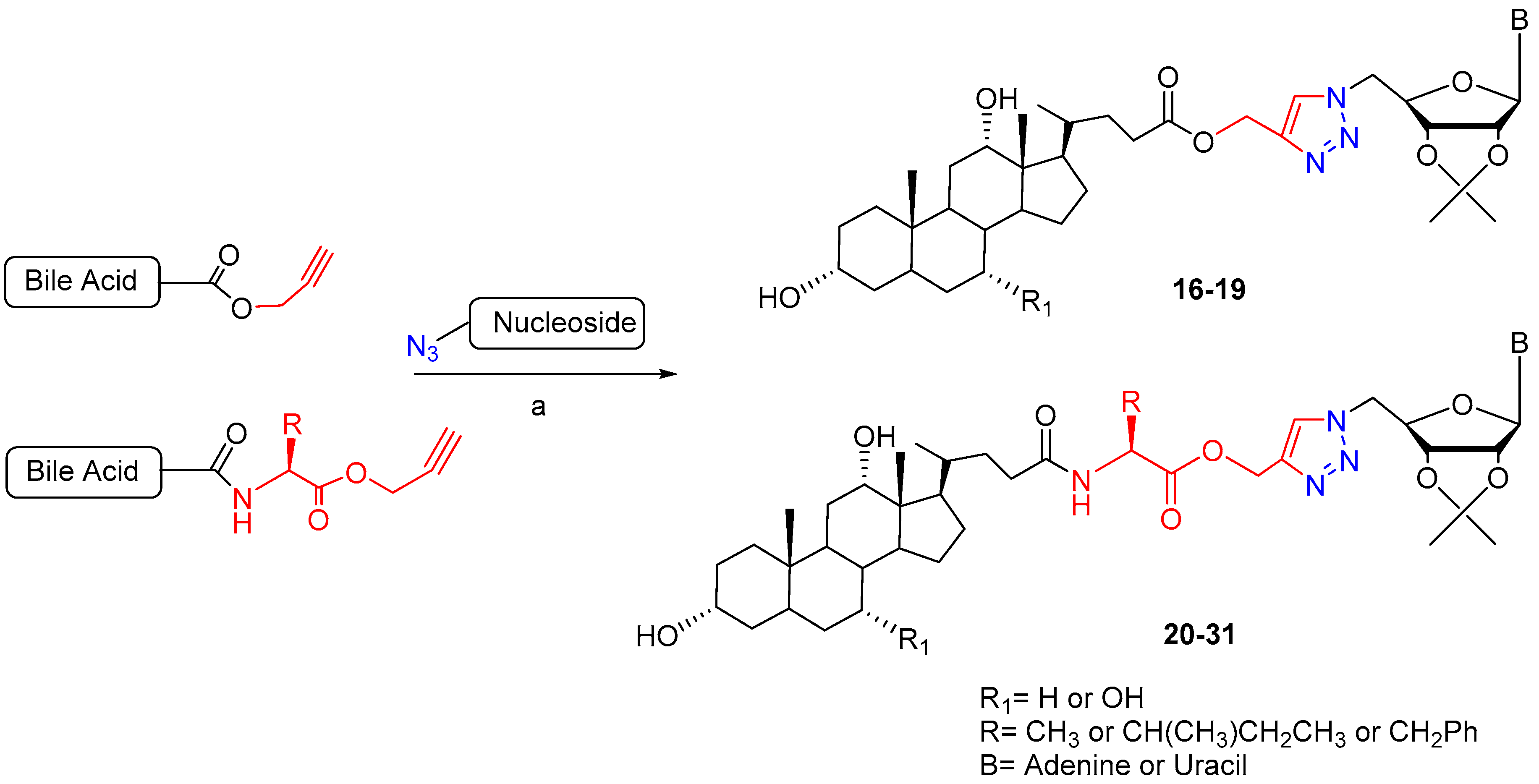
- Perrone, D.; Bortolini, O.; Fogagnolo, M.; Marchesi, E.; Mari, L.; Massarenti, C.; Navacchia, M.L.; Sforza, F.; Varani, K.; Capobianco, M.L. Synthesis and in vitro cytotoxicity of deoxyadenosine–bile acid conjugates linked with 1,2,3-triazole. New J. Chem. 2013, 37, 3559–3567. [Google Scholar] [CrossRef]
- Navacchia, M.L.; Marchesi, E.; Mari, L.; Chinaglia, N.; Gallerani, E.; Gavioli, R.; Capobianco, M.L.; Perrone, D. Rational design of nucleoside–bile acid conjugates incorporating a triazole moiety for anticancer evaluation and SAR exploration. Molecules 2017, 22, 1710. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, D.S.; Krishna, V.S.; Sriram, D.; Yogeeswari, P.; Sakhuja, R. Clickable conjugates of bile acids and nucleosides: Synthesis, characterization, in vitro anticancer and antituberculosis studies. Steroids 2018, 139, 35–44. [Google Scholar] [CrossRef]
- Marchesi, E.; Chinaglia, N.; Capobianco, M.L.; Marchetti, P.; Huang, T.-E.; Weng, H.-C.; Guh, J.-H.; Hsu, L.-C.; Perrone, D.; Navacchia, M.L. Dihydroartemisinin–bile acid hybridization as an effective approach to enhance dihydroartemisinin anticancer activity. ChemMedChem 2019, 14, 779–787. [Google Scholar] [CrossRef]
- Perrone, D.; Navacchia, M.L.; University of Ferrara, Ferrara, Italy. Unpublished Personal Data. 2020.
- Huang, T.-E.; Deng, Y.-N.; Hsu, J.-L.; Leu, W.-J.; Marchesi, E.; Capobianco, M.L.; Marchetti, P.; Navacchia, M.L.; Guh, J.-H.; Perrone, D.; et al. Evaluation of the anticancer activity of a bile acid-dihydroartemisinin hybrid ursodeoxycholic-dihydroartemisinin in hepatocellular carcinoma cells. Front. Pharmacol. 2020, 11, 599067. [Google Scholar] [CrossRef] [PubMed]
- Letis, A.S.; Seo, E.-J.; Nikolaropoulos, S.S.; Efferth, T.; Giannis, A.; Fousteris, M.A. Synthesis and cytotoxic activity of new artemisinin hybrid moleculesagainst human leukemia cells. Biorg. Med. Chem. 2017, 25, 3357–3367. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhao, T.; Cheng, D.; Chu, C.; Tong, S.; Yan, J.; Li, Q.-Y. Synthesis and biological activity of some bile acid-based camptothecin analogues. Molecules 2014, 19, 3761–3776. [Google Scholar] [CrossRef] [Green Version]
- Xiao, L.; Yu, E.; Yue, H.; Li, Q. Enhanced liver targeting of camptothecin via conjugation with deoxycholic acid. Molecules 2019, 24, 1179. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Zhang, H.; Mu, L.; Yang, X. Artemisinins as anticancer drugs: Novel therapeutic approaches, molecular mechanisms, and clinical trials. Front. Pharmacol. 2020, 11, 529881. [Google Scholar] [CrossRef]
- Aminake, M.N.; Mahajan, A.; Kumar, V.; Hans, R.; Wiesner, L.; Taylor, D.; de Kock, C.; Grobler, A.; Smith, P.J.; Kirschner, M.; et al. Synthesis and evaluation of hybrid drugs for a potential HIV/AIDS-malaria combination therapy. Bioorg. Med. Chem. 2012, 20, 5277–5289. [Google Scholar] [CrossRef]
- Majer, F.; Sharma, R.; Mullins, C.; Keogh, L.; Phipps, S.; Duggan, S.; Kelleher, D.; Keely, S.; Long, A.; Radics, G.; et al. New highly toxic bile acids derived from deoxycholic acid, chenodeoxycholic acid and lithocholic acid. Bioorg. Med. Chem. 2014, 22, 256–268. [Google Scholar] [CrossRef]
- Mujahid, A.R.; Evens, A.M.; Tallman, M.S.; Nelson, B.P.; Rosen, S.T. T-cell non-Hodgkin lymphoma. T-cell non-Hodgkin lymphoma. Blood 2006, 107, 1255–1264. [Google Scholar]
- Chao, C.N.; Huang, Y.L.; Lin, M.C.; Fang, C.Y.; Shen, C.H.; Chen, P.L.; Wang, M.; Chang, D.; Tseng, C.E. Inhibition of human diffuse large B-cell lymphoma growth by JC polyomavirus-like particles delivering a suicide gene. J. Transl. Med. 2015, 13, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Guo, X.; Yue, W.; Wang, J.; Yang, J.; Chen, J. Artemether suppresses cell proliferation and induces apoptosis in diffuse large B cell lymphoma cells. Exp. Ther. Med. 2017, 14, 4083–4090. [Google Scholar]
- Breuer, E.; Efferth, T. Treatment of iron-loaded veterinary sarcoma by Artemisia annua. Biochem. Pharmacol. 2012, 83, 1278–1289. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Luo, Z.; Xiang, T.; Wang, K.; Li, J.; Wang, P. Dihydroartemisinin induces endoplasmic reticulum stress- mediated apoptosis in HepG2 human hepatoma cells. Tumori 2011, 97, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Im, E.; Yeo, C.; Lee, H.J.; Lee, E.O. Dihydroartemisinin induced caspase-dependent apoptosis through inhibiting the specificity protein 1 pathway in hepatocellular carcinoma SK-Hep-1 cells. Life Sci. 2018, 192, 286–292. [Google Scholar] [CrossRef] [PubMed]
- Qin, G.; Zhao, C.; Zhang, L.; Hongyu, L.; Yingyao, Q.; Liuying, C.; Shengnan, W.; Xiaoping, W.; Tongsheng, C. Dihydroartemisinin induces apoptosis preferentially via a Bim-mediated intrinsic pathway in hepatocarcinoma cells. Apoptosis 2015, 20, 1072–1086. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-Q.; Li, W.-Q.; Morris-Natschke, S.L.; Qian, K.; Yang, L.; Zhu, G.-X.; Wu, X.-B.; Chen, A.-L.; Zhang, S.Y.; Song, Z.L.; et al. Perspectives on biologically active camptothecin derivatives. Med. Res. Rev. 2015, 35, 753–789. [Google Scholar] [CrossRef] [Green Version]
- Navacchia, M.L.; Fraix, A.; Chinaglia, N.; Gallerani, E.; Perrone, D.; Cardile, V.; Graziano, A.C.E.; Capobianco, M.L.; Sortino, S. NO Photoreleaser-deoxyadenosine and -bile acid derivative bioconjugates as novel potential photochemotherapeutics. ACS Med. Chem. Lett. 2016, 7, 939–943. [Google Scholar] [CrossRef] [Green Version]
- Sreekanth, V.; Bansal, S.; Motiani, R.K.; Kundu, S.; Muppu, S.K.; Majumdar, T.D.; Panjamurthy, K.; Sengupta, S.; Bajaj, A. Design, synthesis, and mechanistic investigations of bile acid−tamoxifen conjugates for breast cancer therapy. Bioconjugate Chem. 2013, 24, 1468–1484. [Google Scholar] [CrossRef]
- Zhang, D.; Li, D.; Shang, L.; Hea, Z.; Suna, J. Transporter-targeted cholic acid-cytarabine conjugates for improved oral absorption. Int. J. Pharm. 2016, 511, 161–169. [Google Scholar]
- Agarwal, D.S.; Singh, R.P.; Lohitesh, K.; Jha, P.N.; Chowdhury, R.; Sakhuja, R. Synthesis and evaluation of bile acid amides of α-cyanostilbenes as anticancer agents. Mol. Divers. 2018, 22, 305–321. [Google Scholar] [CrossRef]
- Agarwal, D.S.; Anantaraju, H.S.; Sriram, D.; Yogeeswari, P.; Nanjegowda, S.H.; Mallu, P.; Sakhuja, R. Synthesis, characterization and biological evaluation of bile acidaromatic/heteroaromatic amides linked via amino acids as anti-cancer agents. Steroids 2016, 107, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, A.W.; Schoenfisch, M.H. Nitric oxide release: Part II. therapeutic applications. Chem. Soc. Rev. 2012, 41, 3742–3752. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, H. Solid tumor physiology and hypoxia-induced chemo/adio-resistance: Novel strategy for cancer therapy: Nitric oxide donor as a therapeutic enhancer. Nitric Oxide 2008, 19, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Pallavicini, M.G. Cytosine arabinoside: Molecular, pharmacokinetic and cytokinetic considerations. Pharmacol. Ther. 1984, 25, 207–238. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, Y.; Chen, H.; Yao, X.; Xiao, Y.; Zeng, X.; Zheng, Q.; Wei, Y.; Song, C.; Zhang, Y. Synthetic resveratrol derivatives and their biological activities: A review. Open J. Med. Chem. 2015, 5, 97–105. [Google Scholar] [CrossRef] [Green Version]



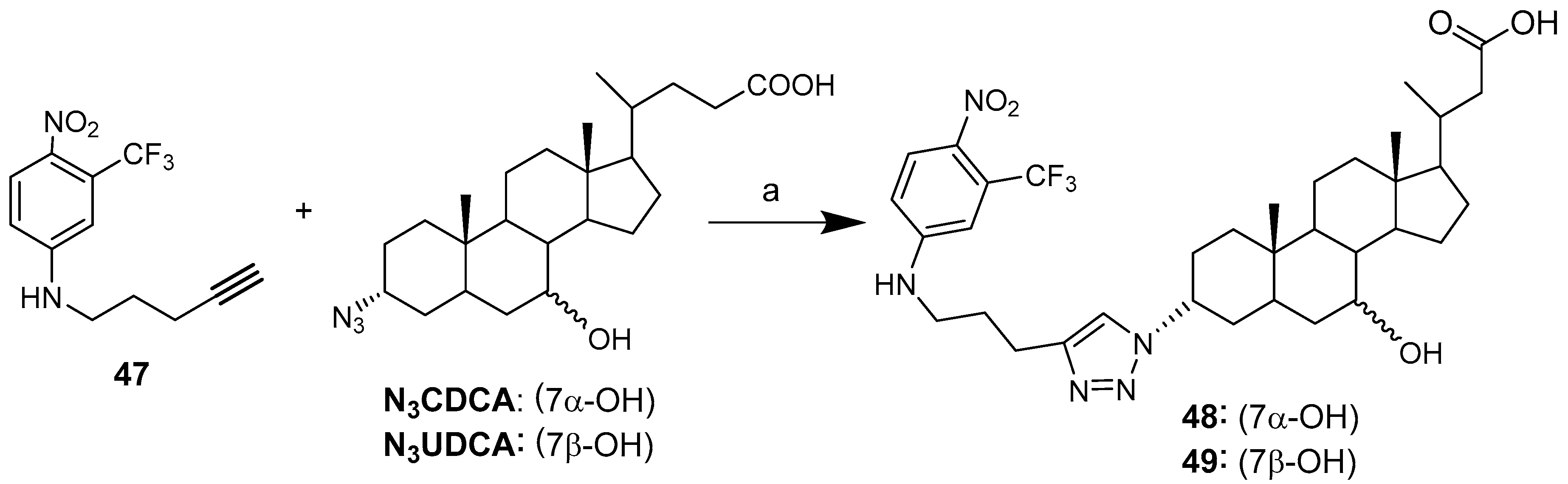

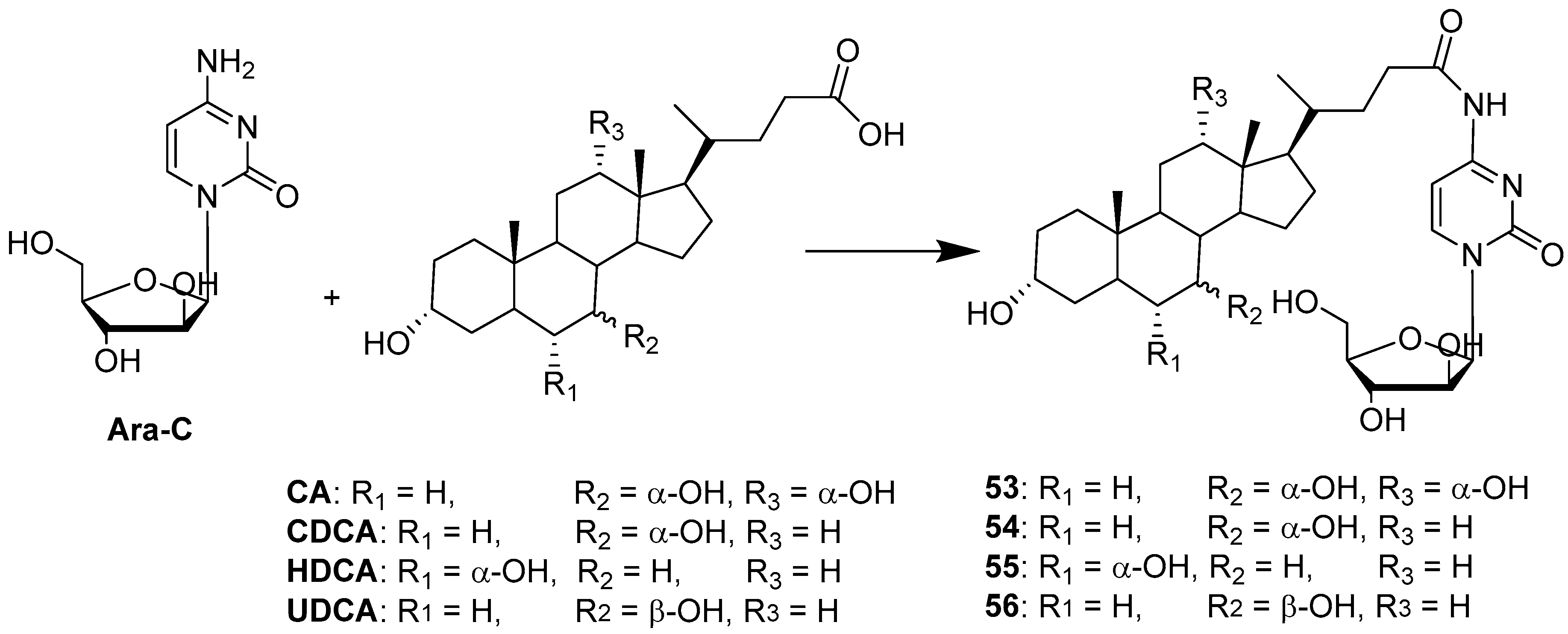

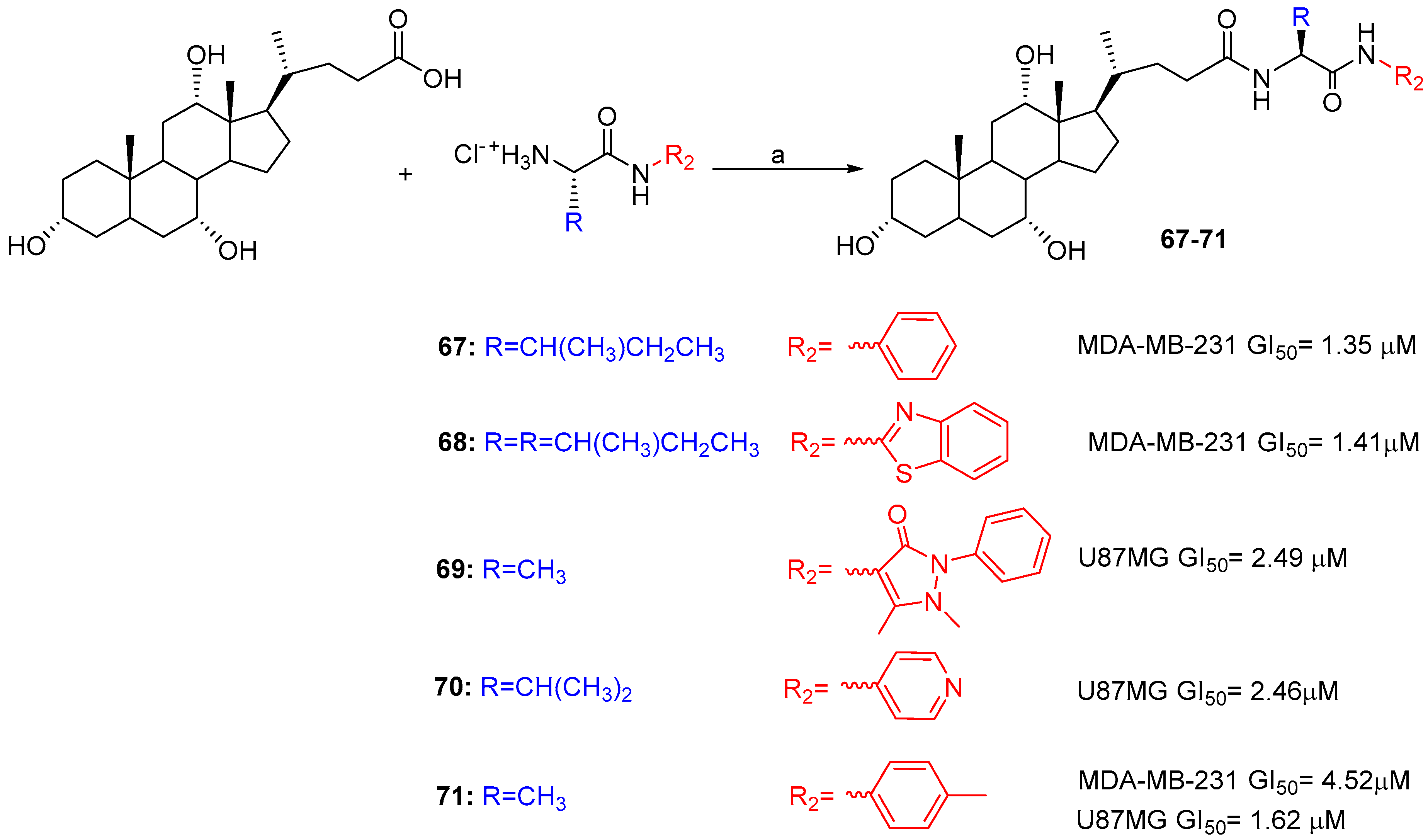

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hybrid | Conjugation Type | Cancer Cell Line | Mechanism/Event | Ref. |
|---|---|---|---|---|
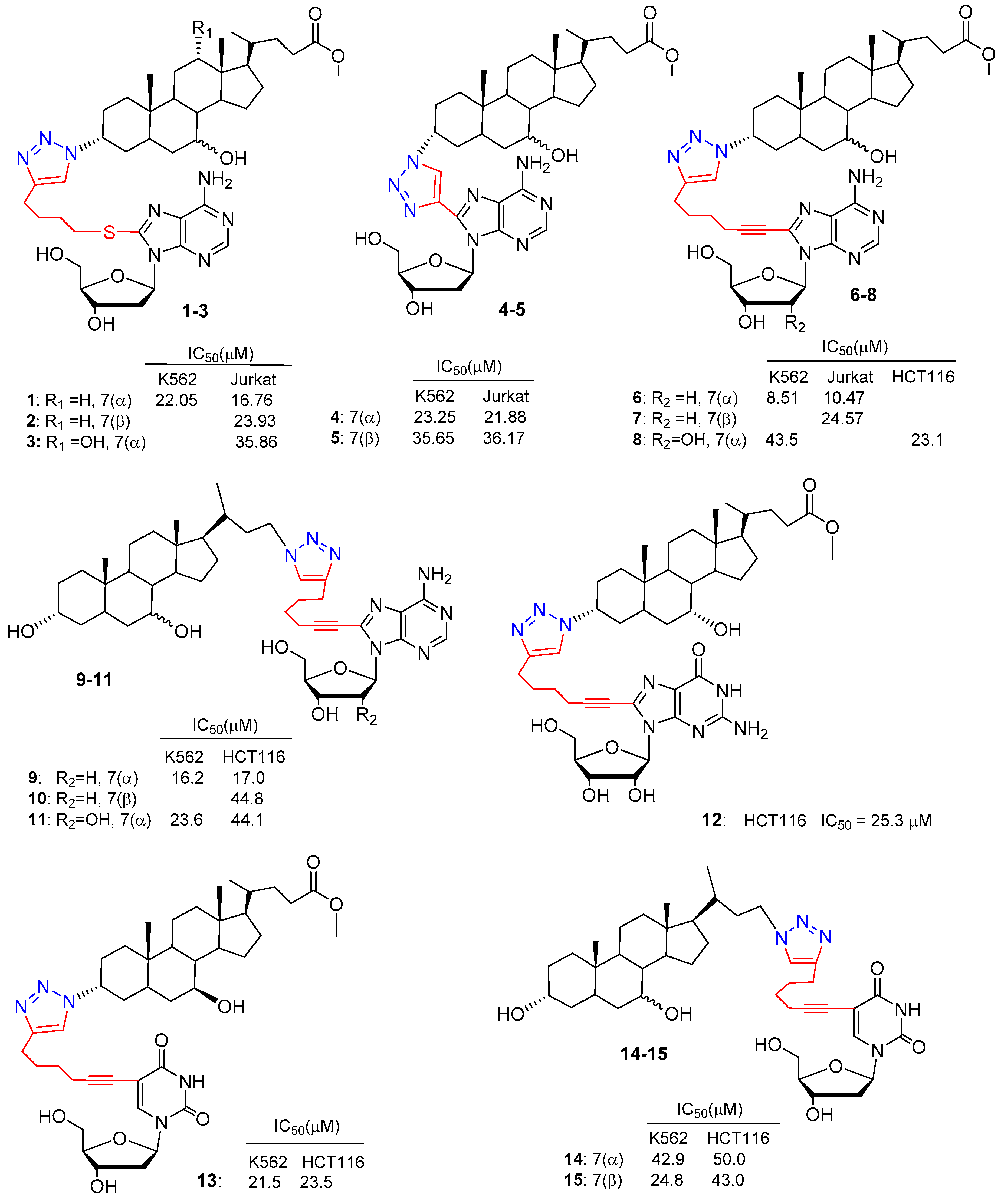
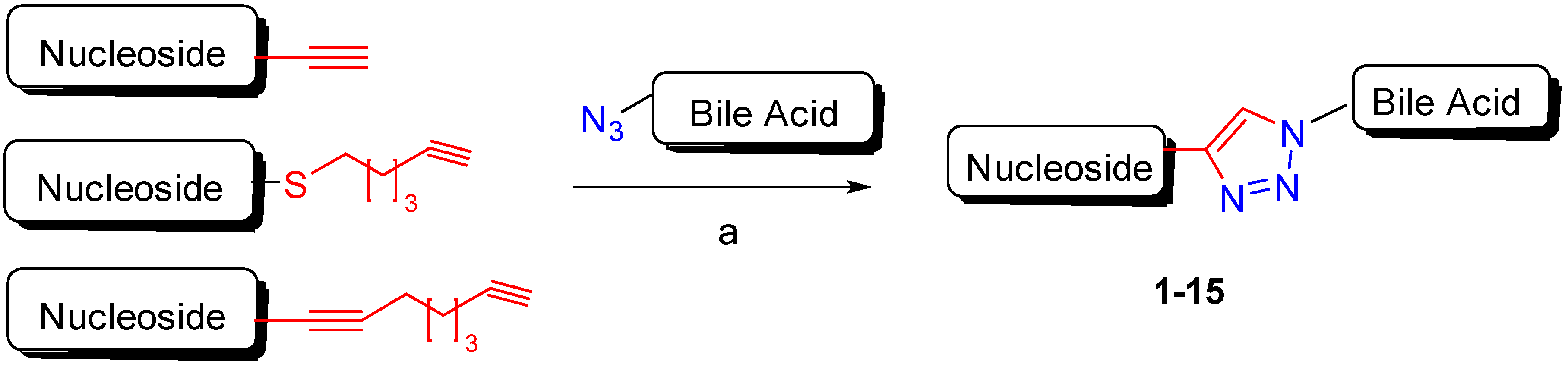
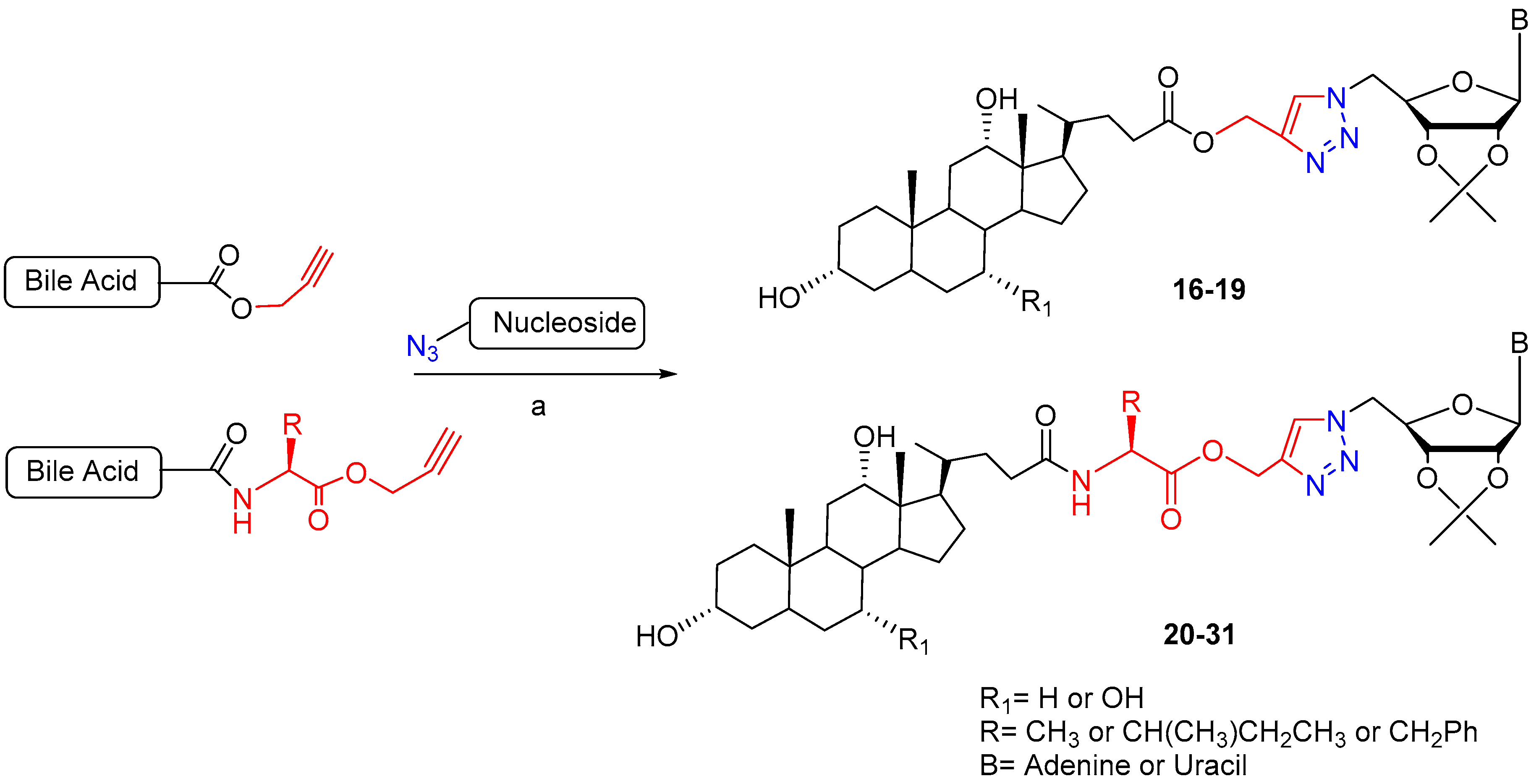
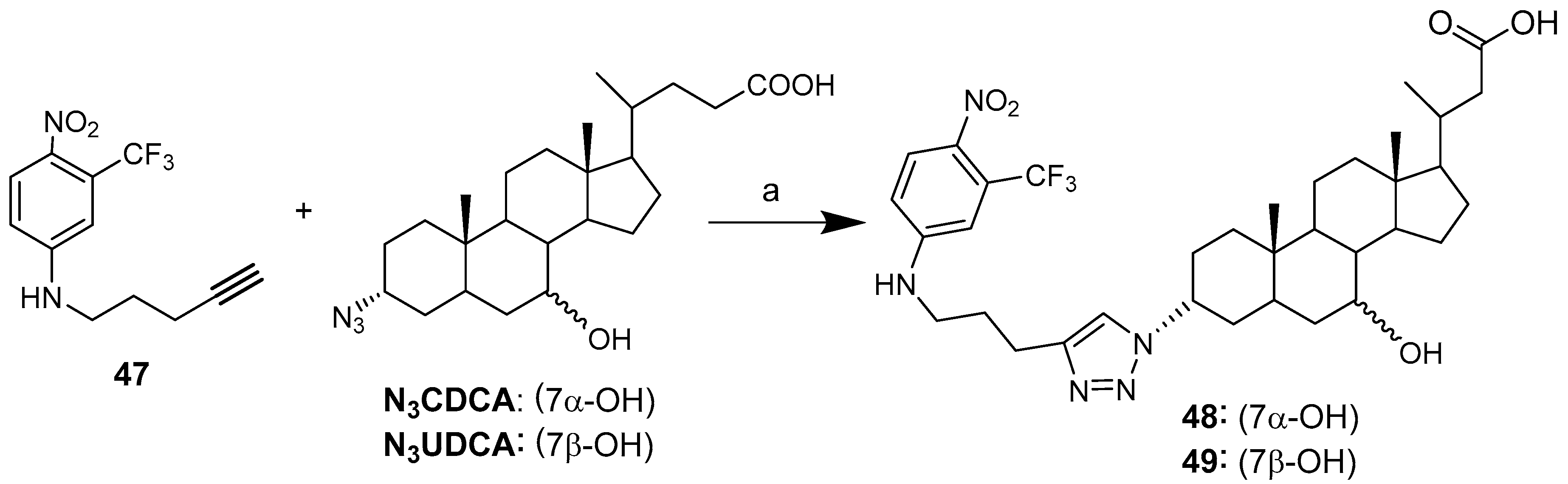
| BA-nucleosides | Click | K562, Jurkat, HCT-116, MCF-7, IMR-32. | Induce apoptosis, better selectivity than Cisplatin. | [28,29,30] |
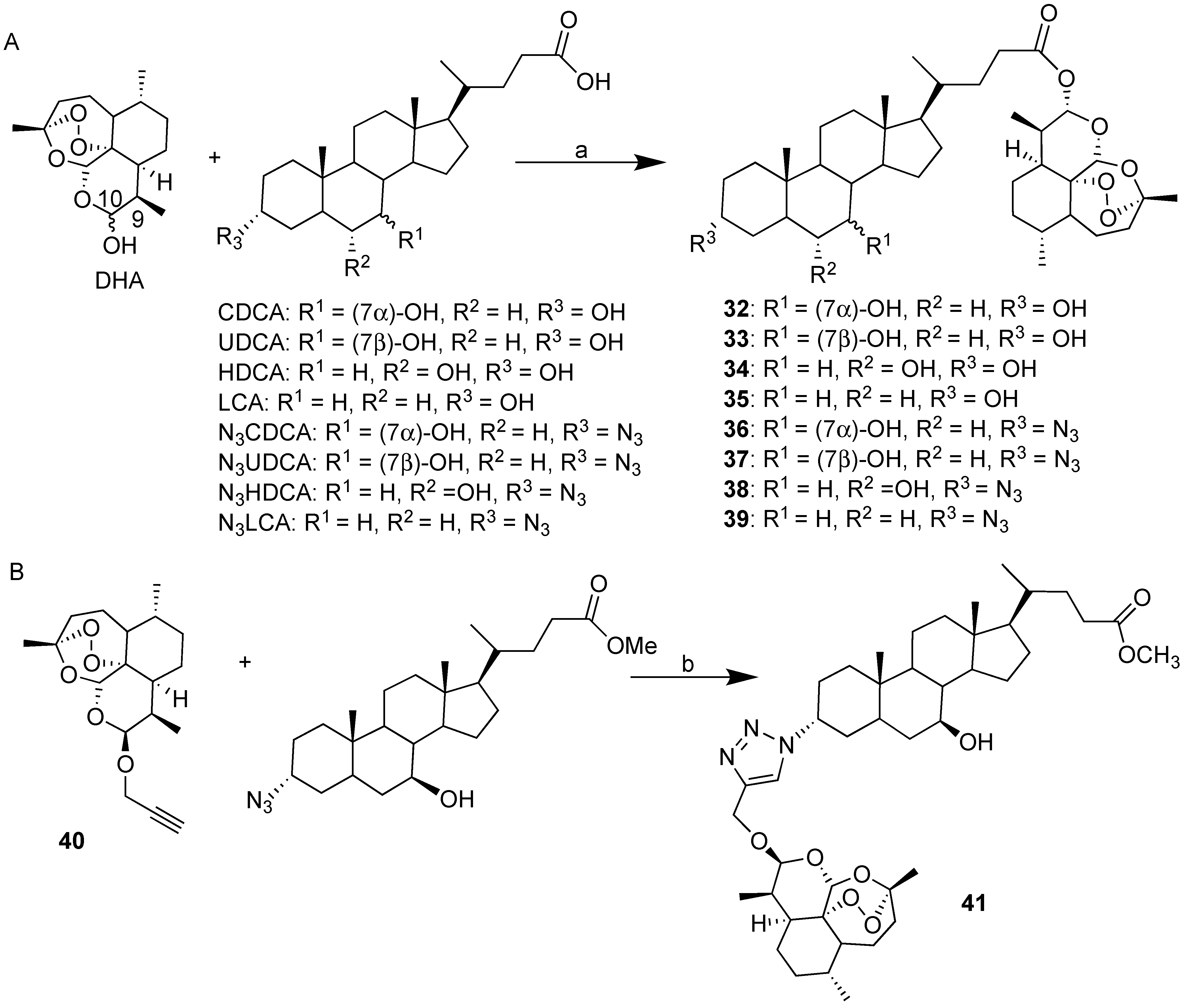
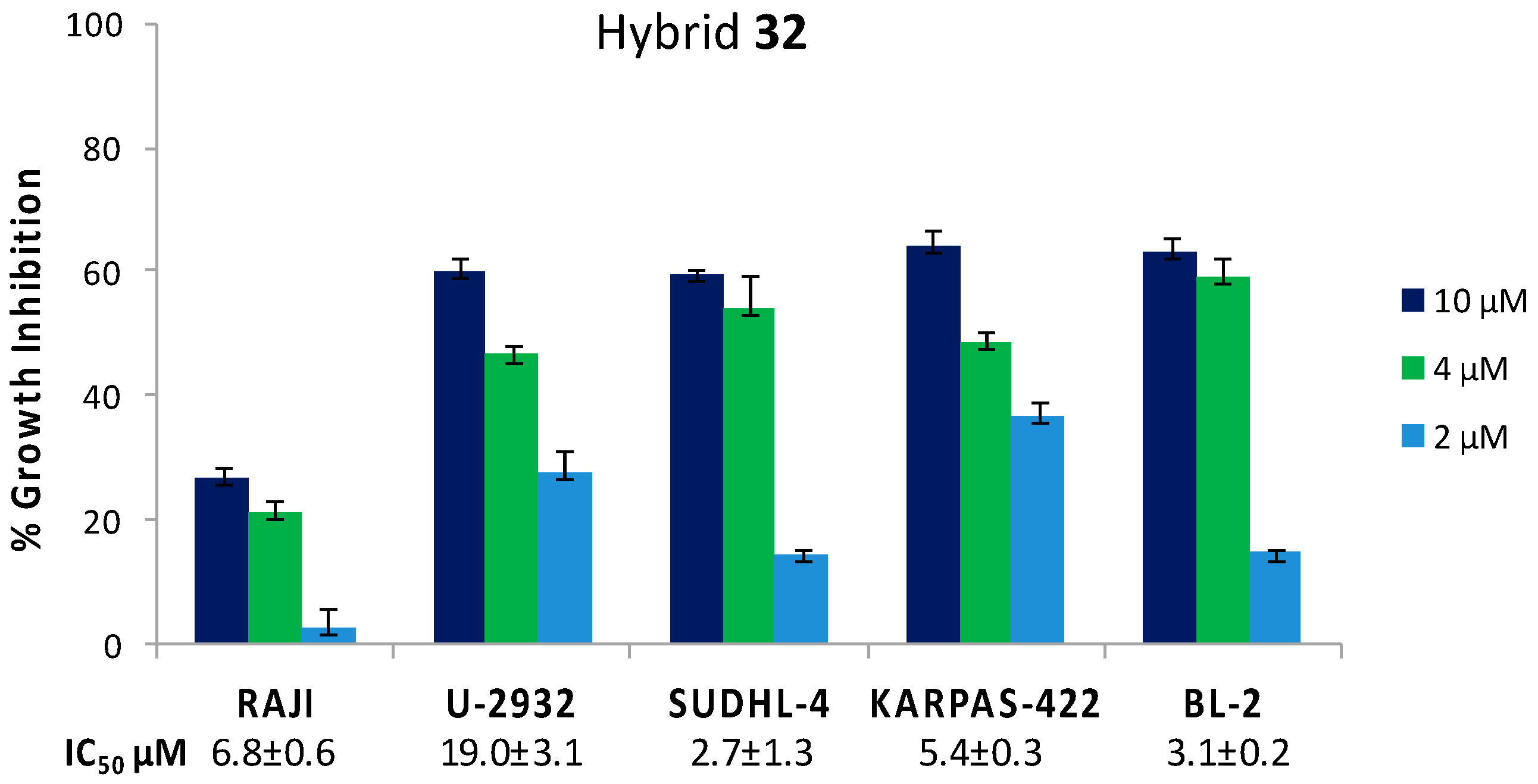
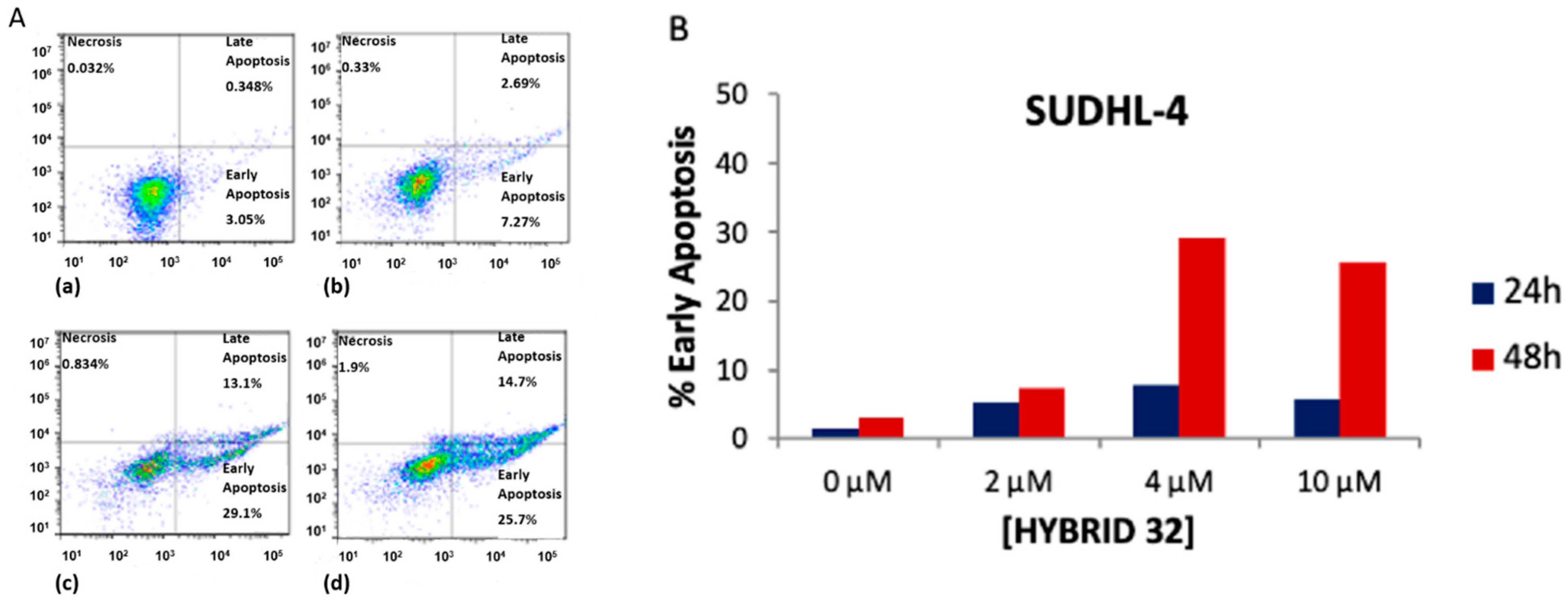
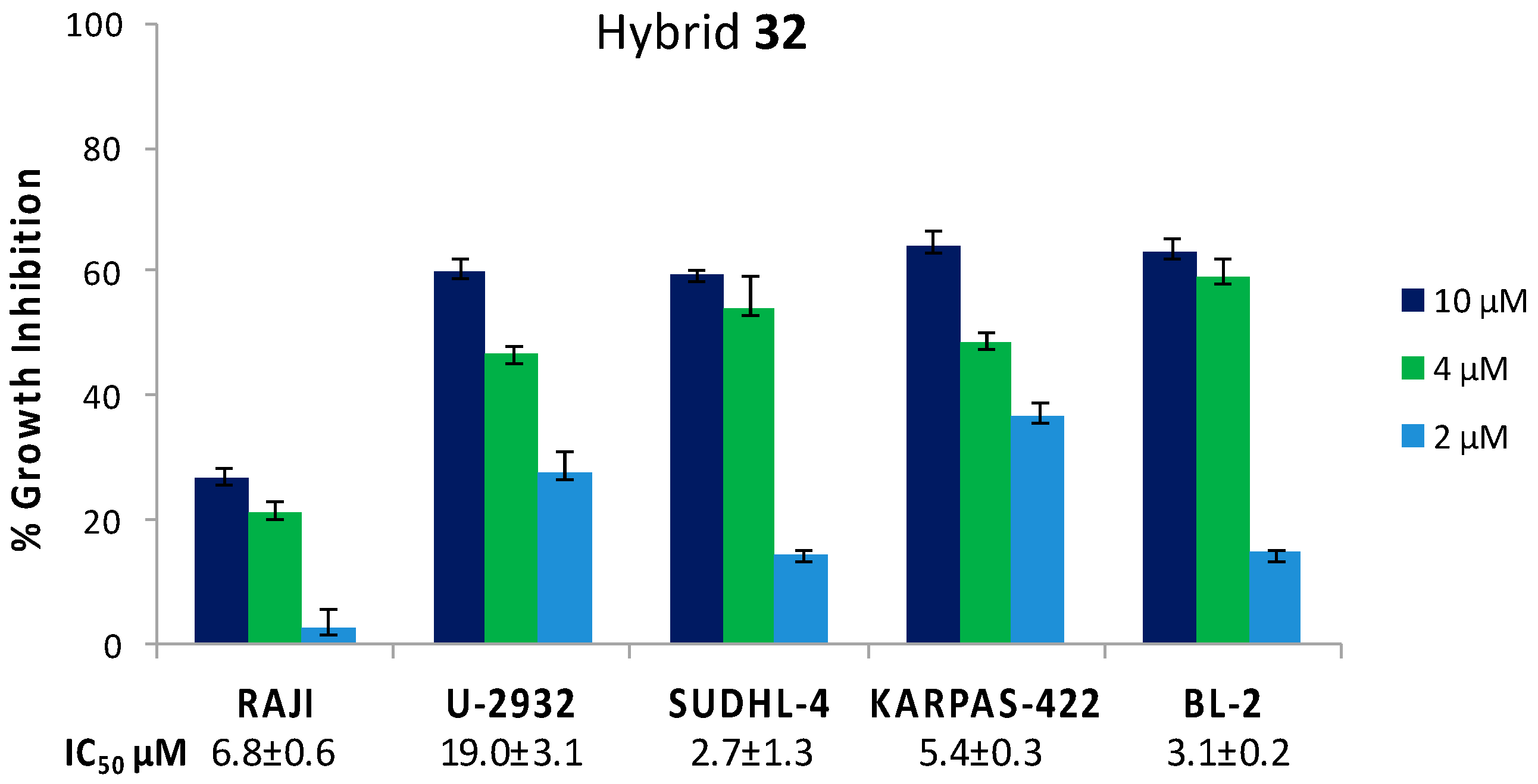
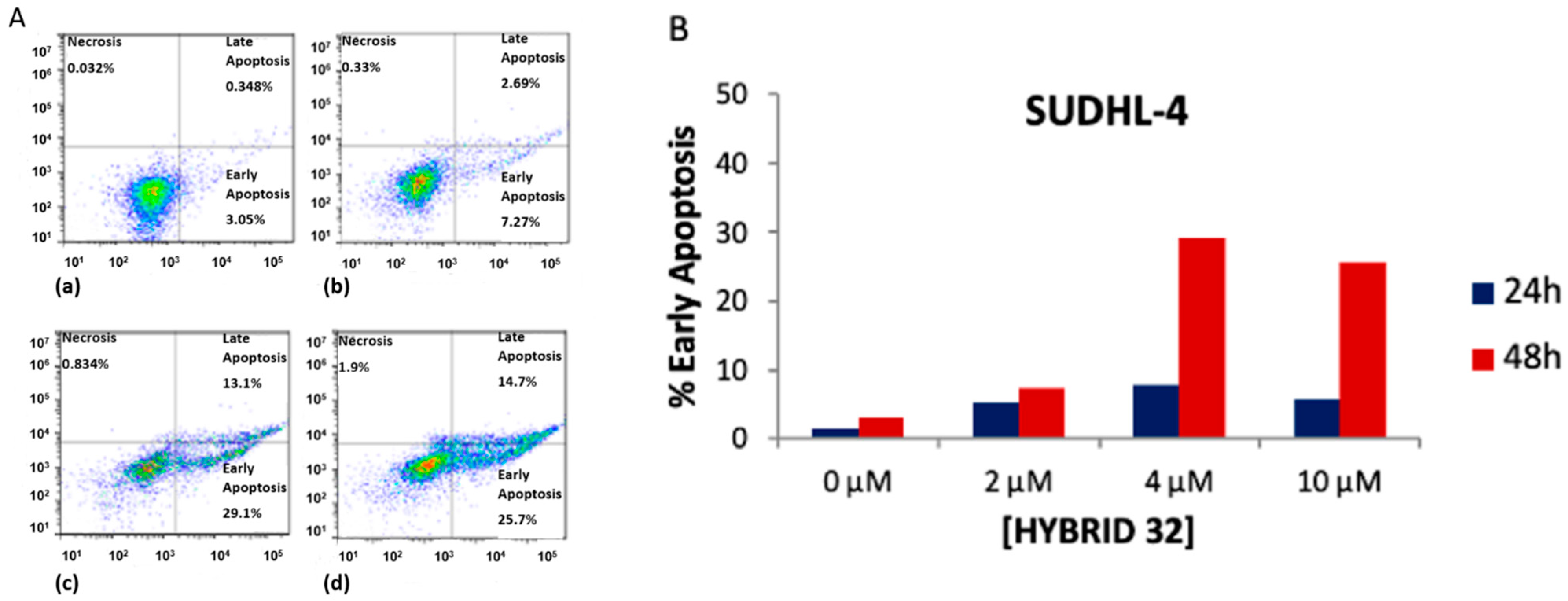
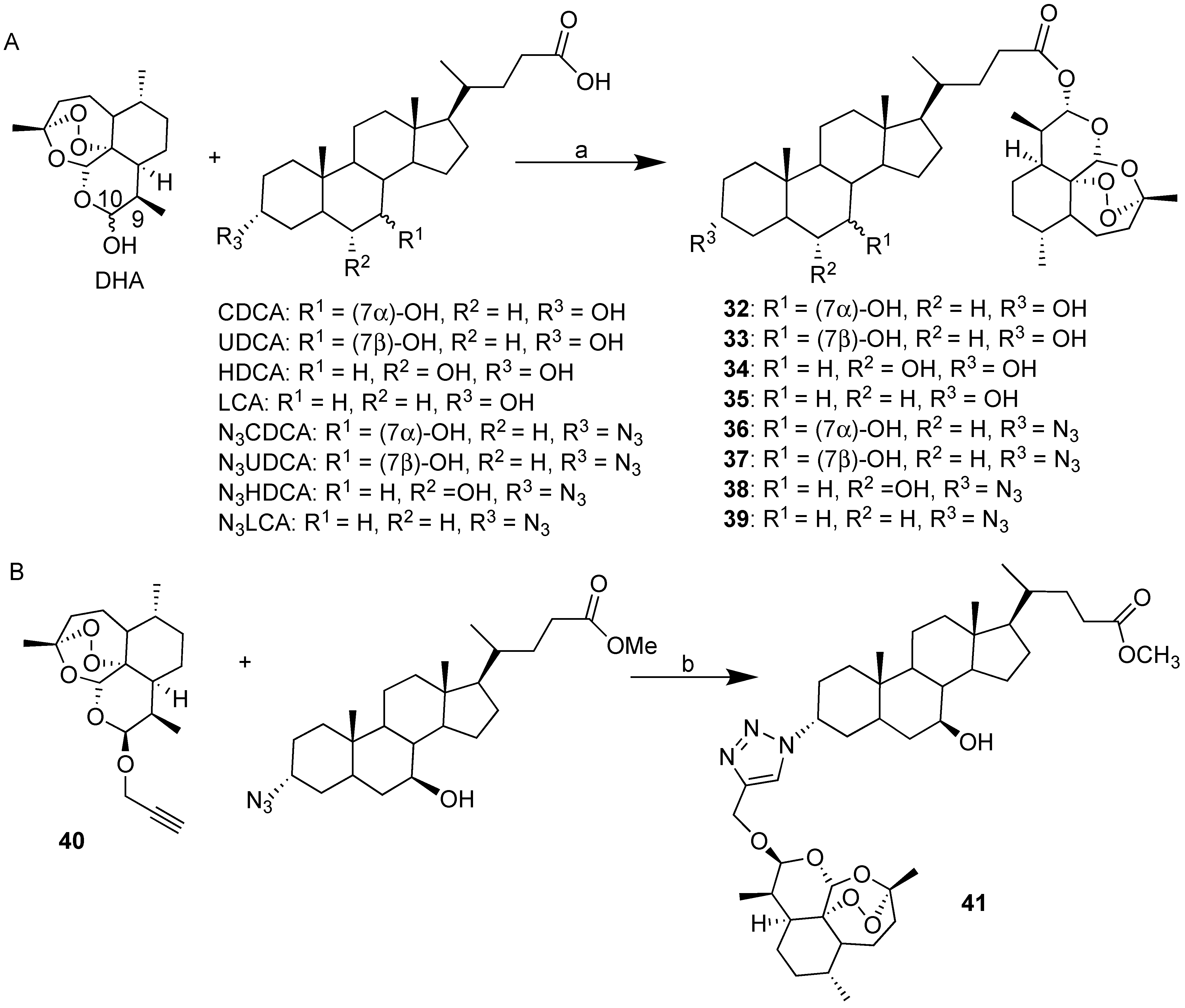
| BA-DHA | Click/Condensation | K562, RAJI, U-2932, SUDHL-4, KARPAS-422, BL-2, HL-60, HepG2, Huh-7. | Improved cytotoxicity, induce apoptosis and G0/G1 arrest, ROS. | [31,32,33] |
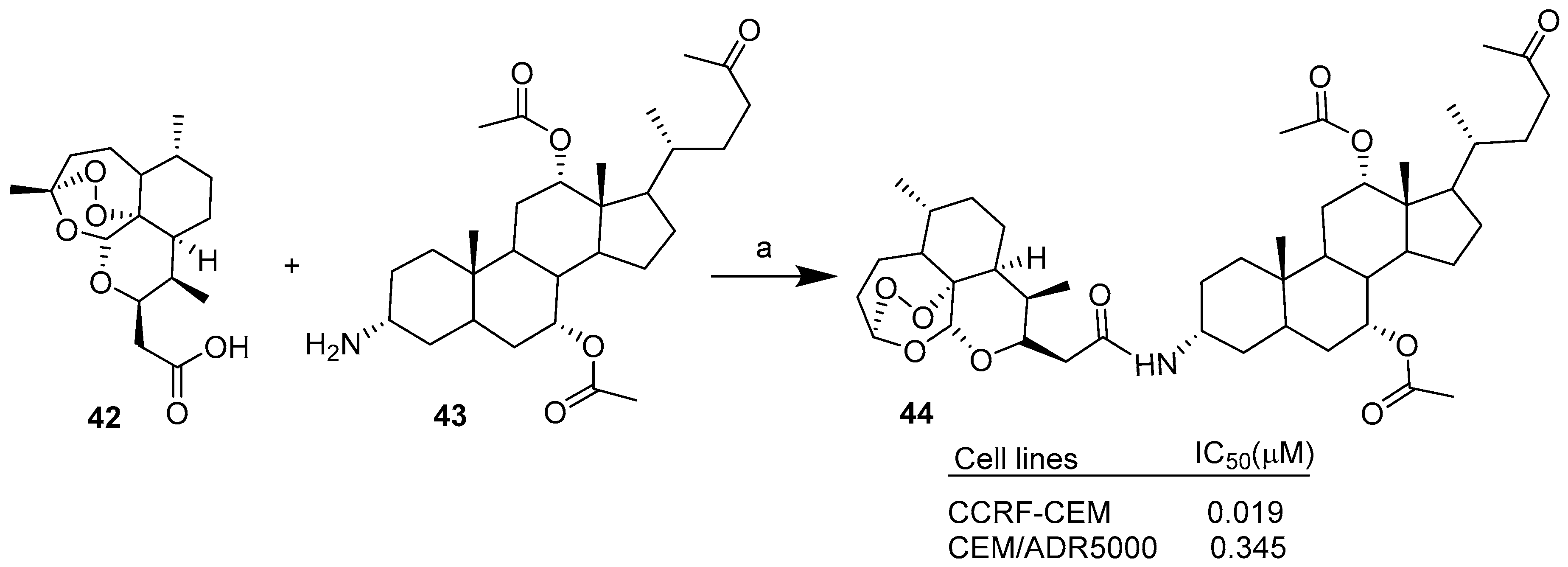
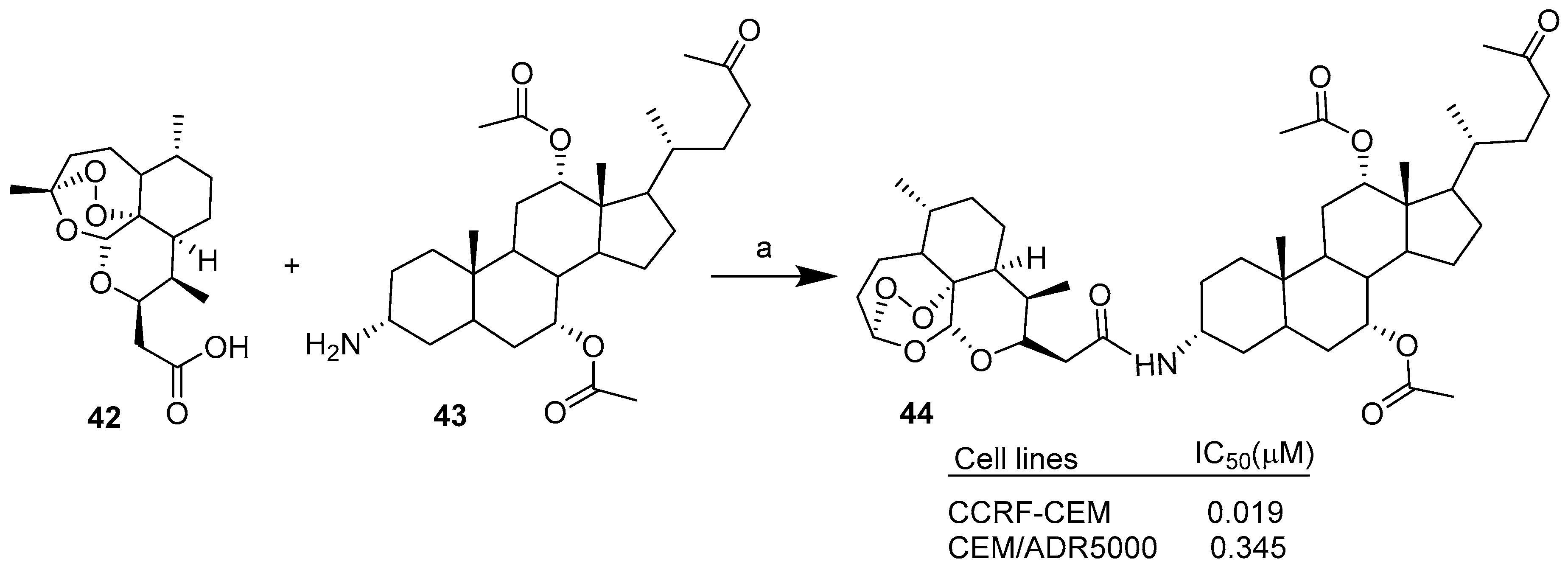
| Condensation | CCRF-CEM, CEM/ADR5000. | Improved cytotoxicity, comparable/better cytotoxicity than Doxorubicin. | [34] | |
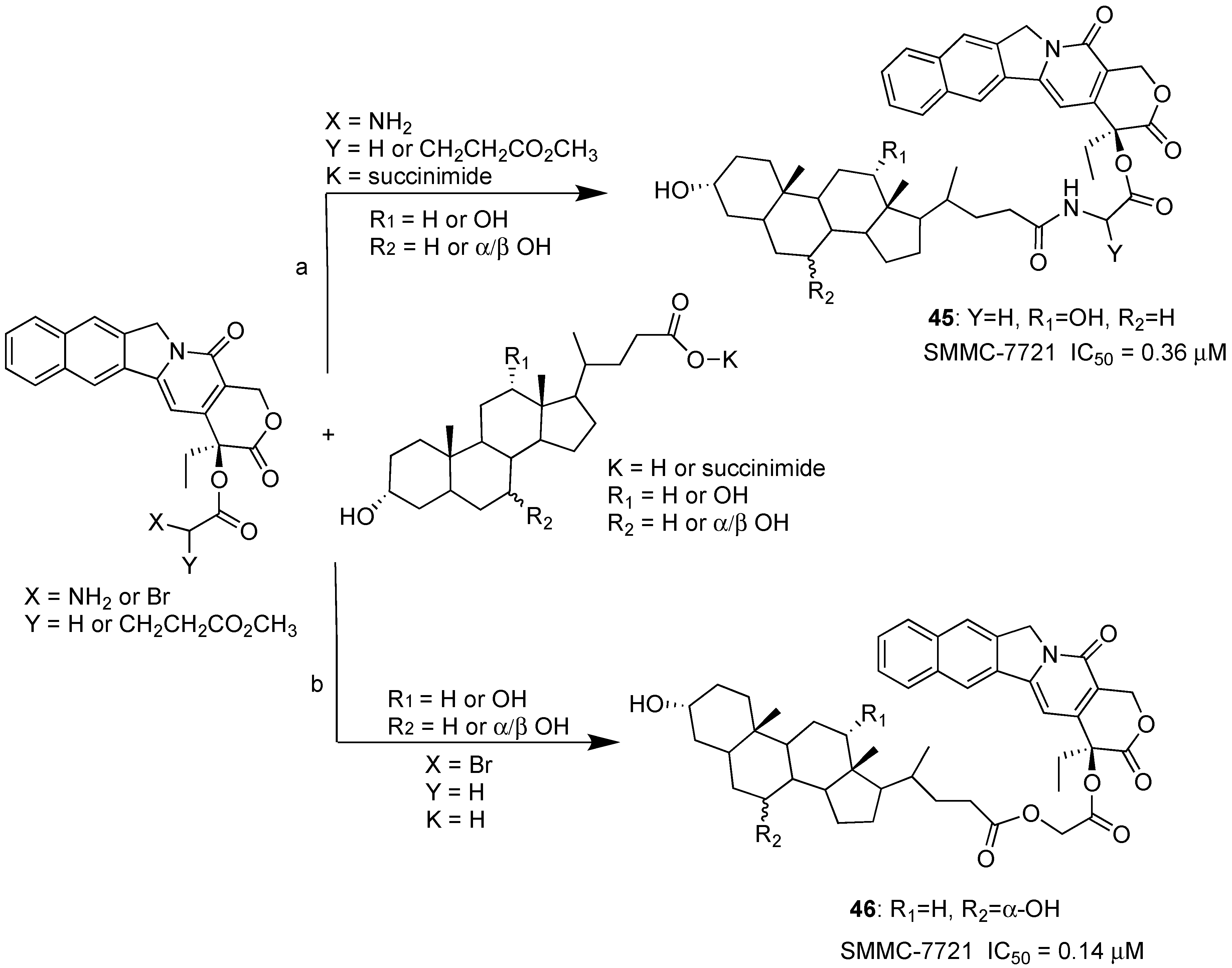
| BA-CPT | Condensation | MCF-7, HCT-116, SMMC-7721. | Better inhibition, enhanced stability. | [35,36] |
| Compound | IC50 [µM] | |||
|---|---|---|---|---|
| PC-3 | MCF-7 | IMR-32 | ||
| 16 | R1 = OH, B = A | 16.35 | 17.06 | 17.45 |
| 17 | R1 = H, B = A | 13.50 | 16.76 | 16.06 |
| 18 | R1 = OH, B = U | 17.89 | 8.08 | 16.8 |
| 19 | R1 = H, B = U | 16.69 | >100 | 16.68 |
| 20 | R1 = OH, B = A, R = CH3 | 31.47 | 18.15 | 82.93 |
| 21 | R1 = H, B = A, R = CH3 | 16.12 | >100 | 16.01 |
| 22 | R1 = OH, B = A, R = CH(CH3)CH2CH3 | 16.41 | 13.76 | 17.12 |
| 23 | R1 = H, B = A, R = CH(CH3)CH2CH3 | 17.06 | 16.39 | 41.04 |
| 24 | R1 = OH, B = A, R = CH2Ph | 14.23 | 14.96 | 19.53 |
| 25 | R1 = H, B = A, R = CH2Ph | 17.63 | 20.09 | 14.42 |
| 26 | R1 = OH, B = U, R = CH3 | 17.16 | 14.30 | 14.30 |
| 27 | R1 = H, B = U, R = CH3 | 16.38 | >100 | 31.69 |
| 28 | R1 = OH, B = U, R = CH(CH3)CH2CH3 | 18.01 | >100 | 18.23 |
| 29 | R1 = H, B = U, R = CH(CH3)CH2CH3 | >100 | 17.33 | 15.38 |
| 30 | R1 = OH, B = U, R = CH2Ph | 16.24 | >100 | 8.71 |
| 31 | R1 = H, B = U, R = CH2Ph | 16.17 | >100 | 18.59 |
| HepG2 [31] | Huh-7 [33] | HL-60 [31] | ||||
|---|---|---|---|---|---|---|
| IC50 µM | DHA/Hybrid | IC50 µM | DHA/Hybrid | IC50 µM | DHA/Hybrid | |
| DHA | 21 ± 2 | - | 39.96 ± 1.31 | - | 2.0 ± 0.4 | - |
| 32 | 3.8 ± 0.4 | 5.5 | 14.41 ± 2.18 | 2.8 | 0.5 ± 0.06 | 5.5 |
| 33 | 1.8 ± 0.2 | 11.7 | 2.16 ± 0.39 | 18.5 | 0.19 ± 0.03 | 10.5 |
| 34 | 2.0 ± 0.2 | 10.5 | 6.03 ± 0.68 | 6.62 | 0.35 ± 0.06 | 5.7 |
| 35 | 5.7 ± 0.7 | 3.7 | 5.97 ± 0.74 | 6.69 | 0.41 ± 0.05 | 4.9 |
| 36 | 20 ± 2 | 1.1 | 50.30 ± 2.45 | 0.78 | 0.68 ± 0.01 | 2.9 |
| 37 | 2.2 ± 0.4 | 9.5 | 4.13 ± 0.84 | 9.68 | 0.24 ± 0.02 | 8.3 |
| 38 | 14.2 ± 0.7 | 1.5 | 15.92 ± 1.28 | 2.72 | 1.7 ± 0.2 | 1.2 |
| 39 | 4.7 ± 0.4 | 4.7 | 5.08 ± 0.44 | 8.18 | 0.35 ± 0.04 | 5.7 |
| 41 | 1.7 ± 0.2 | 12.4 | - | - | 0.328 ± 0.002 | 6.1 |
| Hybrid | Conjugation Type | Cancer Cell Line | Mechanism/Event | Ref. |
|---|---|---|---|---|
| BA-NOdonor | Click | K562, HTC116. | Improved cytotoxicity and cytoselectivity. | [48] |
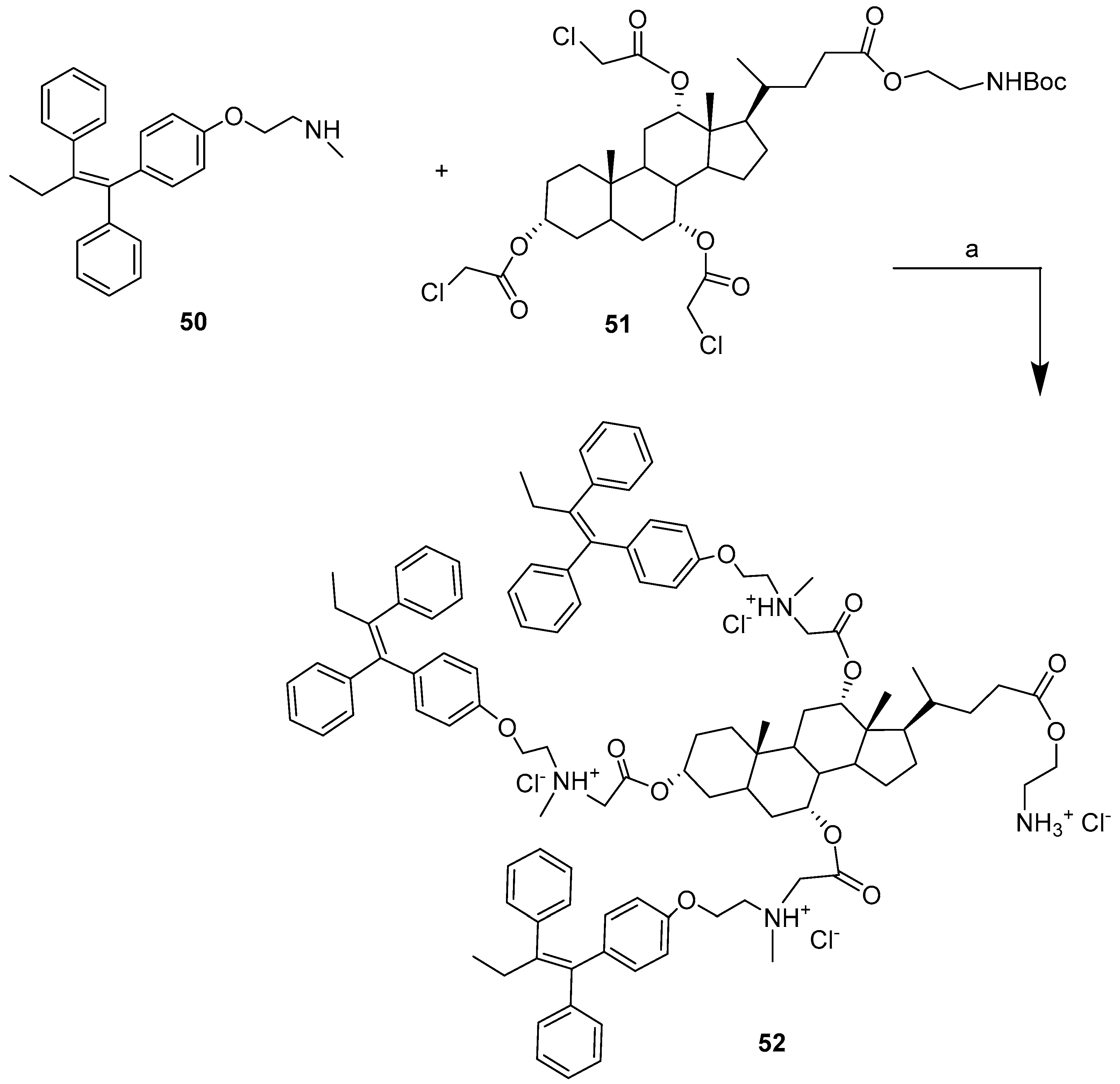
| BA-TAM | Condensation | 4T1, MCF-7, T47D. | Better inhibition. | [49] |
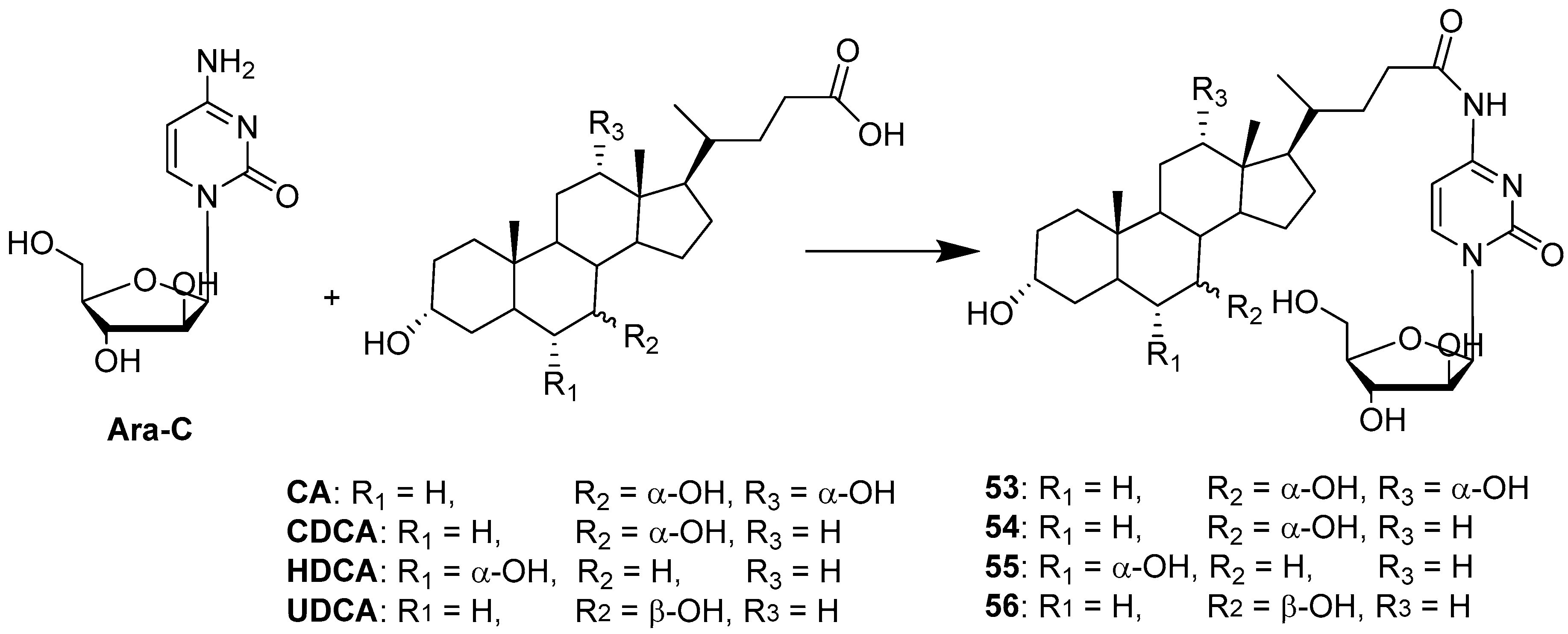
| BA-Ara-C | Condensation | HepG2 | Increased bioavailability via AOTP-mediated transport. | [50] |
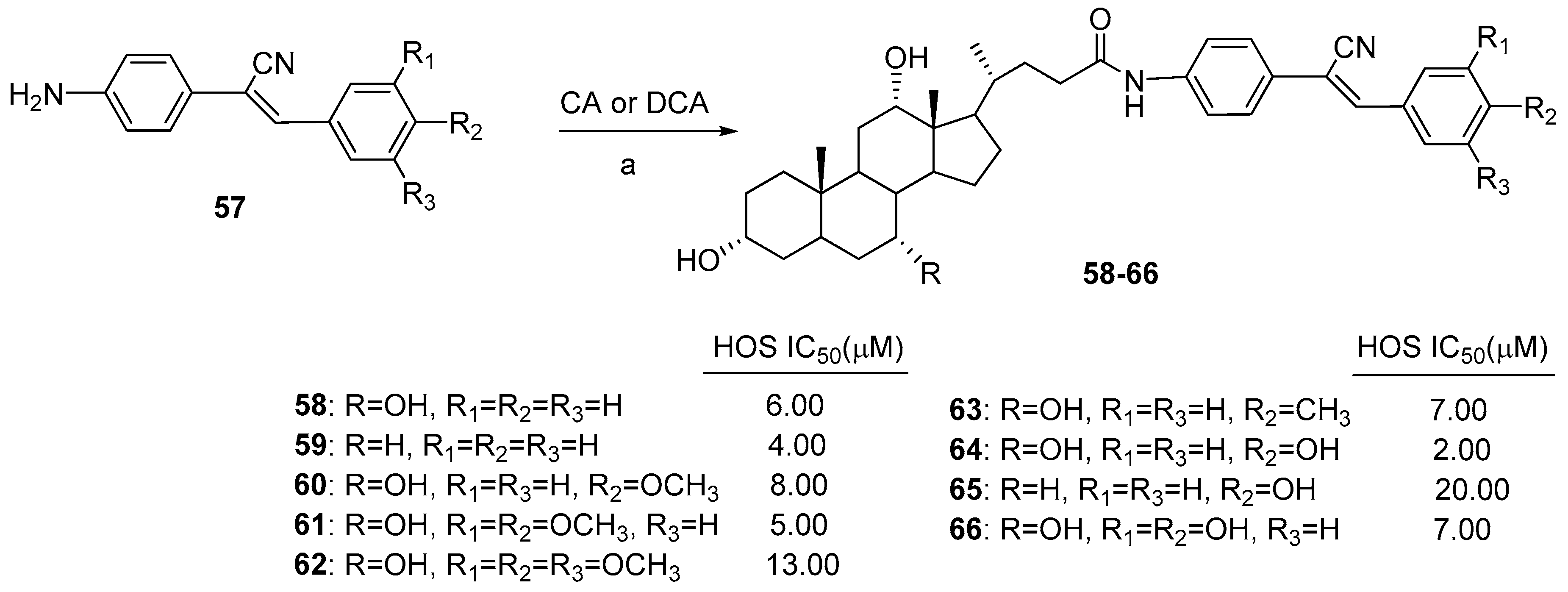
| BA-α-Cyanostilbene | Condensation | HOS | Induce apoptosis and G1 arrest. | [51] |
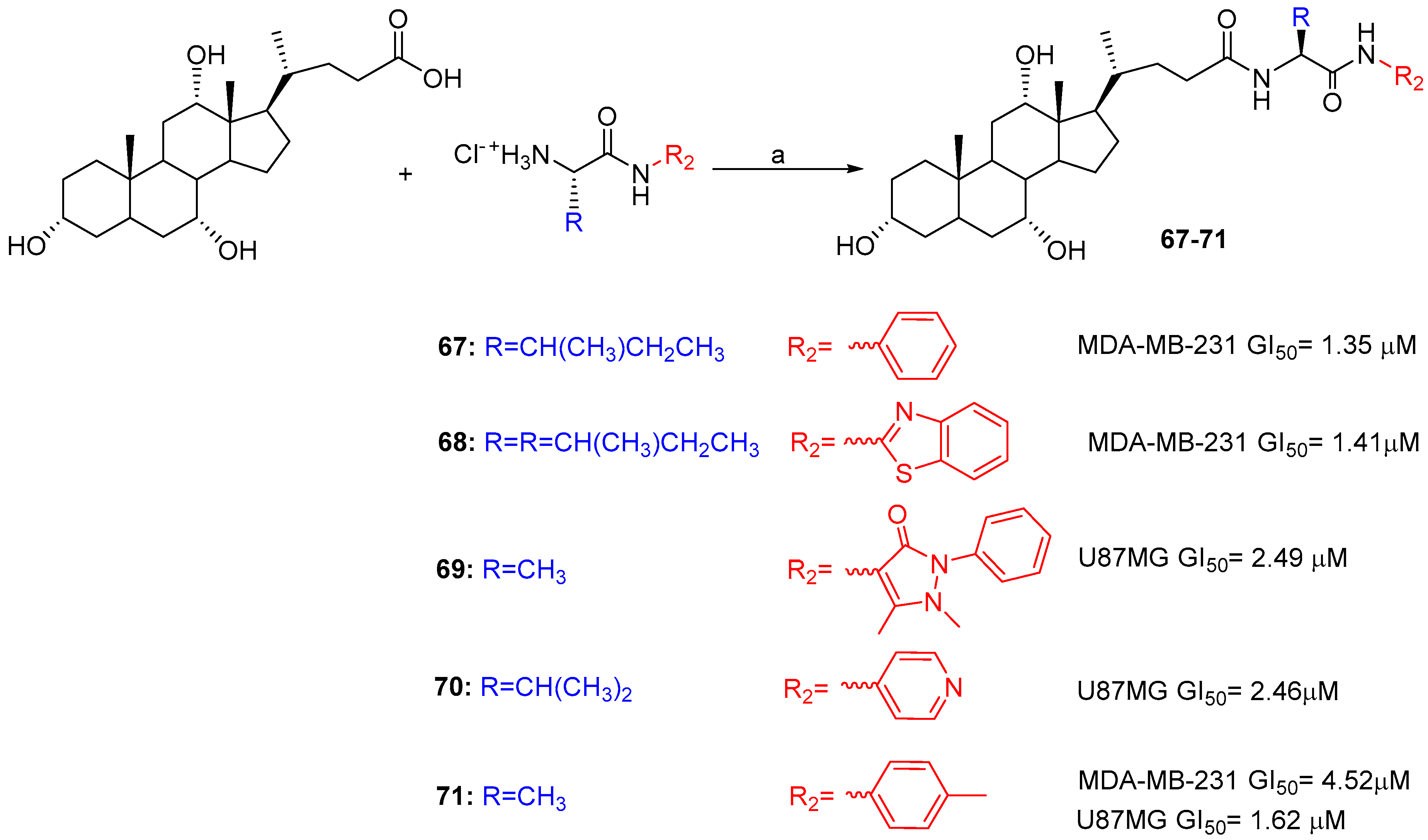
| BA-Aryl/heteroaryl | Condensation | MDA-MB-231, U87. | Comparable/better cytotoxicity than Cisplatin and Doxorubicin | [52] |
| IC50 µM | |||
|---|---|---|---|
| Compound | K562 | HTC116 | Fibroblast |
| 48 | 24.0 ± 1.2 | 21.0 ± 0.8 | 32.0 ± 1.0 |
| 49 | 31.6 ± 1.6 | 14.0 ± 1.0 | >>100 |
| Cisplatin | 5.40 ± 1.0 | 8.5 ± 1.2 | 25.4 ± 3.5 |
| IC50 µM | ||||
|---|---|---|---|---|
| Compound | 4T1 | MCF-7 | T47D | MDA-MB 231 |
| TAM | 12.25 ± 1.92 | 18.25 ± 4.19 | 22.41 ± 2.15 | 19.41 ± 4.47 |
| 52 | 5.28 ± 4.32 | 8.1 ± 3.82 | 9.42 ± 4.21 | 17.55 ± 5.85 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Navacchia, M.L.; Marchesi, E.; Perrone, D. Bile Acid Conjugates with Anticancer Activity: Most Recent Research. Molecules 2021, 26, 25. https://doi.org/10.3390/molecules26010025
Navacchia ML, Marchesi E, Perrone D. Bile Acid Conjugates with Anticancer Activity: Most Recent Research. Molecules. 2021; 26(1):25. https://doi.org/10.3390/molecules26010025
Chicago/Turabian StyleNavacchia, Maria Luisa, Elena Marchesi, and Daniela Perrone. 2021. "Bile Acid Conjugates with Anticancer Activity: Most Recent Research" Molecules 26, no. 1: 25. https://doi.org/10.3390/molecules26010025
APA StyleNavacchia, M. L., Marchesi, E., & Perrone, D. (2021). Bile Acid Conjugates with Anticancer Activity: Most Recent Research. Molecules, 26(1), 25. https://doi.org/10.3390/molecules26010025