
Stable Bicyclic Functionalized Nitroxides: The Synthesis of Derivatives of Aza-nortropinone–5-Methyl-3-oxo-6,8-diazabicyclo[3.2.1]-6-octene 8-oxyls



Abstract
: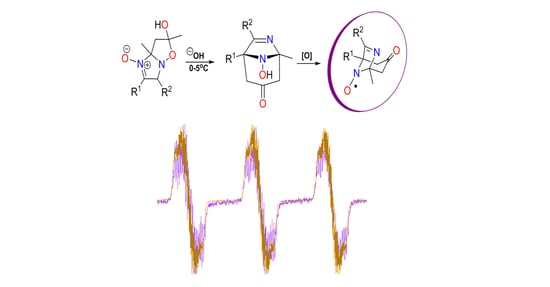
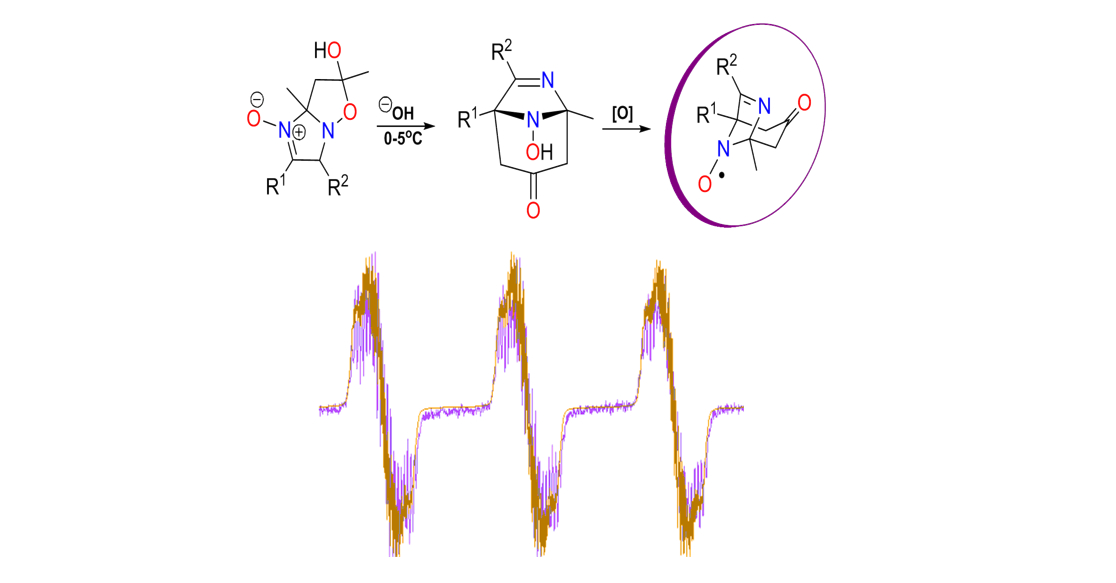{kind=link}
{kind=link}
{kind=link}
{kind=link}
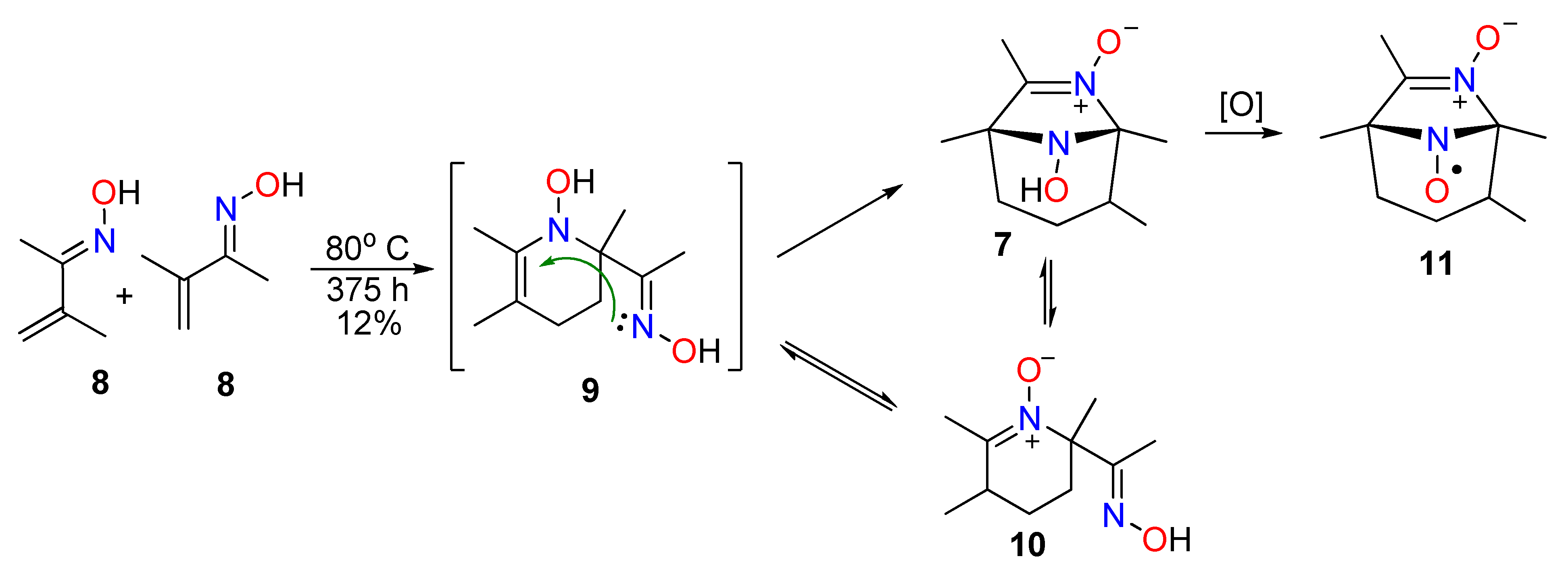{kind=link}
1. Introduction
2. Results
2.1. The Synthesis of Bicyclic Nitroxides
2.2. EPR Study of New Bicyclic Nitroxides 6
3. Materials and Methods
3.1. General Information
3.2. Synthesis
3.2.1. Imidazo[1,2-b]isoxazoles 2a,c,d
3.2.2. The General Synthetic Procedure for the Recyclization of Imidazo[1,2-b]isoxazoles 2a,c,d to 8-Hydroxy-5-methyl-3-oxo-6,8-diazabicyclo[3,2,1]-6-octenes 5a,c,d
3.2.3. The General Procedure for the Oxidation of 8-Hydroxy-6,8-diazabicyclo[3.2.1]oct-6-en-3-ones 5a,c,d
3.2.4. The General Procedure for the Reduction of NRs 6a,c,d
3.2.5. Base-Promoted Recyclization of 8-Hydroxy-6,8-diazabicyclo[3.2.1]oct-6-en-3-one 5d into Tetrahydropyridine-4-one 3d
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Volodarsky, L.B.; Reznikov, V.A.; Ovcharenko, V.I. Synthetic Chemistry of Stable Nitroxides; CRC Press: Boca Raton, FL, USA, 1994; pp. 1–240. [Google Scholar]
- Volodarsky, L.B.; Grigor’ev, I.A.; Dikanov, S.A.; Reznikov, V.A.; Shchukin, G.I. Imidazoline Nitroxides. Synthesis and Properties; CRC Press: Boca Raton, FL, USA, 1988; Volume 1, pp. 1–222. [Google Scholar]
- Keana, J.F.W. Newer aspects of the synthesis and chemistry of nitroxide spin labels. Chem. Rev. 1978, 78, 37–64. [Google Scholar] [CrossRef]
- Karoui, H.; Le Moigne, F.; Ouari, O.; Tordo, P. Nitroxide Radicals: Properties, Synthesis and Applications. In Stable Radicals: Fundamentals and Applied Aspects of Odd-Electron Compounds; Hicks, R.G., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2010; pp. 173–229. [Google Scholar] [CrossRef]
- Tretyakov, E.V.; Ovcharenko, V.I. The chemistry of nitroxide radicals in the molecular design of magnets. Russ. Chem. Rev. 2009, 78, 971–1012. [Google Scholar] [CrossRef]
- Haugland, M.M.; Lovett, J.E.; Anderson, E.A. Advances in the synthesis of nitroxide radicals for use in biomolecule spin labelling. Chem. Soc. Rev. 2018, 47, 668–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bognar, B.; Ur, G.; Sar, C.; Hankovszky, O.H.; Hideg, K.; Kalai, T. Synthesis and Application of Stable Nitroxide Free Radicals Fused with Carbocycles and Heterocycles. Curr. Org. Chem. 2019, 23, 480–501. [Google Scholar] [CrossRef]
- Zaytseva, E.V.; Mazhukin, D.G. Spirocyclic Nitroxides as Versatile Tools in Modern Natural Sciences: From Synthesis to Applications. Part I. Old and New Synthetic Approaches to Spirocyclic Nitroxyl Radicals. Molecules 2021, 26, 677. [Google Scholar] [CrossRef] [PubMed]
- Kirilyuk, I.A.; Mazhukin, D.G. General Approaches to Synthesis of Nitroxides. In Nitroxides: Synthesis, Properties and Applications; Ouari, O., Gigmes, D., Eds.; The Royal Society of Chemistry: Cambridge, UK, 2021; pp. 7–70. [Google Scholar]
- Tebben, L.; Studer, A. Nitroxides: Applications in Synthesis and in Polymer Chemistry. Angew. Chem. Int. Ed. 2011, 50, 5034–5068. [Google Scholar] [CrossRef]
- Ma, Z.; Mahmudov, K.T.; Aliyeva, V.A.; Gurbanov, A.V.; Pombeiro, A.J.L. TEMPO in metal complex catalysis. Coord. Chem. Rev. 2020, 423, 213482. [Google Scholar] [CrossRef]
- Bagryanskaya, E.G.; Marque, S.R.A. Scavenging of Organic C-Centered Radicals by Nitroxides. Chem. Rev. 2014, 114, 5011–5056. [Google Scholar] [CrossRef]
- Khramtsov, V.V.; Zweier, J.L. Functional in vivo EPR Spectroscopy and Imaging Using Nitroxide and Trityl Radicals. In Stable Radicals: Fundamentals and Applied Aspects of Odd-Electron Compounds; Hicks, R.G., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2010; pp. 537–566. [Google Scholar] [CrossRef]
- Bardelang, D.; Hardy, M.; Ouari, O.; Tordo, P. Spin Labels and Spin Probes. In Encyclopedia of Radicals in Chemistry, Biology and Materials; Chatgilialoglu, C., Studer, A., Eds.; Wiley, 2012; pp. 1–51. Available online: https://onlinelibrary.wiley.com/doi/abs/10.1002/9781119953678.rad081 (accessed on 19 May 2021).
- Likhtenshtein, G.I. Nitroxides: Brief. History, Fundamentals, and Recent Developments; Springer: Cham, Switzerland, 2020; pp. 1–316. [Google Scholar] [CrossRef]
- Likhtenshtein, G.I.; Yamauchi, J.; Nakatsuji, S.; Smirnov, A.I.; Tamura, R. Nitroxides: Applications in Chemistry, Biomedicine and Material Science; Wiley: Weinheim, Germany, 2008; pp. 1–419. [Google Scholar]
- Koike, T.; Yasu, Y.; Akita, M. Visible-light-driven Oxidation of 1,3-Dicarbonyl Compounds via Catalytic Disproportionation of TEMPO by Photoredox Catalysis. Chem. Lett. 2012, 41, 999–1001. [Google Scholar] [CrossRef]
- Gentry, E.C.; Rono, L.J.; Hale, M.E.; Matsuura, R.; Knowles, R.R. Enantioselective Synthesis of Pyrroloindolines via Noncovalent Stabilization of Indole Radical Cations and Applications to the Synthesis of Alkaloid Natural Products. J. Am. Chem. Soc. 2018, 140, 3394–3402. [Google Scholar] [CrossRef] [Green Version]
- Liang, K.; Tong, X.; Li, T.; Shi, B.; Wang, H.; Yan, P.; Xia, C. Enantioselective Radical Cyclization of Tryptamines by Visible Light-Excited Nitroxides. J. Org. Chem. 2018, 83, 10948–10958. [Google Scholar] [CrossRef]
- Chen, N.; Xu, H.-C. Electrochemical generation of nitrogen-centered radicals for organic synthesis. Green Synth. Catal. 2021, 2. [Google Scholar] [CrossRef]
- Siu, J.C.; Parry, J.B.; Lin, S. Aminoxyl-Catalyzed Electrochemical Diazidation of Alkenes Mediated by a Metastable Charge-Transfer Complex. J. Am. Chem. Soc. 2019, 141, 2825–2831. [Google Scholar] [CrossRef]
- Graetz, B.; Rychnovsky, S.; Leu, W.-H.; Farmer, P.; Lin, R. C2-Symmetric nitroxides and their potential as enantioselective oxidants. Tetrahedron Asymmetry 2005, 16, 3584–3598. [Google Scholar] [CrossRef]
- Steves, J.E.; Stahl, S.S. Copper(I)/ABNO-Catalyzed Aerobic Alcohol Oxidation: Alleviating Steric and Electronic Constraints of Cu/TEMPO Catalyst Systems. J. Am. Chem. Soc. 2013, 135, 15742–15745. [Google Scholar] [CrossRef] [PubMed]
- Ryland, B.L.; McCann, S.D.; Brunold, T.C.; Stahl, S.S. Mechanism of Alcohol Oxidation Mediated by Copper(II) and Nitroxyl Radicals. J. Am. Chem. Soc. 2014, 136, 12166–12173. [Google Scholar] [CrossRef] [Green Version]
- Rafiee, M.; Miles, K.C.; Stahl, S.S. Electrocatalytic Alcohol Oxidation with TEMPO and Bicyclic Nitroxyl Derivatives: Driving Force Trumps Steric Effects. J. Am. Chem. Soc. 2015, 137, 14751–14757. [Google Scholar] [CrossRef] [Green Version]
- Hickey, D.P.; Schiedler, D.A.; Matanovic, I.; Doan, P.V.; Atanassov, P.; Minteer, S.D.; Sigman, M.S. Predicting Electrocatalytic Properties: Modeling Structure−Activity Relationships of Nitroxyl Radicals. J. Am. Chem. Soc. 2015, 137, 16179–16186. [Google Scholar] [CrossRef]
- Kim, J.; Stahl, S.S. Cu/Nitroxyl-Catalyzed Aerobic Oxidation of Primary Amines into Nitriles at Room Temperature. ACS Catal. 2013, 3, 1652–1656. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.; Stahl, S.S. Efficient and Selective Cu/Nitroxyl-Catalyzed Methods for Aerobic Oxidative Lactonization of Diols. J. Am. Chem. Soc. 2015, 137, 3767–3770. [Google Scholar] [CrossRef]
- Wang, F.; Rafiee, M.; Stahl, S.S. Electrochemical Functional-Group-Tolerant Shono-type Oxidation of Cyclic Carbamates Enabled by Aminoxyl Mediators. Angew. Chem. Int. Ed. 2018, 57, 6686–6690. [Google Scholar] [CrossRef] [PubMed]
- Anthony, D.; Lin, Q.; Baudet, J.; Diao, T. Nickel-Catalyzed Asymmetric Reductive Diarylation of Vinylarenes. Angew. Chem. Int. Ed. 2019, 58, 3198–3202. [Google Scholar] [CrossRef] [PubMed]
- Lennox, A.J.J.; Goes, S.L.; Webster, M.P.; Koolman, H.F.; Djuric, S.W.; Stahl, S.S. Electrochemical Aminoxyl-Mediated α-Cyanation of Secondary Piperidines for Pharmaceutical Building Block Diversification. J. Am. Chem. Soc. 2018, 140, 11227–11231. [Google Scholar] [CrossRef]
- Dupeyre, R.-M.; Rassat, A. Nitroxides. XIX. Norpseudopelletierine-N-oxyl, a New, Stable, Unhindered Free Radical. J. Am. Chem. Soc. 1966, 88, 3180–3181. [Google Scholar] [CrossRef]
- Rassat, A.; Rey, P. Nitroxides: Photochemical Synthesis of Trimethylisoquinuclidine N-Oxyl. Chem. Commun. 1971, 1161–1162. [Google Scholar] [CrossRef]
- Mendenhall, G.D.; Ingold, K.U. Reactions of Bicyclic Nitroxides Involving Reduction of the NO Group. J. Am. Chem. Soc. 1973, 95, 6395–6400. [Google Scholar] [CrossRef]
- Rassat, A.; Ronzaud, J. Nitroxydes-LXXI. Synthese de derives nitroxydes du tropane. Etude par RPE et RMN des interactions hyperfines a longue distance dans ces radicaux. Tetrahedron 1976, 32, 239–244. [Google Scholar] [CrossRef]
- Shibuya, M.; Tomizawa, M.; Sasano, Y.; Iwabuchi, Y. An Expeditious Entry to 9-Azabicyclo[3.3.1]nonane N-Oxyl (ABNO): Another Highly Active Organocatalyst for Oxidation of Alcohols. J. Org. Chem. 2009, 74, 4619–4622. [Google Scholar] [CrossRef]
- Shibuya, M.; Tomizawa, M.; Suzuki, I.; Iwabuchi, Y. 2-Azaadamantane N-Oxyl (AZADO) and 1-Me-AZADO: Highly Efficient Organocatalysts for Oxidation of Alcohols. J. Am. Chem. Soc. 2006, 128, 8412–8413. [Google Scholar] [CrossRef]
- Hayashi, M.; Sasano, Y.; Nagasawa, S.; Shibuya, M.; Iwabuchi, Y. 9-Azanoradamantane N-Oxyl (Nor-AZADO): A Highly Active Organocatalyst for Alcohol Oxidation. Chem. Pharm. Bull. 2011, 59, 1570–1573. [Google Scholar] [CrossRef] [Green Version]
- Grigor’eva, L.N.; Tikhonov, A.Y.; Martin, V.V.; Volodarskii, L.B. Condensation of 1,2-hydroxylaminooximes with acetylacetone. Conversion of tetrahydroimidazo[1,2-b]isoxazoles into derivatives of 2H-imidazole, 1-hydroxypyrrole, and 4-oxotetrahydropyridine. Chem. Heterocycl. Compd. 1990, 26, 637–642. [Google Scholar] [CrossRef]
- Ota, T.; Masuda, S.; Tanaka, H. Cyclodimerization of 3-Methyl-3-buten-2-one Oxime. Chem. Lett. 1981, 10, 411–414. [Google Scholar] [CrossRef]
- Ota, T.; Masuda, S.; Tanaka, H.; Kido, M. Studies on the Polymerization of Acrolein Oxime. XVI. Structure of 8-Hydroxy-1,4,5,7-tetramethyl-6,8-diazabicyclo[3.2.1]oct-6-ene 6-Oxide. Bull. Chem. Soc. Jpn. 1982, 55, 171–173. [Google Scholar] [CrossRef]
- Schulz, M.; Mögel, L.; Römbach, J. Zur Ring-Ketten-Tautomerie und Reaktionen eines bicyclischen 1-Hydroxy-3-imidazolin-3-oxids. J. Prakt. Chem. 1986, 328, 589–596. [Google Scholar] [CrossRef]
- Reinhardt, M.; Schulz, M.; Römbach, J. Radikalabfang. III. [1] ESR-spektroskopische Untersuchung von Radikalreaktionen eines bicyclischen 1 -Hydroxy-3-imidazolin-3-oxids [2]. J. Prakt. Chem. 1986, 328, 597–602. [Google Scholar] [CrossRef]
- Grigor’eva, L.N.; Volodarskii, L.B.; Tikhonov, A.Y. Preparation of aliphatic α-(hydroxyamino) oximes by reaction of α-halo ketones with hydroxylamine. Izv. Sib. Otd. AN SSSR Ser. Khim. 1989, 3, 125–126. [Google Scholar]
- Keana, J.F.W.; Heo, G.S.; Gaughan, G.T. Stereospecific Synthesis of Difunctionalized 2,5-Disubstituted cis-2,5-Dimethylpyrrolidine (Azethoxyl) Nitroxides by Oxidative Cleavage of Protected 8-Azabicyclo[3.2.l]octane Precursors. J. Org. Chem. 1985, 50, 2351. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grigor’eva, L.N.; Tikhonov, A.Y.; Lomanovich, K.A.; Mazhukin, D.G. Stable Bicyclic Functionalized Nitroxides: The Synthesis of Derivatives of Aza-nortropinone–5-Methyl-3-oxo-6,8-diazabicyclo[3.2.1]-6-octene 8-oxyls. Molecules 2021, 26, 3050. https://doi.org/10.3390/molecules26103050
Grigor’eva LN, Tikhonov AY, Lomanovich KA, Mazhukin DG. Stable Bicyclic Functionalized Nitroxides: The Synthesis of Derivatives of Aza-nortropinone–5-Methyl-3-oxo-6,8-diazabicyclo[3.2.1]-6-octene 8-oxyls. Molecules. 2021; 26(10):3050. https://doi.org/10.3390/molecules26103050
Chicago/Turabian StyleGrigor’eva, Larisa N., Alexsei Ya. Tikhonov, Konstantin A. Lomanovich, and Dmitrii G. Mazhukin. 2021. "Stable Bicyclic Functionalized Nitroxides: The Synthesis of Derivatives of Aza-nortropinone–5-Methyl-3-oxo-6,8-diazabicyclo[3.2.1]-6-octene 8-oxyls" Molecules 26, no. 10: 3050. https://doi.org/10.3390/molecules26103050
APA StyleGrigor’eva, L. N., Tikhonov, A. Y., Lomanovich, K. A., & Mazhukin, D. G. (2021). Stable Bicyclic Functionalized Nitroxides: The Synthesis of Derivatives of Aza-nortropinone–5-Methyl-3-oxo-6,8-diazabicyclo[3.2.1]-6-octene 8-oxyls. Molecules, 26(10), 3050. https://doi.org/10.3390/molecules26103050