In Silico and In Vitro Analysis of Major Cannabis-Derived Compounds as Fatty Acid Amide Hydrolase Inhibitors



Abstract
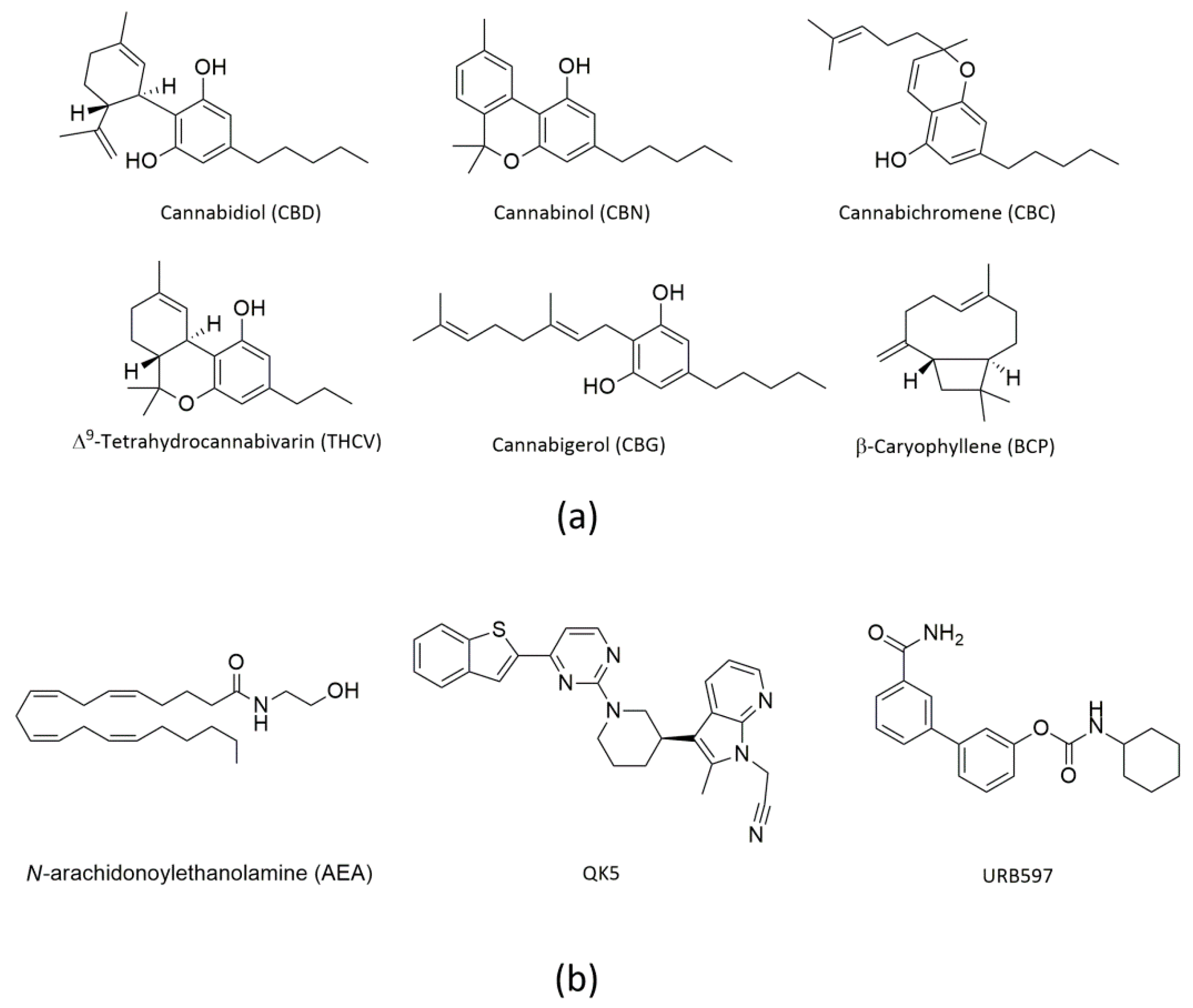
1. Introduction
2. Results
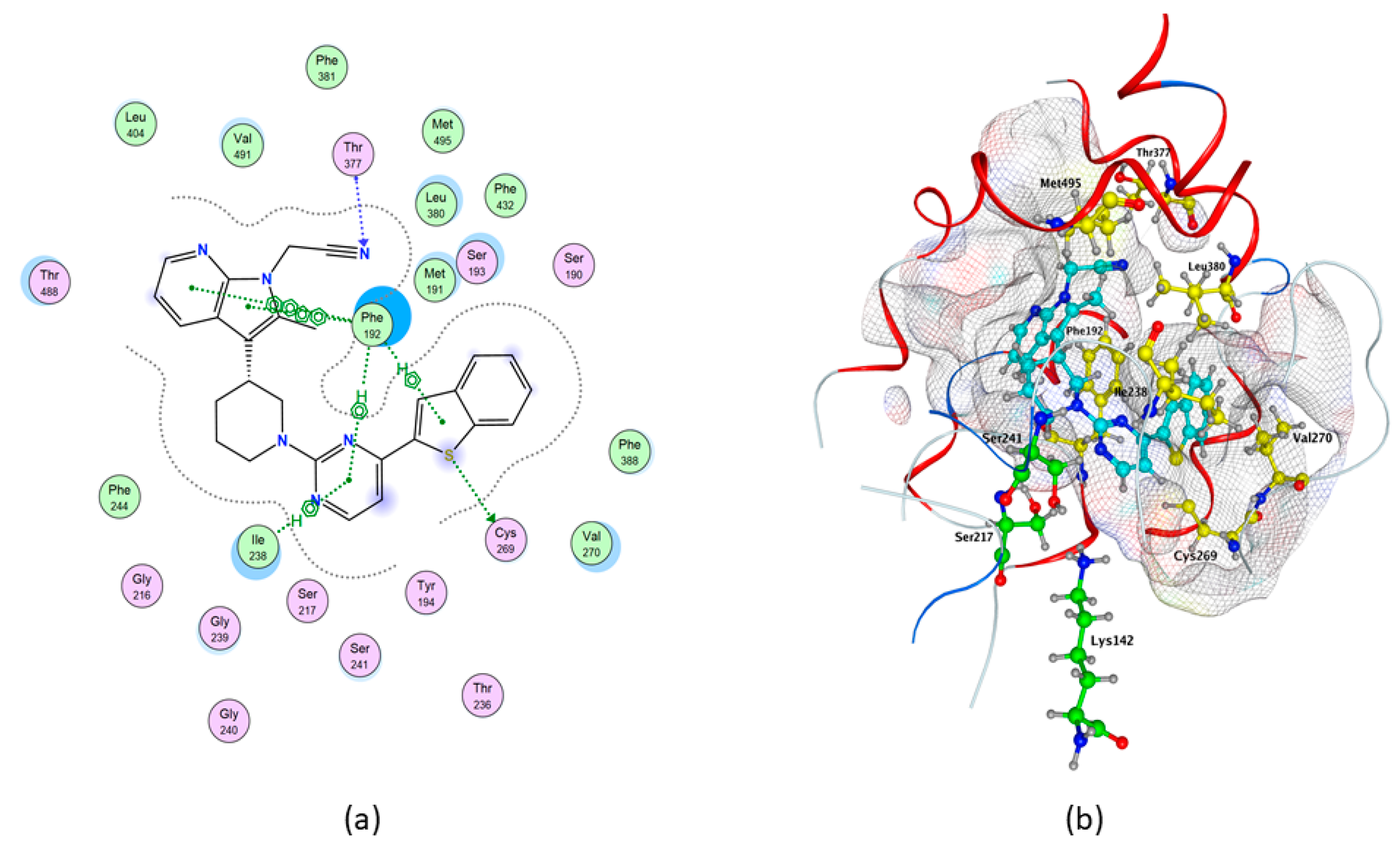
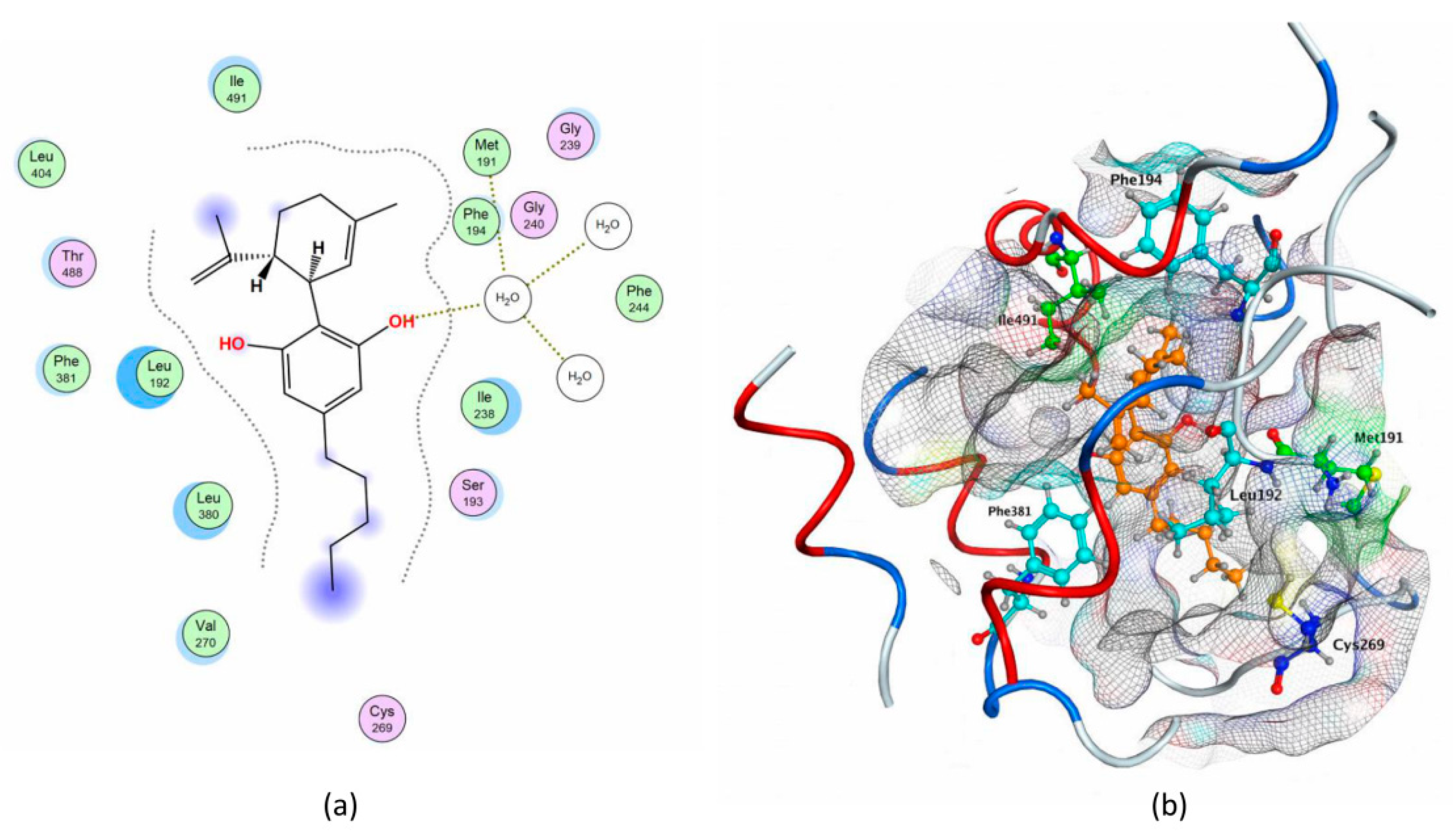
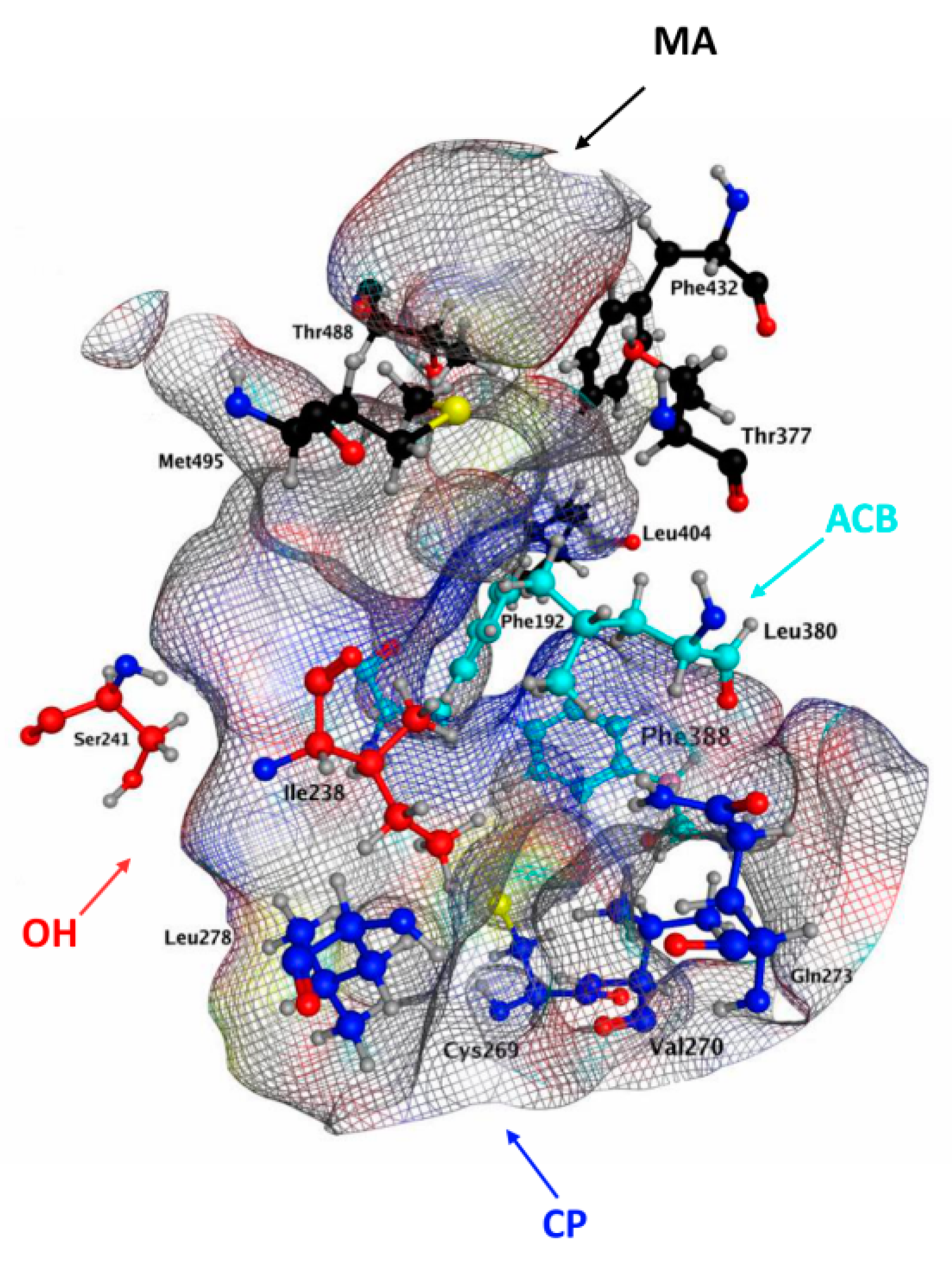
2.1. Molecular Docking of rFAAH
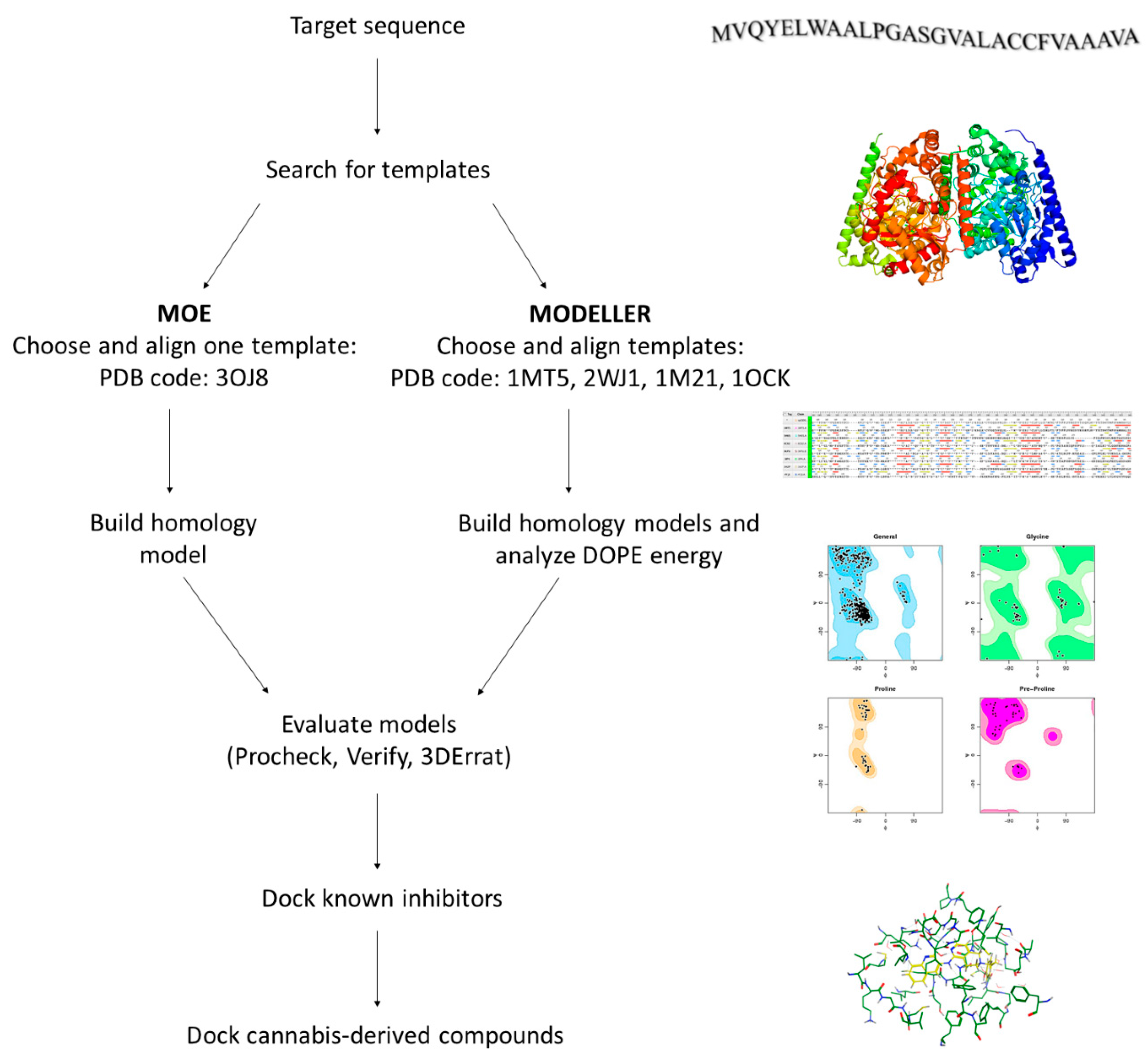
2.2. Homology Modeling of hFAAH
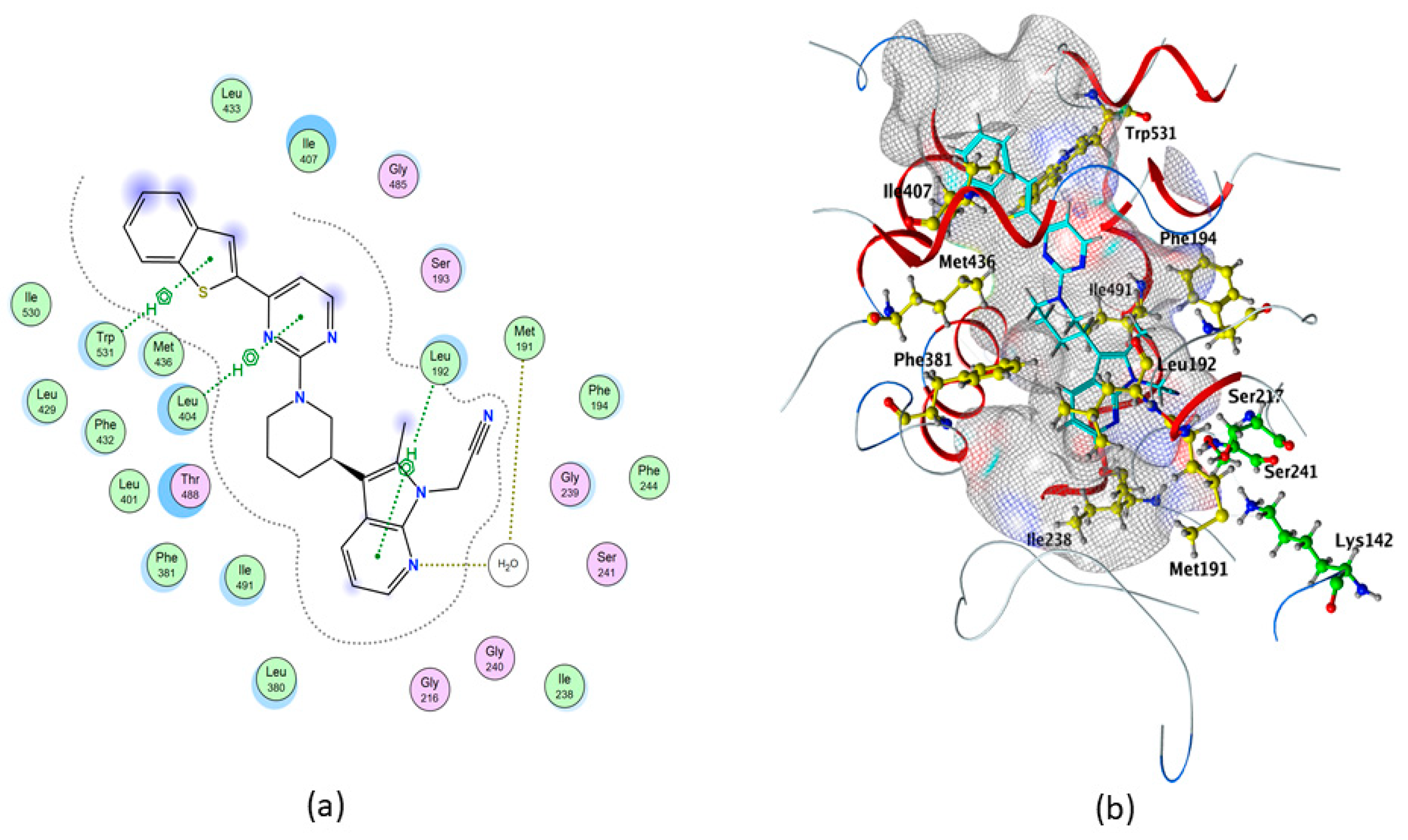
2.3. Molecular Docking of hFAAH
2.4. Activity Assays
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Protein Preparation and Docking Analysis
4.3. Homology Modeling
4.4. FAAH Activity Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cooray, R.; Gupta, V.; Suphioglu, C. Current Aspects of the Endocannabinoid System and Targeted THC and CBD Phytocannabinoids as Potential Therapeutics for Parkinson’s and Alzheimer’s Diseases: A Review. Mol. Neurobiol. 2020, 57, 4878–4890. [Google Scholar] [CrossRef] [PubMed]
- Friedman, D.; French, J.A.; Maccarrone, M. Safety, efficacy, and mechanisms of action of cannabinoids in neurological disorders. Lancet Neurol. 2019, 18, 504–512. [Google Scholar] [CrossRef]
- Patil, A.S.; Mahajan, U.B.; Agrawal, Y.O.; Patil, K.R.; Patil, C.R.; Ojha, S.; Sharma, C.; Goyal, S.N. Plant-derived natural therapeutics targeting cannabinoid receptors in metabolic syndrome and its complications: A review. Biomed. Pharmacother. 2020, 132, 110889. [Google Scholar] [CrossRef] [PubMed]
- Namdar, D.; Anis, O.; Poulin, P.; Koltai, H. Chronological Review and Rational and Future Prospects of Cannabis-Based Drug Development. Molecules 2020, 25, 4821. [Google Scholar] [CrossRef] [PubMed]
- Zagzoog, A.; Mohamed, K.A.; Kim, H.J.J.; Kim, E.D.; Frank, C.S.; Black, T.; Jadhav, P.D.; Holbrook, L.A.; Laprairie, R.B. In vitro and in vivo pharmacological activity of minor cannabinoids isolated from Cannabis sativa. Sci. Rep. 2020, 10, 20405. [Google Scholar] [CrossRef]
- CANNABIDIOL (CBD). Critical Review Report. In Proceedings of the Expert Committee on Drug Dependence, Fortieth Meeting, Geneva, 4–7 June 2018. [Google Scholar]
- Freeman, T.P.; Craft, S.; Wilson, J.; Stylianou, S.; ElSohly, M.; Forti, M.D.; Lynskey, M.T. Changes in delta-9-tetrahydrocannabinol (THC) and cannabidiol (CBD) concentrations in cannabis over time: Systematic review and meta-analysis. Addiction 2020. [Google Scholar] [CrossRef]
- Fiani, B.; Sarhadi, K.J.; Soula, M.; Zafar, A.; Quadri, S.A. Current application of cannabidiol (CBD) in the management and treatment of neurological disorders. Neurol. Sci. 2020, 41, 3085–3098. [Google Scholar] [CrossRef]
- Lattanzi, S.; Zaccara, G.; Russo, E.; La Neve, A.; Lodi, M.A.M.; Striano, P. Practical use of pharmaceutically purified oral cannabidiol in Dravet syndrome and Lennox-Gastaut syndrome. Expert Rev. Neurother. 2020, 1–12. [Google Scholar] [CrossRef]
- Barnett, J.R.; Grinspoon, R.A.; Harisinghani, M.; Caruso, P.A.; Thiele, E.A. The efficacy of cannabidiol on renal angiomyolipoma and subependymal giant cell tumor volume in tuberous sclerosis complex. J. Clin. Neurosci. 2020, 77, 85–88. [Google Scholar] [CrossRef]
- Elsaid, S.; Le Foll, B. The complexity of pharmacology of cannabidiol (CBD) and its implications in the treatment of brain disorders. Neuropsychopharmacology 2020, 45, 229–230. [Google Scholar] [CrossRef]
- Stone, N.L.; Murphy, A.J.; England, T.J.; O’Sullivan, S.E. A systematic review of minor phytocannabinoids with promising neuroprotective potential. Br. J. Pharmacol. 2020, 177, 4330–4352. [Google Scholar] [CrossRef] [PubMed]
- Farha, M.A.; El-Halfawy, O.M.; Gale, R.T.; MacNair, C.R.; Carfrae, L.A.; Zhang, X.; Jentsch, N.G.; Magolan, J.; Brown, E.D. Uncovering the Hidden Antibiotic Potential of Cannabis. ACS Infect. Dis. 2020, 9. [Google Scholar] [CrossRef]
- Weigelt, M.A.; Sivamani, R.; Lev-Tov, H. The therapeutic potential of cannabinoids for integumentary wound management. Exp. Dermatol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Klahn, P. Cannabinoids-Promising Antimicrobial Drugs or Intoxicants with Benefits? Antibiotics 2020, 9, 297. [Google Scholar] [CrossRef] [PubMed]
- Farrimond, J.A.; Whalley, B.J.; Williams, C.M. Cannabinol and cannabidiol exert opposing effects on rat feeding patterns. Psychopharmacology 2012, 223, 117–129. [Google Scholar] [CrossRef]
- Aizpurua-Olaizola, O.; Omar, J.; Navarro, P.; Olivares, M.; Etxebarria, N.; Usobiaga, A. Identification and quantification of cannabinoids in Cannabis sativa L. plants by high performance liquid chromatography-mass spectrometry. Anal. Bioanal. Chem. 2014, 406, 7549–7560. [Google Scholar] [CrossRef]
- Gertsch, J.; Leonti, M.; Raduner, S.; Racz, I.; Chen, J.-Z.; Xie, X.-Q.; Altmann, K.-H.; Karsak, M.; Zimmer, A. Beta-caryophyllene is a dietary cannabinoid. Proc. Natl. Acad. Sci. USA 2008, 105, 9099–9104. [Google Scholar] [CrossRef]
- Liu, H.; Yang, G.; Tang, Y.; Cao, D.; Qi, T.; Qi, Y.; Fan, G. Physicochemical characterization and pharmacokinetics evaluation of β-caryophyllene/β-cyclodextrin inclusion complex. Int. J. Pharm. 2013, 450, 304–310. [Google Scholar] [CrossRef]
- Hashiesh, H.M.; Meeran, M.F.N.; Sharma, C.; Sadek, B.; Kaabi, J.A.; Ojha, S.K. Therapeutic Potential of β-Caryophyllene: A Dietary Cannabinoid in Diabetes and Associated Complications. Nutrients 2020, 12, 2963. [Google Scholar] [CrossRef]
- Fezza, F.; Bari, M.; Florio, R.; Talamonti, E.; Feole, M.; Maccarrone, M. Endocannabinoids, Related Compounds and Their Metabolic Routes. Molecules 2014, 19, 17078–17106. [Google Scholar] [CrossRef]
- Cristino, L.; Bisogno, T.; Di Marzo, V. Cannabinoids and the expanded endocannabinoid system in neurological disorders. Nat. Rev. Neurol. 2020, 16, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Maccarrone, M. Missing pieces to the endocannabinoid puzzle. Trends Mol. Med. 2020, 26, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Maccarrone, M.; Bab, I.; Bíró, T.; Cabral, G.A.; Dey, S.K.; Di Marzo, V.; Konje, J.C.; Kunos, G.; Mechoulam, R.; Pacher, P.; et al. Endocannabinoid signaling at the periphery: 50 years after THC. Trends Pharmacol. Sci. 2015, 36, 277–296. [Google Scholar] [CrossRef] [PubMed]
- Chanda, D.; Neumann, D.; Glatz, J.F.C. The endocannabinoid system: Overview of an emerging multi-faceted therapeutic target. Prostaglandins Leukot. Essent. Fatty Acids 2019, 140, 51–56. [Google Scholar] [CrossRef]
- Mileni, M.; Johnson, D.S.; Wang, Z.; Everdeen, D.S.; Liimatta, M.; Pabst, B.; Bhattacharya, K.; Nugent, R.A.; Kamtekar, S.; Cravatt, B.F.; et al. Structure-guided inhibitor design for human FAAH by interspecies active site conversion. Proc. Natl. Acad. Sci. USA 2008, 105, 12820–12824. [Google Scholar] [CrossRef] [PubMed]
- Cravatt, B.F.; Giang, D.K.; Mayfield, S.P.; Boger, D.L.; Lerner, R.A.; Gilula, N.B. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature 1996, 384, 83–87. [Google Scholar] [CrossRef]
- Fezza, F.; De Simone, C.; Amadio, D.; Maccarrone, M. Fatty acid amide hydrolase: A gate-keeper of the endocannabinoid system. Subcell. Biochem. 2008, 49, 101–132. [Google Scholar] [CrossRef]
- Patricelli, M.P.; Lashuel, H.A.; Giang, D.K.; Kelly, J.W.; Cravatt, B.F. Comparative Characterization of a Wild Type and Transmembrane Domain-Deleted Fatty Acid Amide Hydrolase: Identification of the Transmembrane Domain as a Site for Oligomerization †. Biochemistry 1998, 37, 15177–15187. [Google Scholar] [CrossRef]
- Bracey, M.H.; Hanson, M.A.; Masuda, K.R.; Stevens, R.C.; Cravatt, B.F. Structural Adaptations in a Membrane Enzyme That Terminates Endocannabinoid Signaling. Science 2002, 298, 1793–1796. [Google Scholar] [CrossRef]
- Palermo, G.; Campomanes, P.; Cavalli, A.; Rothlisberger, U. Anandamide hydrolysis in FAAH reveals a dual strategy for efficient enzyme-assisted amide bond cleavage via nitrogen inversion. J. Phys. Chem. B 2015, 13. [Google Scholar] [CrossRef]
- Palermo, G. Computational insights into function and inhibition of fatty acid amide hydrolase. Eur. J. Med. Chem. 2015, 12. [Google Scholar] [CrossRef] [PubMed]
- Palermo, G.; Bauer, I.; Campomanes, P.; Cavalli, A.; Armirotti, A.; Girotto, S.; Rothlisberger, U.; De Vivo, M. Keys to lipid selection in fatty acid amide hydrolase catalysis: Structural flexibility, gating residues and multiple binding pockets. PLoS Comput. Biol. 2015, 11, e1004231. [Google Scholar] [CrossRef] [PubMed]
- Mei, G.; Venere, A.D.; Gasperi, V.; Nicolai, E.; Masuda, K.R.; Finazzi-Agrò, A.; Cravatt, B.F.; Maccarrone, M. Closing the gate to the active site: Effect of the inhibitor methoxyarachidonoylfluorophosphonate on the conformation and membrane binding of fatty acid amide hydrolase. J. Biol. Chem. 2007, 282, 3829–3836. [Google Scholar] [CrossRef] [PubMed]
- Palermo, G.; Campomanes, P.; Neri, M.; Piomelli, D.; Cavalli, A.; Rothlisberger, U.; De Vivo, M. Wagging the Tail: Essential Role of Substrate Flexibility in FAAH Catalysis. J. Chem. Theory Comput. 2013, 9, 1202–1213. [Google Scholar] [CrossRef] [PubMed]
- Patricelli, M.P.; Cravatt, B.F. Characterization and Manipulation of the Acyl Chain Selectivity of Fatty Acid Amide Hydrolase. Biochemistry 2001, 40, 6107–6115. [Google Scholar] [CrossRef]
- Otrubova, K.; Ezzili, C.; Boger, D.L. The discovery and development of inhibitors of fatty acid amide hydrolase (FAAH). Bioorg. Med. Chem. Lett. 2011, 21, 4674–4685. [Google Scholar] [CrossRef]
- Lodola, A.; Sirirak, J.; Fey, N.; Rivara, S.; Mor, M.; Mulholland, A.J. Structural Fluctuations in Enzyme-Catalyzed Reactions: Determinants of Reactivity in Fatty Acid Amide Hydrolase from Multivariate Statistical Analysis of Quantum Mechanics/Molecular Mechanics Paths. J. Chem. Theory Comput. 2010, 6, 2948–2960. [Google Scholar] [CrossRef]
- Otrubova, K.; Cravatt, B.F.; Boger, D.L. Design, Synthesis, and Characterization of α-Ketoheterocycles That Additionally Target the Cytosolic Port Cys269 of Fatty Acid Amide Hydrolase. J. Med. Chem. 2014, 57, 1079–1089. [Google Scholar] [CrossRef]
- Bertolacci, L.; Romeo, E.; Veronesi, M.; Magotti, P.; Albani, C.; Dionisi, M.; Lambruschini, C.; Scarpelli, R.; Cavalli, A.; Garau, G. A Binding Site for Nonsteroidal Anti-inflammatory Drugs in Fatty Acid Amide Hydrolase. J. Am. Chem. Soc. 2013, 4. [Google Scholar] [CrossRef]
- Fazio, D.; Criscuolo, E.; Piccoli, A.; Barboni, B.; Fezza, F.; Maccarrone, M. Advances in the discovery of fatty acid amide hydrolase inhibitors: What does the future hold? Expert Opin. Drug Discov. 2020, 15, 765–778. [Google Scholar] [CrossRef]
- Di Venere, A.; Dainese, E.; Fezza, F.; Angelucci, B.C.; Rosato, N.; Cravatt, B.F.; Finazzi-Agrò, A.; Mei, G.; Maccarrone, M. Rat and human fatty acid amide hydrolases: Overt similarities and hidden differences. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2012, 1821, 1425–1433. [Google Scholar] [CrossRef] [PubMed]
- Muhammed, M.T.; Aki-Yalcin, E. Homology modeling in drug discovery: Overview, current applications, and future perspectives. Chem. Biol. Drug Des. 2019, 93, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Chudyk, E.I.; Dyguda-Kazimierowicz, E.; Langner, K.M.; Sokalski, W.A.; Lodola, A.; Mor, M.; Sirirak, J.; Mulholland, A.J. Nonempirical Energetic Analysis of Reactivity and Covalent Inhibition of Fatty Acid Amide Hydrolase. J. Phys. Chem. B 2013, 117, 6656–6666. [Google Scholar] [CrossRef] [PubMed]
- Bisogno, T.; Maccarrone, M. Latest advances in the discovery of fatty acid amide hydrolase inhibitors. Expert Opin. Drug Discov. 2013, 8, 509–522. [Google Scholar] [CrossRef]
- Petrocellis, L.D.; Ligresti, A.; Moriello, A.S.; Allarà, M.; Bisogno, T.; Petrosino, S.; Stott, C.G.; Marzo, V.D. Effects of cannabinoids and cannabinoid-enriched Cannabis extracts on TRP channels and endocannabinoid metabolic enzymes. Br. J. Pharmacol. 2011, 163, 1479–1494. [Google Scholar] [CrossRef]
- Elmes, M.W.; Kaczocha, M.; Berger, W.T.; Leung, K.; Ralph, B.P.; Wang, L.; Sweeney, J.M.; Miyauchi, J.T.; Tsirka, S.E.; Ojima, I.; et al. Fatty Acid-binding Proteins (FABPs) Are Intracellular Carriers for Δ9-Tetrahydrocannabinol (THC) and Cannabidiol (CBD). J. Biol. Chem. 2015, 290, 8711–8721. [Google Scholar] [CrossRef]
- Gustin, D.J.; Ma, Z.; Min, X.; Li, Y.; Hedberg, C.; Guimaraes, C.; Porter, A.C.; Lindstrom, M.; Lester-Zeiner, D.; Xu, G.; et al. Identification of potent, noncovalent fatty acid amide hydrolase (FAAH) inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 2492–2496. [Google Scholar] [CrossRef]
- Burley, S.K.; Berman, H.M.; Bhikadiya, C.; Bi, C.; Chen, L.; Di Costanzo, L.; Christie, C.; Dalenberg, K.; Duarte, J.M.; Dutta, S.; et al. RCSB Protein Data Bank: Biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic Acids Res. 2019, 47, D464–D474. [Google Scholar] [CrossRef]
- Chemical Computing Group (CCG) | Research. Available online: https://www.chemcomp.com/Research-Citing_MOE.htm (accessed on 29 November 2020).
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef]
- Labahn, J.; Neumann, S.; Büldt, G.; Kula, M.-R.; Granzin, J. An Alternative Mechanism for Amidase Signature Enzymes. J. Mol. Biol. 2002, 322, 1053–1064. [Google Scholar] [CrossRef]
- Shin, S.; Yun, Y.S.; Koo, H.M.; Kim, Y.S.; Choi, K.Y.; Oh, B.-H. Characterization of a Novel Ser-cisSer-Lys Catalytic Triad in Comparison with the Classical Ser-His-Asp Triad. J. Biol. Chem. 2003, 278, 24937–24943. [Google Scholar] [CrossRef]
- Mileni, M.; Garfunkle, J.; DeMartino, J.K.; Cravatt, B.F.; Boger, D.L.; Stevens, R.C. Binding and Inactivation Mechanism of a Humanized Fatty Acid Amide Hydrolase by α-Ketoheterocycle Inhibitors Revealed from Cocrystal Structures. J. Am. Chem. Soc. 2009, 131, 10497–10506. [Google Scholar] [CrossRef]
- Ezzili, C.; Mileni, M.; McGlinchey, N.; Long, J.Z.; Kinsey, S.G.; Hochstatter, D.G.; Stevens, R.C.; Lichtman, A.H.; Cravatt, B.F.; Bilsky, E.J.; et al. Reversible Competitive α-Ketoheterocycle Inhibitors of Fatty Acid Amide Hydrolase Containing Additional Conformational Constraints in the Acyl Side Chain: Orally Active, Long-Acting Analgesics. J. Med. Chem. 2011, 54, 2805–2822. [Google Scholar] [CrossRef]
- Larsson, P.; Wallner, B.; Lindahl, E.; Elofsson, A. Using multiple templates to improve quality of homology models in automated homology modeling. Protein Sci. 2008, 17, 990–1002. [Google Scholar] [CrossRef]
- Shen, M.; Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. Publ. Protein Soc. 2006, 15, 2507–2524. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Bowie, J.U.; Luthy, R.; Eisenberg, D. A method to identify protein sequences that fold into a known three-dimensional structure. Science 1991, 253, 164–170. [Google Scholar] [CrossRef]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef]
- Pontius, J.; Richelle, J.; Wodak, S.J. Deviations from standard atomic volumes as a quality measure for protein crystal structures. J. Mol. Biol. 1996, 121–136. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Y.; Lv, J. An Effective Cumulative Torsion Angles Model for Prediction of Protein Folding Rates. Protein Pept. Lett. 2020, 27, 321–328. [Google Scholar] [CrossRef]
- Lovell, S.C.; Davis, I.W.; Arendall, W.B.; de Bakker, P.I.W.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Cα geometry: ϕ,ψ and Cβ deviation. Proteins Struct. Funct. Bioinform. 2003, 50, 437–450. [Google Scholar] [CrossRef]
- Montero, C.; Campillo, N.E.; Goya, P.; Páez, J.A. Homology models of the cannabinoid CB1 and CB2 receptors. A docking analysis study. Eur. J. Med. Chem. 2005, 40, 75–83. [Google Scholar] [CrossRef]
- Jurcik, A.; Bednar, D.; Byska, J.; Marques, S.M.; Furmanova, K.; Daniel, L.; Kokkonen, P.; Brezovsky, J.; Strnad, O.; Stourac, J.; et al. CAVER Analyst 2.0: Analysis and visualization of channels and tunnels in protein structures and molecular dynamics trajectories. Bioinformatics 2018, 34, 3586–3588. [Google Scholar] [CrossRef]
- Petrocellis, L.D.; Orlando, P.; Moriello, A.S.; Aviello, G.; Stott, C.; Izzo, A.A.; Marzo, V.D. Cannabinoid actions at TRPV channels: Effects on TRPV3 and TRPV4 and their potential relevance to gastrointestinal inflammation. Acta Physiol. 2012, 204, 255–266. [Google Scholar] [CrossRef]
- De Almeida, D.L.; Devi, L.A. Diversity of molecular targets and signaling pathways for CBD. Pharmacol. Res. Perspect. 2020, 8, e00682. [Google Scholar] [CrossRef]
- Esposito, G.; Scuderi, C.; Valenza, M.; Togna, G.I.; Latina, V.; De Filippis, D.; Cipriano, M.; Carratù, M.R.; Iuvone, T.; Steardo, L. Cannabidiol Reduces Aβ-Induced Neuroinflammation and Promotes Hippocampal Neurogenesis through PPARγ Involvement. PLoS ONE 2011, 6, e28668. [Google Scholar] [CrossRef]
- Bisogno, T.; Hanuš, L.; Petrocellis, L.D.; Tchilibon, S.; Ponde, D.E.; Brandi, I.; Moriello, A.S.; Davis, J.B.; Mechoulam, R.; Marzo, V.D. Molecular targets for cannabidiol and its synthetic analogues: Effect on vanilloid VR1 receptors and on the cellular uptake and enzymatic hydrolysis of anandamide. Br. J. Pharmacol. 2001, 134, 845–852. [Google Scholar] [CrossRef]
- Russo, E.B.; Burnett, A.; Hall, B.; Parker, K.K. Agonistic Properties of Cannabidiol at 5-HT1a Receptors. Neurochem. Res. 2005, 30, 1037–1043. [Google Scholar] [CrossRef]
- Zhao, Y.-S.; Zheng, Q.-C.; Zhang, H.-X.; Chu, H.-Y.; Sun, C.-C. Homology modelling and molecular dynamics study of human fatty acid amide hydrolase. Mol. Simul. 2009, 35, 1201–1208. [Google Scholar] [CrossRef]
- McConkey, B.J.; Sobolev, V.; Edelman, M. The performance of current methods in ligand–protein docking. Curr. Sci. 2002, 83, 845–856. [Google Scholar]
- Davis, I.W.; Baker, D. RosettaLigand Docking with Full Ligand and Receptor Flexibility. J. Mol. Biol. 2009, 385, 381–392. [Google Scholar] [CrossRef]
- Consortium, T.U. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef]
- Gattinoni, S.; De Simone, C.; Dallavalle, S.; Fezza, F.; Nannei, R.; Amadio, D.; Minetti, P.; Quattrociocchi, G.; Caprioli, A.; Borsini, F.; et al. Enol carbamates as inhibitors of fatty acid amide hydrolase (FAAH) endowed with high selectivity for FAAH over the other targets of the endocannabinoid system. ChemMedChem 2010, 5, 357–360. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | S (kcal/mol) | Polar Interaction | Residues | Atom Compound | Atom Receptor | Distance (Å) | E (kcal/mol) |
|---|---|---|---|---|---|---|---|
| QK5 | −10.3374 | H-acceptor | H2O | N | O | 3.22 | −1.1 |
| pi–H | Leu192 | 6-ring | C | 4.31 | −0.8 | ||
| pi–H | Leu404 | 6-ring | C | 3.86 | −0.9 | ||
| pi–H | Trp531 | 5-ring | C | 3.66 | −0.6 | ||
| CBG | −8.9776 | - | - | - | - | - | - |
| CBD | −8.2035 | H-donor | H2O/Met191 | O | O | 3.21 | −0.9 |
| CBC | −7.9880 | pi–H | Leu380 | 6-ring | C | 3.78 | −0.5 |
| CBN | −7.7719 | H-donor | Met436 | O | S | 3.29 | −2.6 |
| pi–H | Phe381 | 6-ring | C | 4.59 | −0.7 | ||
| THCV | −7.6375 | - | - | - | - | - | - |
| BCP | −6.4016 | - | - | - | - | - | - |
| Cavities | Residues r/h | QK5 | CBG | CBD | CBC | CBN | THCV | BCP | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| rFAAH | hFAAH | rFAAH | hFAAH | rFAAH | hFAAH | rFAAH | hFAAH | rFAAH | hFAAH | rFAAH | hFAAH | rFAAH | hFAAH | ||
| MA | Asp403 | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| Arg486 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| Ile407 | ✔ | - | ✔ | - | - | - | - | - | - | - | - | - | - | - | |
| MA/ACB | Leu/Phe192 | ✔ | ✔ | - | - | ✔ | - | ✔ | ✔ | ✔ | ✔ | ✔ | ✔ | ✔ | ✔ |
| Phe/Tyr194 | ✔ | - | - | - | ✔ | - | - | - | - | - | - | - | ✔ | - | |
| Phe381 | ✔ | ✔ | - | - | ✔ | - | ✔ | - | ✔ | - | ✔ | - | - | - | |
| Phe432 | - | - | - | - | - | - | ✔ | - | - | - | ✔ | - | - | - | |
| Met436 | ✔ | - | ✔ | - | - | - | - | - | ✔ | - | ✔ | - | - | - | |
| Trp 531 | ✔ | - | ✔ | - | - | - | - | - | ✔ | - | - | - | - | - | |
| OH | Ile 238 | ✔ | ✔ | - | ✔ | - | ✔ | ✔ | ✔ | - | ✔ | - | ✔ | ✔ | ✔ |
| Gly239 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| Gly240 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| Ser241 | - | ✔ | - | - | - | - | - | - | - | ✔ | - | - | - | - | |
| OIR | Met191 | ✔ | - | - | ✔ | ✔ | ✔ | - | ✔ | - | ✔ | - | ✔ | - | ✔ |
| Ile/Val491 | ✔ | ✔ | - | ✔ | ✔ | ✔ | ✔ | - | ✔ | - | ✔ | ✔ | ✔ | - | |
| Val/Met495 | - | ✔ | - | - | - | - | - | - | ✔ | - | - | - | - | - | |
| Ser218 | - | - | - | - | - | - | - | - | - | - | - | - | - | - | |
| CP | Thr236 | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| Cys269 | - | ✔ | - | ✔ | ✔ | ✔ | - | ✔ | - | ✔ | - | ✔ | - | ✔ | |
| Compounds | S (kcal/mol) | Interaction | Residues | Atom Compound | Atom Receptor | Distance (Å) | E (kcal/mol) |
|---|---|---|---|---|---|---|---|
| QK5 | −10.3235 | H-donor | Cys 269 | S | S | 3.46 | −1.1 |
| H-acceptor | Thr 377 | N | C | 3.73 | −0.5 | ||
| pi–H | Phe 192 | 6-ring | C | 4.28 | −0.6 | ||
| Phe 192 | 5-ring | C | 4.2 | −1.1 | |||
| Ile238 | 6-ring | N | 4.37 | −1.7 | |||
| Ile238 | C | 3.7 | −0.7 | ||||
| pi–pi | Phe 192 | 5-ring | 6-ring | 3.86 | 0 | ||
| Phe 192 | 6-ring | 6-ring | 3.97 | 0 | |||
| CBG | −8.8566 | - | - | - | - | - | - |
| CBC | −8.8508 | pi–H | Ile238 | 6-ring | C | 4.23 | −0.7 |
| CBN | −8.7723 | H-donor | Ser241 | O | O | 3.22 | −0.5 |
| H-pi | Phe192 | C | 6-ring | 4.23 | −0.5 | ||
| pi–H | Phe192 | 6-ring | C | 4.26 | −1 | ||
| Ile238 | C | 3.53 | −0.5 | ||||
| Ile238 | N | 4.33 | −0.8 | ||||
| CBD | −8.3664 | - | - | - | - | - | - |
| THCV | −7.8611 | pi–H | Phe192 | 6-ring | C | 4.35 | −0.7 |
| Ile238 | N | 4.21 | −0.5 | ||||
| Ile238 | C | 3.49 | −0.5 | ||||
| BCP | −6.0123 | - | - | - | - | - | - |
| Compound | IC50 (µM) Towards rFAAH | IC50 (µM) Towards hFAAH |
|---|---|---|
| CBD | 43.5 ± 1.5 | >100 |
| CBN | 60.0 ± 10.0 | ~100 |
| CBC | ~100 | >100 |
| CBG | ~100 | >100 |
| THCV | >100 | >100 |
| BCP | >100 | >100 |
| S (Kcal/Mol) | Total Key Interactions | Polar Key Interactions | % In Vitro Inhibition (At 100 µm) | Binding Cavities | |
|---|---|---|---|---|---|
| QK5 | −10.3374 | 9 | 3 | N.A. | MA, OH, MA/ACB, Met191 |
| CBD | −8.2035 | 6 | 1 | 94 | MA/ACB, CP, Met191 |
| CBN | −7.7719 | 6 | 2 | 65 | MA/ACB |
| CBG | −8.9776 | 3 | 0 | 53 | MA, MA/ACB |
| CBC | −7.988 | 5 | 0 | 51 | OH, MA/ACB |
| THCV | −7.6375 | 5 | 0 | 30 | MA/ACB |
| BCP | −6.4016 | 4 | 0 | 18 | OH, MA/ACB |
Sample Availability: Samples of the compounds are not available. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Criscuolo, E.; De Sciscio, M.L.; Fezza, F.; Maccarrone, M. In Silico and In Vitro Analysis of Major Cannabis-Derived Compounds as Fatty Acid Amide Hydrolase Inhibitors. Molecules 2021, 26, 48. https://doi.org/10.3390/molecules26010048
Criscuolo E, De Sciscio ML, Fezza F, Maccarrone M. In Silico and In Vitro Analysis of Major Cannabis-Derived Compounds as Fatty Acid Amide Hydrolase Inhibitors. Molecules. 2021; 26(1):48. https://doi.org/10.3390/molecules26010048
Chicago/Turabian StyleCriscuolo, Emanuele, Maria Laura De Sciscio, Filomena Fezza, and Mauro Maccarrone. 2021. "In Silico and In Vitro Analysis of Major Cannabis-Derived Compounds as Fatty Acid Amide Hydrolase Inhibitors" Molecules 26, no. 1: 48. https://doi.org/10.3390/molecules26010048
APA StyleCriscuolo, E., De Sciscio, M. L., Fezza, F., & Maccarrone, M. (2021). In Silico and In Vitro Analysis of Major Cannabis-Derived Compounds as Fatty Acid Amide Hydrolase Inhibitors. Molecules, 26(1), 48. https://doi.org/10.3390/molecules26010048