Abstract
Due to their structural similarity with natural α-amino acids, α-aminophosphonic acid derivatives are known biologically active molecules. In view of the relevance of tetrasubstituted carbons in nature and medicine and the strong dependence of the biological activity of chiral molecules into their absolute configuration, the synthesis of α-aminophosphonates bearing tetrasubstituted carbons in an asymmetric fashion has grown in interest in the past few decades. In the following lines, the existing literatures for the synthesis of optically active tetrasubstituted α-aminophosphonates are summarized, comprising diastereoselective and enantioselective approaches.
1. Introduction
α-Amino acids are a key structure in living organisms as the essential part of proteins and peptides. Many α-amino acid derivatives are used in daily life, such as the sweetener aspartame, penicillin-derived antibiotics or antihypertensive enalapril. Due to the relevance of α-amino acids in nature, a vast number of methods for the synthesis of natural and non-natural α-amino acids have been developed [1,2,3]. Within the most relevant α-amino acid mimetics, α-aminophosphonic acids are the result of a bioisosteric substitution of the planar carboxylic acid by a phosphonic acid group in α-amino acid structures (Figure 1).
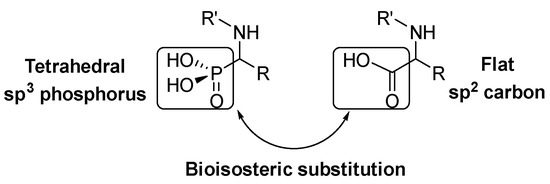
Figure 1.
Structural analogy of α-amino acids and α-aminophosphonic acids.
This isosteric replacement is of great interest since, due to the tetrahedral configuration of the phosphorus atom, α-aminophosphonic acid derivatives can behave as stable analogues of the transition state for the cleavage of peptides, thus inhibiting enzymes involved in proteolysis processes and, consequently, they display assorted biological activities [4,5,6,7]. In particular, a number of α-aminophosphonic acid derivatives have found applications as agrochemicals [8,9], as well as antimicrobial [10,11,12], antioxidant [13,14,15] or anticancer agents (Figure 2) [16,17,18].
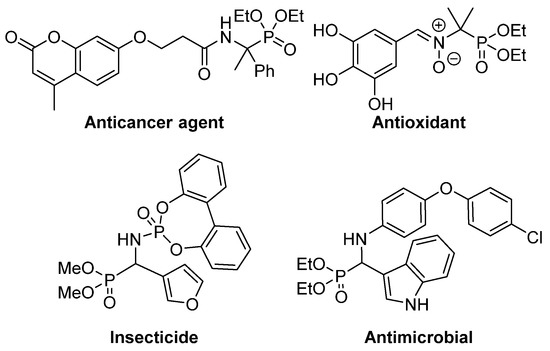
Figure 2.
Some examples of biologically active α-aminophosphonic acid derivatives.
The thalidomide disaster was a shocking revelation of the strong dependence of the biological activity of chiral substrates into their absolute configuration. This dependence is also evident for α-aminophosphonic acid derivatives and, as examples, (R)-phospholeucine exhibits a stronger activity as leucine-peptidase inhibitor than its enantiomer (Figure 3) [19,20], and the phosphopeptide (S),(R)-alaphosphalin has a more efficient antibiotic activity than its other three possible isomers [21,22].
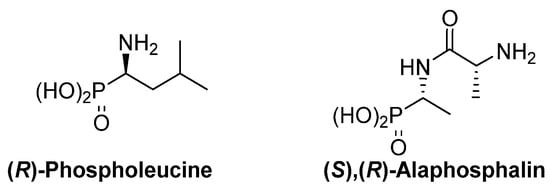
Figure 3.
Most active isomers of phospholeucine and alaphosphalin.
α-Aminophosphonic acids are usually obtained from the hydrolysis of their phosphonate esters and, for this reason, the development of efficient synthetic methodologies to access enantioenriched α-aminophosphonates has become an imperative task in organic chemistry. The existing literature to date in this field is mostly related to the synthesis of trisubstituted α-aminophosphonates and the examples illustrating asymmetric strategies leading to the formation of the tetrasubstituted substrates are scarce [23,24]. The efficient formation of quaternary centers is known as a critical challenge in organic synthesis [25,26,27] and the formation of tetrasubstituted centers from ketimines was for a long time unachievable. The poor electrophilic character of the ketimine group and the additional steric hindrance on the substrate that results in a decreased reactivity are the two main challenges to overcome. In addition, the enantiotopic faces of ketimine substrates are not as easily discriminated as those of aldimines if asymmetric syntheses are required [28].
The existing methods for the synthesis of tetrasubstituted α-aminophosphonates can be classified in three main groups, depending on the type of bond created in the key reaction leading to their formation Figure 4). The first of those approaches implies the use of strategies that entail C-C bond formation, either through the addition of carbon nucleophiles to α-phosphorylated imines (Figure 4a) or functionalization of α-aminophosphonate anions with electrophiles (Figure 4b). In addition, the most straightforward method for the synthesis of α-aminophosphonates comprises reactions that imply C-P bond formation through the addition of phosphorus nucleophiles to ketimines (Figure 4c). Another alternative to these routes consists of processes that involve C-N bond formation, which are carried out mainly through electrophilic amination reactions (Figure 4d).
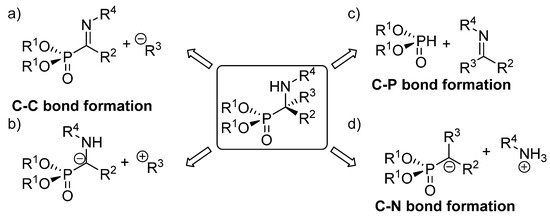
Figure 4.
Asymmetric synthetic approaches to tetrasubstituted α-aminophosphonates: (a) addition of nucleophiles to phosphorylated imines; (b) addition of α aminophosphonates to electrophiles; (c) hydrophosphonylation of imines; (d) electrophilic amination.
In the following lines of this review, the existing methodologies regarding the asymmetric synthesis of tetrasubstituted α-aminophosphonates are summarized. The synthetic routes for the preparation of these compounds are classified into diastereoselective and enantioselective methodologies and grouped by the type of bond formed in the key step.
2. Diastereoselective Synthesis of Tetrasubstituted α-Aminophosphonic Acid Derivatives
2.1. C-C Bond Formation
A simple strategy for the preparation of tetrasubstituted α-aminophosphonates is the functionalization of tetrasubstituted α-aminophosphonates, taking advantage of the acidic nature of the hydrogen atom adjacent to the phosphorus substituent. Using this approach, Seebach described in 1995 the first example of a stereoselective synthesis of tetrasubstituted α-aminophosphonic acids [29]. As shown in Scheme 1, racemic imidazolidinone 2 is first obtained starting from glycine ester 1, by formation of the amide derivative with dimethylamine and subsequent condensation of the amino group with pivalaldehyde. Then, a kinetic resolution using (R)-mandelic acid 3 is carried out, allowing the isolation of diastereomeric salt (R, R)-4, which is treated first with NaOH and then with Boc2O and DMAP to yield enantiomerically pure imidazolidinone 5 [30].
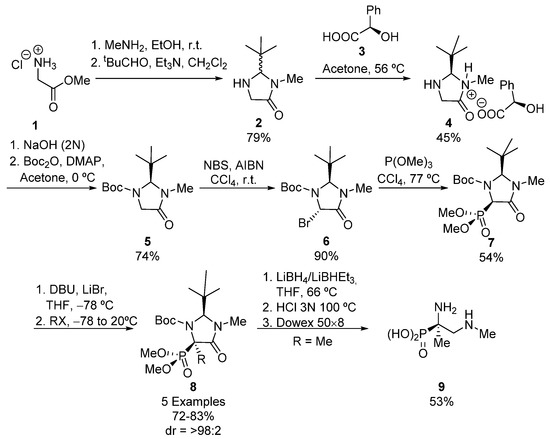
Scheme 1.
First stereoselective synthesis of tetrasubstituted α-aminophosphonic acids.
Then bromide 6 is formed via radical halogenation, and the subsequent Arbuzov reaction with trimethylphosphite leads to a single isomer of tertiary α-aminophosphonate 7 in moderate yield [31]. Next, in the key step, compound 7 is treated with a strong base and an alkyl, allyl or benzyl halide, leading to the formation of tetrasubstituted α-aminophosphonates 8 in good yields and excellent diastereoselectivities (72–83%, >98:2 dr). In all cases, the electrophile reagent approaches from the less hindered face of imidazolidinone ring, in an anti-addition. In order to obtain the acyclic α-aminophosphonic acid derivative 9, the authors performed a reduction of the amide carbonyl in 8 and a hydrolysis of the resulting intermediate in aqueous HCl [29].
In addition, the authors extended their methodology to the use of imidazolidines, instead of amides 7, obtaining, after the addition of benzyl, methyl or allyl halides, cis tetrasubstituted α-aminophosphonates in moderate yields and diastereoselectivities (33–53%, 1.3:1–3.8:1 dr). However, the use of ethyl, propyl and butyl halides gave trans products in better yields and diastereomeric ratios (45–60%, 1:17 ≤ 1:50 dr).
A similar procedure was developed by Davis for the synthesis of pyrrolidine-derived α-aminophosphonates 17 (Scheme 2). In this case, sulfinyl imine 10 is attacked by the enolate derived from ethyl acetate, to obtain N-sulfinyl β-amino ester 11 in a diastereoselective fashion. Then, the addition of lithium methyl phosphonate leads to N-sulfinyl δ-amino β-ketophosphonate 12 in very good yield. Next, the sulfinyl protecting group is easily removed and replaced with a Boc group, obtaining amide 13 which, after treatment with 4-acetamidobenzenesulfonyl azide, provides the corresponding α-diazo derivative 14. Finally, the treatment of α-diazophosphonate 14 in the presence of Rh2(OAc)4 catalyst yields cis pyrrolidine phosphonate 15, as the major diastereoisomer (68%, 81:19 dr) [32]. Tertiary α-aminophosphonate 15 can be functionalized, with retention of the configuration, using a strong base and allyl bromide, providing tetrasubstituted α-aminophosphonate 16. Although substrate 16 is obtained as a mixture of rotamers, the removal of N-Boc group renders pyrrolidin-3-one 17 as a single isomer. Additionally, acyclic α-amino α-ketophosphonate 18 can be also prepared after a ring-opening process via a Pd-catalyzed hydrogenation [33].
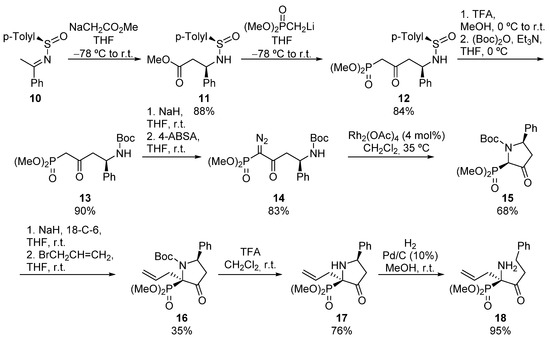
Scheme 2.
Stereoselective synthesis of pyrrolidine-derived α-aminophosphonate 17.
Following the same principle, Amedjkouh described the synthesis of bicyclic α-aminophosphonate 24 (Scheme 3). In this case, the synthetic methodology starts with the preparation of oxazolopyrrolidine phosphonate 23 from (R)-phenylglycinol (19), benzotriazole (20), and 2,5-dimethoxytetrahydrofuran (21), obtaining enantiomerically pure oxazolopyrrolidine 22, by formation of succinaldehyde from the hydrolysis of furan derivative 21 and subsequent multicomponent reaction with substrates 19 and 20. Then, tertiary oxazolopyrrolidine phosphonate 23 is formed through Arbuzov reaction of oxazolopyrrolidine 22 and triethyl phosphite [34,35]. The treatment of α-aminophosphonate 23 with butyllithium followed by the addition of alkyl halides results in the formation of tetrasubstituted α-aminophosphonates 24 with total retention of the configuration, and yields that are moderate to good, when using aliphatic halides (35–81%), but low if benzyl halide is used (10%). Finally, the elimination of the chiral auxiliary via catalytic hydrogenation affords optically pure phosphoproline derivative 25 [35].
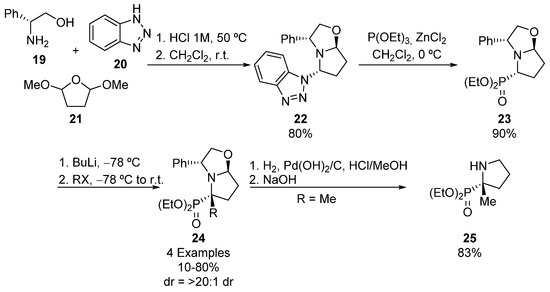
Scheme 3.
Synthesis of tetrasubstituted α-aminophosphonates 24 and 25.
According to the authors, the high diastereoselectivity observed for this transformation is related to a proposed transition state TS1 (Figure 5). Thus, the lithium ion is coordinated with the phosphonate oxygen and the tertiary nitrogen atoms, forming a five-membered ring pseudocycle, where the σ* P=O acceptor orbital lies in parallel to the lone pair of the anion, which is therefore stabilized by hyperconjugation. Under this conformation, the functionalization with the alkyl group occurs with retention of configuration.
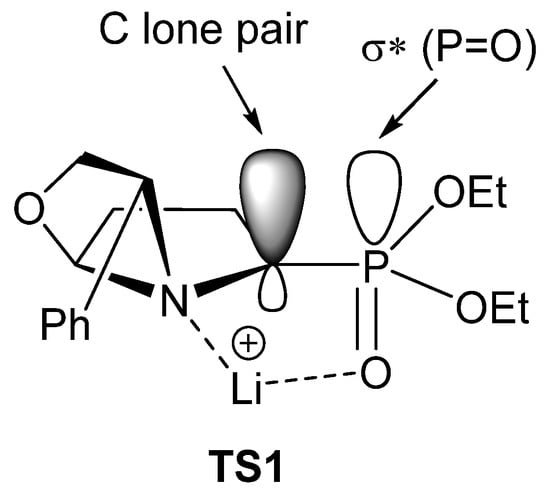
Figure 5.
Proposed transition state for the preparation of α-aminophosphonates 24 (* denotes antibonding molecular orbital).
Another possibility for the asymmetric formation of tetrasubstituted α-aminophosphonates implying C-C bond formation relies on the addition of carbon nucleophiles to chiral ketimines. In this context, our research group described in 2013 a methodology for the preparation of tetrasubstituted α-aminophosphonates through the addition of carbon nucleophiles to α-phosphorated ketimines 29 (Scheme 4) [36]. First, Pudovic reaction of TADDOL-derived chiral phosphite 26 with imine 27 affords trisubstituted α-aminophosphonate 28 as a mixture of diastereoisomers (93%, 1:1 dr). α-Aminophosphonate 28 is then transformed into the enantiopure chiral ketimine 29 through a formal oxidation, consisting of an initial chlorination, followed by a β-elimination of hydrogen chloride using a polymeric base. Then, organometallic species react fast with α-iminophosphonate 29, delivering tetrasubstituted α-aminophosphonates 30 in very good yields. Better results in terms of diastereoselectivity are obtained in this reaction when aliphatic Grignard reagents (R = Me, 80%, 94:6 dr) are used if compared to the aromatic substrate R = 2-naphthyl, 81%, 55:45 dr). Additionally, the hydrolysis of the tosyl and chiral auxiliary group affords tetrasubstituted α-aminophosphonic acid (S)-31.
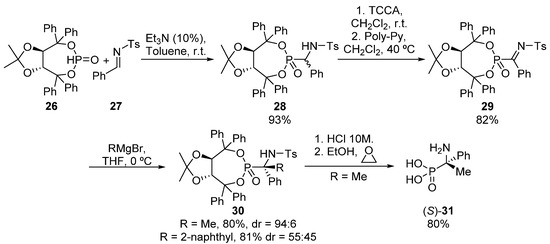
Scheme 4.
Synthesis of tetrasubstituted α-aminophosphonates from ketimines 29.
In the proposed transition state, depicted in Figure 6, the phosphorus-containing seven-membered ring adopts a more stable boat conformation, which is fixed by the trans configuration of the five-membered fused ring. Under this conformation, the two heteroatoms must embrace the more stable equatorial orientation, forcing the two hydrogen atoms to the axial positions. According to this model, the nucleophilic attack to the Re-face is substantially favored, due to the presence of the axial phenyl groups blocking the Si face.
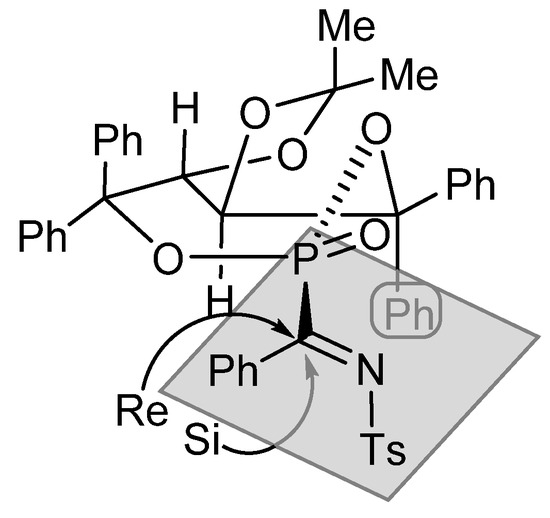
Figure 6.
Proposed model of addition of carbon nucleophiles to ketimines 29.
Menthol-derived phosphonate imines 32 are another kind of useful chiral electrophiles used in the diastereoselective preparation of tetrasubstituted α-aminophosphonates (Scheme 5). For example, the use of proline (I) as a chiral catalyst in the addition of acetone (33) gives α-aminophosphonate 34 in good yield and diastereoselectivity (86%, 1:30 dr). The authors also describe the addition of other nucleophiles to ketimines 32, such as pyrroles 35, indole (37), and nitromethane, to yield α-aminophosphonate derivatives 36, 38 and 39, although in these cases, they observe lower diastereoselectivities [37].
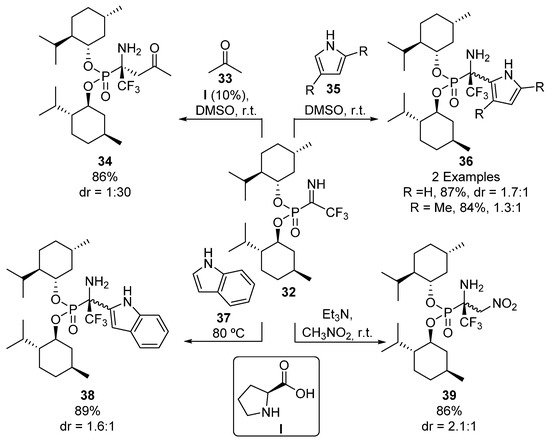
Scheme 5.
Synthesis of tetrasubstituted α-aminophosphonates from menthol-derived phosphonate imines 32.
The same research group used also chiral N-methylbenzylimines 41 in cycloaddition reactions (Scheme 6) [38] for the asymmetric preparation of tetrasubstituted α-aminophosphonates 43. In his case, the synthesis of chiral imine 41 comprises an initial treatment of (S)-1-phenylethylamine ((S)-40) with trifluoroacetic acid and triphenylphosphine, in the presence of trimethylamine in order to form haloimine 41. Then, the Arbuzov reaction with triethyl phosphite, yields the corresponding fluorinated α-ketiminophosphonate 42. Upon treatment with diazomethane, ketimine 42 undergoes a cycloaddition reaction that leads the formation of triazoline-derived α-aminophosphonate 43 in good yield but with moderate diastereoselectivity (83%, 2.5:1 dr). Although the authors did not assign the configuration of the major isomer, both can be separated through chromatography.
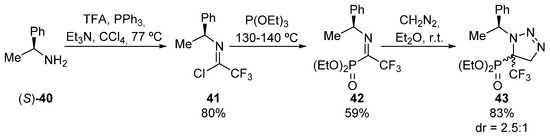
Scheme 6.
Synthesis of triazoline-derived α-aminophosphonate 43.
2.2. C-P Bond Formation
Another common strategy for the preparation of tetrasubstituted α-iminophosphonates comprises the addition of phosphorus nucleophiles to ketimines. For example, Davis described in 2001 the use of p-toluensulfinyl imines 44 as starting materials for the diastereoselective preparation of tetrasubstituted α-aminophosphonates 45 (Scheme 7) [39]. In this report, chiral ketimines 44 are treated with lithium diethyl phosphite at low temperature to obtain α-aminophosphonates 45 in excellent yields and diastereoselectivities (71–97%, 82:18–99:1 dr). In the last step, enantiomerically pure α-aminophosphonic acids 46 can be isolated by simple hydrolysis using hydrochloric acid.
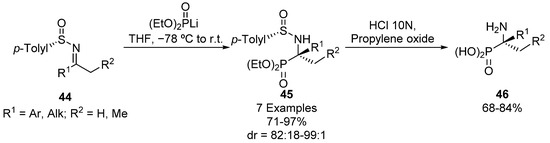
Scheme 7.
Diastereoselective preparation of tetrasubstituted α-aminophosphonates 45.
The high degree of diastereoselectivity in this reaction is explained by the authors as a transition state where there is a chelation of the lithium cation to both, the sulfinyl and phosphite oxygen atoms, in a seven-membered twisted-chair transition state. As shown in Figure 7, TS3 is favored because, under this conformation, the bulky aryl group adopts an energetically favored equatorial position if compared to TS4, where the aromatic substituent group must assume an energetically less favorable axial position.
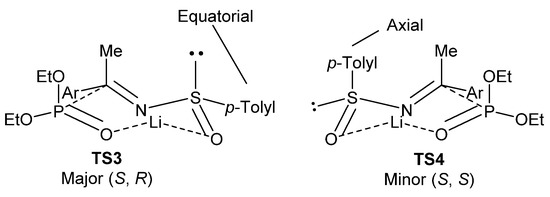
Figure 7.
Proposed transition states for the formation of tetrasubstituted α-aminophosphonates 45.
Following a similar methodology, the same research group describes also the preparation of tetrasubstituted phosphoproline derivative 50 (Scheme 8) [40]. In this case, the treatment of oxo-sulfinimine 47 with lithium diethyl phosphite gives α-aminophosphonate 48, which after treatment with hydrochloric acid, forms cyclic tetrasubstituted α-aminophosphonate 49 in good yield. Then, a syn addition of molecular hydrogen from the less hindered face yields phosphoproline derivative 50 with 50% enantiomeric excess.
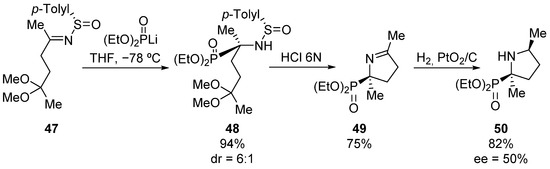
Scheme 8.
Preparation of cyclic tetrasubstituted α-aminophosphonate 50.
Based on the same principle, Yuan’s group used tert-butylsulfinyl imines 51 as chiral auxiliaries for the synthesis of tetrasubstituted α-aminophosphonates 52, via nucleophilic addition of phosphites (Scheme 9) [41,42]. Substrates 52 are obtained with high yields and diastereoselectivities in all cases (70–85%, 86:14 ≥ 98:2 dr), using different alkyl phosphites (R2 = Me, Et, n-Pr) and several alkyl or aromatic substituents in sulfinimine 51. In an identical way as described by Davis, substrates 52 can be transformed into α-aminophosphonic acids 53 by a simple hydrolysis. For this transformation, the authors propose the plausible transition state TS5, where the potassium cation is chelated to the sulfinyl and phosphonate oxygens, and the nucleophilic attack of the phosphite nucleophile occurs from the less hindered face, opposite to the tert-butyl group.
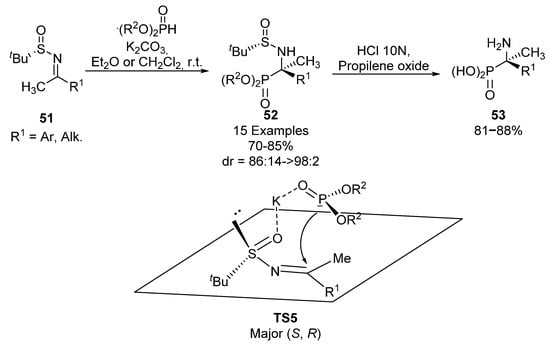
Scheme 9.
Sulfinimine-mediated synthesis of α-aminophosphonates 52.
A few years later, Ellman’s research group described a modification of this reaction, using potassium bis(trimethylsilyl)amide (KHMDS), which favors the solubility obtaining in this way α-aminophosphonates 52 with better reaction conversions and in excellent yields but lower diastereoselectivities (88–95%, 12:1–99:1 dr) [43].
In addition, Yuan’s group used this strategy, employing chloro-substituted sulfinyl imines 54 for the preparation of three-, four- and five-membered cyclic tetrasubstituted α-aminophosphonates 56 (Scheme 10) [42]. The reaction proceeds efficiently using different imines 54 (R = Me, Ph; n = 1, 2, 3) and heterocyclic substrates 56 can be obtained through intermediate 55 in good yields and diastereoselectivities (75–83%, 71:29–92:8 dr). In this transformation, a strong dependence of the diasteroselectivity on the size of the cycle is observed, obtaining an excellent dr (92:8) for a three-membered cyclic substrate, while a drop into the diastereoselectivity is observed for the four-membered derivative (89:11 dr) and even lower dr values are obtained for five-membered heterocycles (71:29 dr).
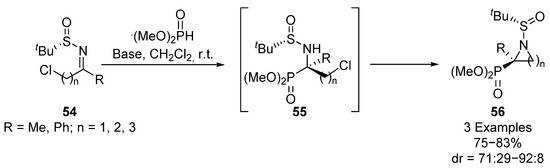
Scheme 10.
Sulfinimine-mediated synthesis of cyclic α-aminophosphonates 56.
Likewise, in 2014, Liu and colleagues extended this strategy to the nucleophilic addition of diphenyl phosphite to fluorine-substituted α,β-unsaturated sulfinimines 57, in this case in the presence of a rubidium catalyst (Scheme 11) [44]. Allyl α-aminophosphonates 58 are obtained in good yields and diastereoselectivities (56–87%, 75:25–92:8 dr) with different fluoroalkyl substituents. In addition, the selective deprotection of the sulfinyl group in acidic media, to produce α-aminophosphonate 59 in good yield, is described.
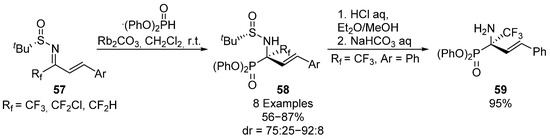
Scheme 11.
Synthesis of fluorine substituted α-aminophosphonates 58 derived from α,β-unsaturated sulfinyl imines 57.
In 2017, Cramer reported an additional example of an asymmetric addition of phosphorus nucleophiles to imines for the preparation of tetrasubstituted α-aminophosphonates (Scheme 12) [45]. In this work, first chiral imine 61 is obtained in good yield and enantiomeric excess (90%, 97% ee) from imidoyl chloride 60 in the presence of a palladium, catalyst II and CsOAc. Then, via a boron trifluoride-mediated hydrophosphonylation reaction of imine 61, α-aminophosphonate 62 is formed in good yield as a single diastereoisomer.
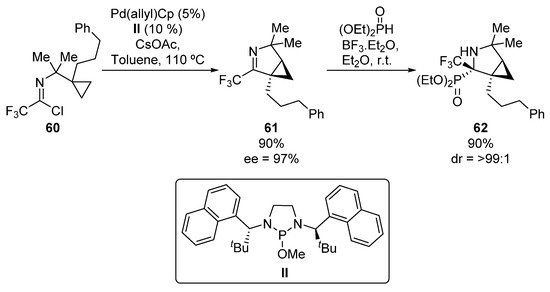
Scheme 12.
Synthesis of α-aminophosphonates 62.
A related strategy for the asymmetric induction in the preparation of tetrasubstituted α-aminophosphonates consists of the use of acetal-derived iminium salts as electrophiles. In 2000, Fadel and colleagues detailed a one-pot synthesis of cyclopropane α-aminophosphonates 65 (R1 = Me) using this methodology (Scheme 13) [46]. In this example, the sequence starts with the cyclization of bromoester 63 (R1 = Me) in the presence of sodium and TMSCl, to obtain silylated acetal 64. Then, deprotected hemiacetal intermediate 66 is formed by an alcoholysis in presence of a catalytic amount of an acid source (TMSCl or AcOH), followed by the reaction with (S)-1-phenylethylamine ((S)-40), to furnish α-amino alcohols 67. Under acidic conditions, intermediate 67 is converted into iminium species 68, which undergo a nucleophilic addition of phosphite (R2 = Me, Et) from the less hindered face of the C=N bond, to provide finally diastereoisomeric phosphonates 65 (R1 = Me) in good yields and diastereoselectivities (60–82%, 80:20–88:12 dr).
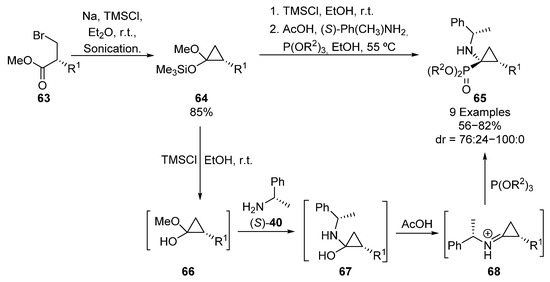
Scheme 13.
Synthesis of cyclopropane α-aminophosphonates 65.
A few years later, the authors extended the scope of this reaction to differently substituted acetals 64 (R1 = Et, Bn, iPr, tBu), obtaining cyclopropane-derived α-aminophosphonates 65 in good yields and excellent diastereoselectivities (56–78%, 76:24–100:1 dr). Remarkably, the use of a tert-butyl substituent provided a single diastereoisomer (100:1 dr) [47].
Following the same approach, Faldel’s group described later the synthesis of spirocyclic α-aminophosphonates 70 (Scheme 14) [48]. Starting also from silylated acetal 64, deprotected acetal intermediate 66 is again formed by an alcoholysis and then, iminium species 72 is obtained by reaction with (R)-phenylglycinol under acidic conditions. The subsequent nucleophilic addition of triethyl phosphite gives spirophosphonate 69, by means of an intramolecular transesterification, with good yield and diastereoselectivities (71%, 89:11 dr). However, the use of norephedrine as a chiral source results in a drop in both yield and diastereomeric ratio (36%, 78:22 dr). It must be pointed out that substrates 69 are obtained as a mixture of epimers (80:20), due to the presence of an additional chiral center at the phosphorus atom. The major diastereoisomer can be isolated and further transformed into enantiopure cyclopropane-derived aminophosphonic acid 70, by an initial hydrogenolysis reaction, followed by hydrolysis of phosphonate group.
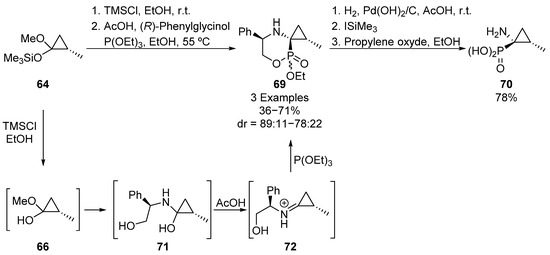
Scheme 14.
Synthesis of spirocyclic α-aminophosphonates 69.
In 2007, Fadel described also a similar process, in this case using heterocyclic iminium salts (Scheme 15) [49]. In this approach, N-Boc-protected piperidinone 73 is treated with (S)-1-phenylethylamine ((S)-40) in presence of acetic acid, followed by the nucleophilic addition of triethyl phosphite, leading to the formation of tetrasubstituted α-aminophosphonates 74 and 75 as an inseparable mixture of diastereoisomers (75%, 60:40 dr).
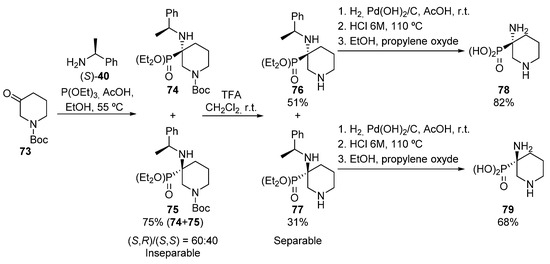
Scheme 15.
Synthesis of cyclic α-aminophosphonates 78 and 79.
Then, after the hydrolysis of the N-Boc protecting group, diastereoisomers 76 and 77 are formed, which in this case can be separated. In addition, the hydrogenolysis and hydrolysis reactions of each diastereoisomer gives enantiopure α-aminophosphonic acids 78 and 79.
Along the same line, the same research group described the preparation of bicyclic tetrasubstituted α-aminophosphonates 84 starting from ketone acetals 80 (Scheme 16) [50]. The esterification reaction of acetals 80 with (S)-phenylalanine derivative 81, which acts as chiral auxiliary, leads to intermediate 82, which is cyclized to form imine substrates 83 as a mixture of diastereoisomers. The formation of iminium cation in the presence of triethyl phosphite gives bicyclic tetrasubstituted α-aminophosphonates 84 in good yields and excellent diastereoselectivities (46–77%, 89:11–99:1 dr). These α-aminophosphonates are transformed into cyclic serine analogues 87 through their oxidation, and further hydrolysis of the intermediate imine 85/enamine 86 mixture.
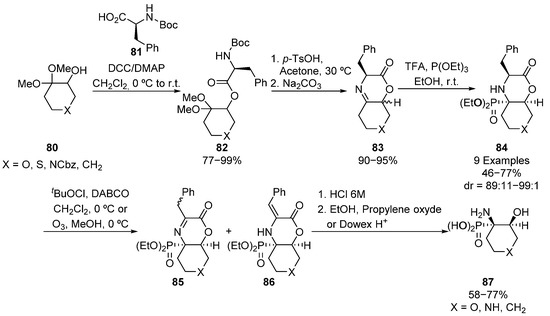
Scheme 16.
Synthesis of bicyclic tetrasubstituted α-aminophosphonates 84.
In this case, the authors justify the high level of diastereoselectivity by an equilibrium between two iminium epimers in TS6 and TS7 through the parent enamine salt (Figure 8). It is estimated that the chair–boat conformation (TS6) is favored in 2.14–5.56 kcal/mol relative to the epimeric twist boat–boat conformation (TS7), resulting in the kinetic addition of the phosphite to the less hindered Si-face of the iminium species in TS6.
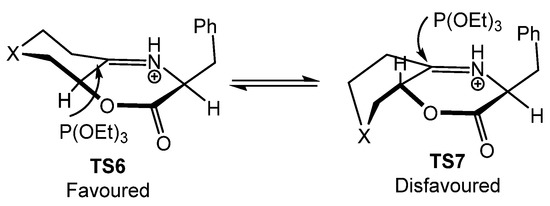
Figure 8.
Proposed transition states for the formation of α-aminophosphonates 84.
In addition, the use of bicyclic iminium salts has been reported for the asymmetric preparation of cyclic α-aminophosphonates 90 (Scheme 17). Chiral cyclic imines 89 are synthesized from diamine 88 and ketoesters and their subsequent treatment in toluene with dialkyl phosphites gives tetrasubstituted α-aminophosphonates 90 in high yields and diastereoselectivities (78–95%, 68:32–98:2 dr) [51,52]. However, if imines 89 are activated with bromotrimethylsilane, they are supposed to form an iminium ion, which is reactive towards tris(trimethylsilyl) phosphite and then, α-aminophosphonic acid derivatives 91 can be obtained in high yields and diastereoselectivities (70–99%, 85:15–98:2 dr) [51].
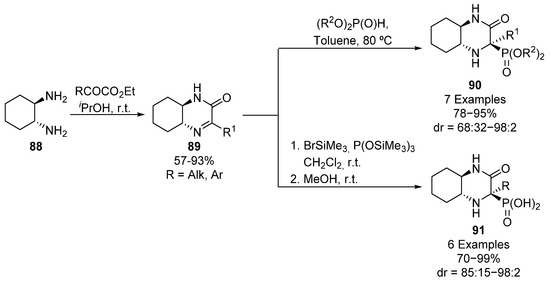
Scheme 17.
Synthesis of bicyclic α-aminophosphonates 90 and α-aminophosphonic acid derivatives 91.
Other useful strategy for the diastereoselective synthesis of tetrasubstituted α-aminophosphonates with C-P bond formation, complementary to the hydrophosphonylation of chiral imines, is the addition of chiral phosphorus nucleophiles to activated ketimines. For example, in 2011, Chen and Miao used a multicomponent Kabachnik–Fields reaction of phosphorylated chiral nucleophile 92, diethyl phosphoramidate 93 and ketone 94 to obtain α-aminophosphonate 95 in a diastereoselective fashion (Scheme 18) [53]. The authors propose that the nucleophilic addition in TS9 is expected to be less favored, compared to the addition proposed in TS8, where the chiral dioxaphospholanedicarboxylate 92, which plays a crucial role in the control of the diastereoselectivity, reacts from the sterically less hindered face.
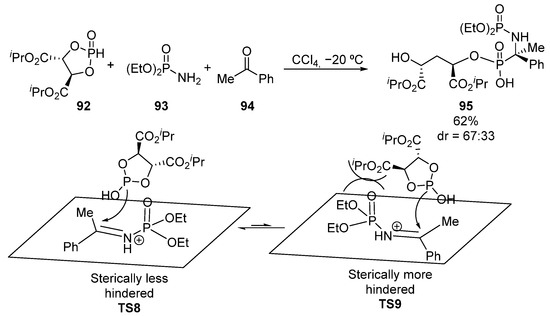
Scheme 18.
Multicomponent Kabachnik–Fields reaction for the synthesis of α-aminophosphonates 95.
A different methodology for the asymmetric formation of tetrasubstituted α-aminophosphonates that involves C-P bond formation was reported by Hammershmidt, where a phosphoramidate-α-aminophosphonate rearrangement is described, leading to the formation of diverse α-aminophosphonates 98 in moderate to good yields and excellent stereocontrol (38–80%, 96–99% ee) (Scheme 19) [54,55]. This route involves N-Boc protection of phosphoramides 96, and metalation with sec-butyllithium to form the corresponding carbanion 99. The rearrangement of the phosphorous substituent and the final quenching with acetic acid provides tetrasubstituted α-aminophosphonates 98.
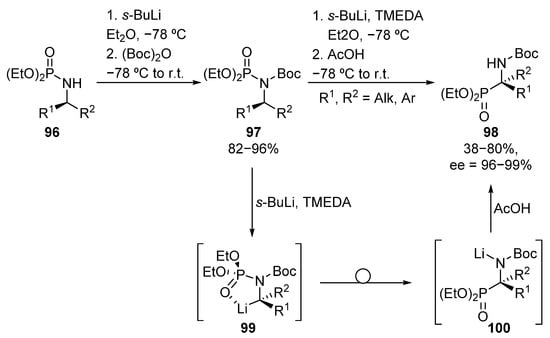
Scheme 19.
Synthesis of α-aminophosphonates 98 via phosphoramidate-α-aminophosphonate rearrangement.
2.3. C-N Bond Formation
The introduction of nitrogen reagents into the skeleton of phosphonates is also an alternative methodology that can be useful for the preparation of tetrasubstituted α-aminophosphonates. In this regard, in 1999 Davis and colleagues applied successfully the aza-Darzens reaction for this purpose (Scheme 20) [56]. Starting from chiral sulfinyl imine 101 and diethyl 1-chloroethylphosphonate (102), initially, a mixture of three isomers of α-chloro-β-amino adducts 103–105 is obtained.

Scheme 20.
Aza-Darzens reaction for the synthesis of (R)- and (S)-107.
The major isomer 103 can be isolated after chromatography and then, in the presence of sodium hydride, enantiomerically pure aziridine 106 is obtained. After the elimination of the chiral auxiliary group with TFA, followed by ring-opening via hydrogenolysis, enantiopure tetrasubstitued α-aminophosphonate (R)-107 is obtained. For the preparation of the opposite enantiomer, the side mixture of α-chloro-β-amino adducts 104 and 105 is used. After the hydrolysis of the chiral auxiliary group and the subsequent ring-opening of the corresponding aziridine intermediate, tetrasubstitued α-aminophosphonate (S)-107 is obtained.
Continuing with strategies that entail C-N bond formation, Curtius rearrangement is also a useful method for the introduction of amino groups starting from carboxylic acids, which can be used for the preparation of tetrasubstitued α-aminophosphonates. For example, in 1999, Le Corre used enantiomerically pure chiral sulfate 108 and phosphorylated malonate derivative 109 for the preparation of cyclopropane phosphonate 110 (Scheme 21) [57].
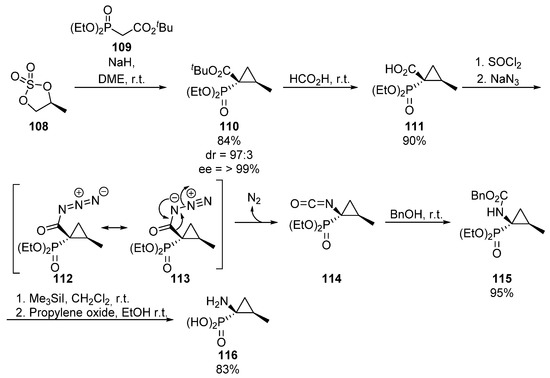
Scheme 21.
Curtius rearrangement for the synthesis of α-aminophosphonate 116.
Then, the ester group is hydrolyzed, to obtain carboxylic acid substituted phosphonate 111 which, after activation of the acid with thionyl chloride and the addition of sodium azide, leads to acyl azide species 112/113. At this point, Curtius rearrangement gives isocyanate 114, which is captured by means of the in situ addition of benzyl alcohol, to afford N-protected amino ester 115. Finally, the benzyl protecting group is hydrolyzed yielding cyclopropane-derived tetrasubstituted α-aminophosphonate 116.
Ito’s group also used Curtius rearrangement for the preparation of tetrasubstitued α-aminophosphonate 123 (Scheme 22) [58]. The rhodium-catalyzed conjugate addition of cyanophosphonate 117 to acrolein (118) leads to the formation of aldehyde 119 with high yield and enantiomeric excess (80%, 92% ee). Compound 119 is then treated with phosphonium ylide 120, and the newly formed C=C bond, prepared through the Wittig olefination, is directly hydrogenated to obtain cyanophosphoate 121. The acidic hydrolysis of the nitrile moiety in this substrate followed by an in situ esterification of the carboxylic acid intermediate with diazomethane affords phophorated methyl ester 122. Finally, the methoxycarbonyl group in 122 is selectively hydrolyzed under basic conditions, and the resulting carboxylate treated with diphenyl phosphoroazidate, which by means of a Curtius rearrangement followed by trapping with benzyl alcohol, affords tetrasubstitued α-aminophosphonate 123 (81%, 88% ee).
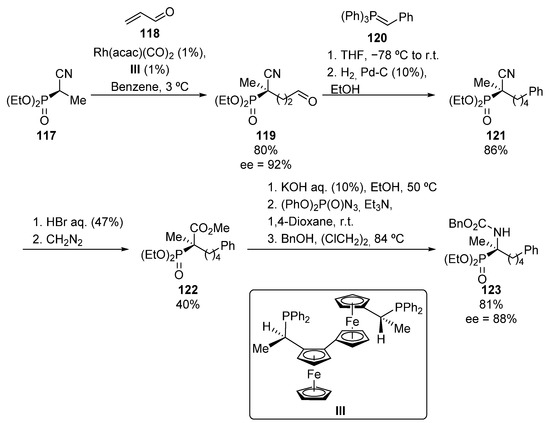
Scheme 22.
Curtius rearrangement applied to the synthesis of α-aminophosphonate 123.
Following a similar approach, a few years later, Krawczyk and colleagues reported an analogous reaction, in which the synthetic route starts with the reaction between cyclic sulfate 124 and ethyl diethoxyphosphorylacetate 125, to afford phosphorated ester 126 as a single diastereoisomer (Scheme 23) [59]. In order to perform the Curtius rearrangement, first the ester group needs to be hydrolyzed to form carboxylic acid 127, and then the addition of diphenylphosphoryl azide (DPPA) affords isocyanate 128, which is immediately captured as carbamate 129 by the in situ addition of ethanol. In addition, the benzyl and carbamateprotecting groups can be eliminated via hydrogenolysis and hydrolysis, respectively, affording enantiopure α-aminophosphonic acid derivative (1R,2S)-130 in excellent yield.
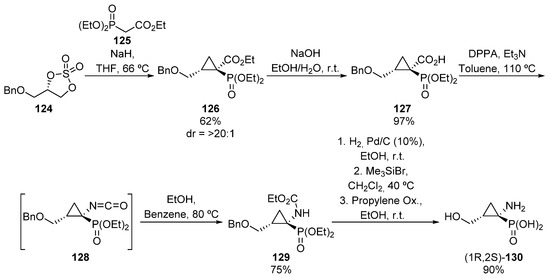
Scheme 23.
Synthesis of α-aminophosphonic acid derivative (1R,2S)-130.
For the preparation of the opposite enantiomer, (1S,2R)-130, the authors used a complementary strategy as depicted in Scheme 24. In this case, the synthesis of racemic lactone 132 is performed by treatment of epibromohydrin 131 with malonate derivative 125 in the presence of sodium hydride. Then, the reaction of lactone 132 with (R)-1-phenylethylamine 40 gives products (1S,2R,1′R)-133 and (1R,2S,1′R)-133 as a mixture of diastereoisomers, which can be separated by column chromatography. Once pure (1S,2R,1′R)-133 is isolated, the hydrolysis of the amine in presence of sulfuric acid yields enantiomerically pure lactone (1S,5R)-134, which is then treated with saturated methanolic ammonia followed by an acylation reaction to obtain amide 135. The lead tetraacetate-mediated Hoffmann rearrangement in tert-butyl alcohol gives carbamate 136, which in presence of potassium carbonate yields N-Boc aminocyclopropane phosphonate 137. Finally, the sequential treatment of 137 with TFA and bromotrimethylsilane affords α-aminophosphonic acid derivative (1S,2R)-130 in excellent yield [59].
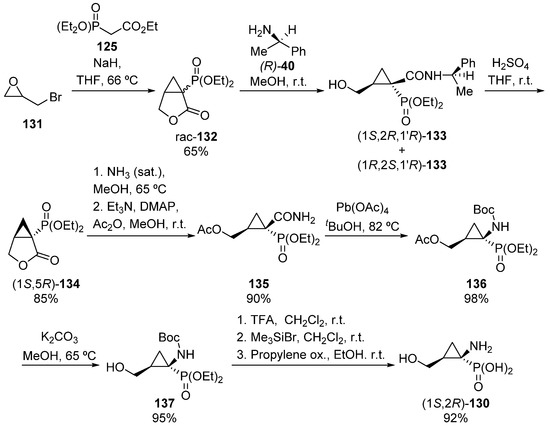
Scheme 24.
Synthesis of α-aminophosphonic acid derivative (1S,2R)-130.
3. Enantioselective Synthesis of Tetrasubstituted α-Aminophosphonic Acid Derivatives
3.1. C-C Bond Formation
The first example of an enantioselective synthesis of tetrasubstituted chiral α-amino phosphonates was reported in 1999 by Ito’s group (Scheme 25) [60]. The reaction consists of an asymmetric palladium–IV-catalyzed allylation of racemic β-keto-α-aminophosphonates 138 that allows the obtaining of optically active α-amino phosphonates 140 in moderate to good yields and enantiocontrol (27–87%, 46–88% ee). In addition, the authors also report the subsequent diastereoselective reduction of the ketone moiety, affording β-hydroxy-α-amino phosphonates 141. Remarkably, when the reaction is carried out in methanol at low temperature, using sodium or tetra n-butylammonium borohydrides as reducing reagents, the 141-syn isomer is obtained (74–89%, 74:26 to 82:18 syn/anti ratio). In contrast, the reaction in tert-butyl alcohol at 50 °C, using sodium borohydride, affords the opposite isomer (78%, 15:85 syn/anti ratio).
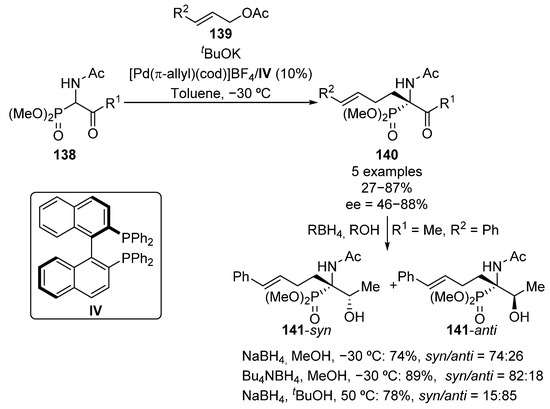
Scheme 25.
First enantioselective synthesis of tetrasubstituted α-aminophosphonates.
Due to their strong nucleophilic character, the use of nitroalkane enolates has been widely extended in organic chemistry in the functionalization of aldehydes or imines since the Henry reaction was reported in 1895. Several examples regarding the enantioselective nucleophilic addition of α-nitrophosphonates to different electrophile reagents for the synthesis of α-aminophosphonates have been reported in recent years. In this context, the first example of this reaction was reported in 2008 by Johnston and colleagues (Scheme 26) [61]. The reaction consists of a Brönsted acid V-catalyzed addition of trisubstituted nitrophosphonates 142 to N-Boc aldimines 143, which leads to chiral phosphonates 144 in yields ranging from 48% to 86% and with high stereocontrol (up to 94:6 dr, up to 99% ee).
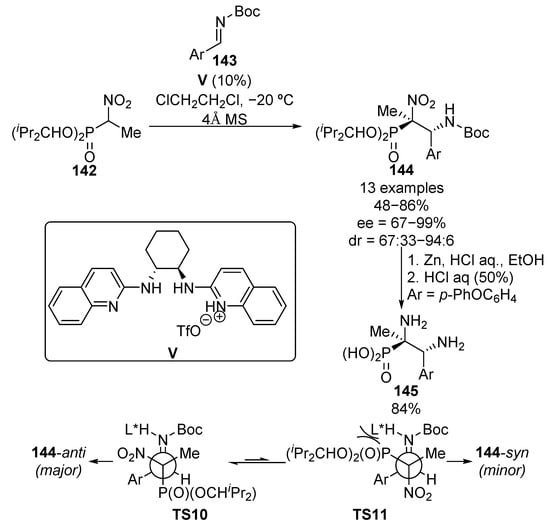
Scheme 26.
Stereoselective addition of α-nitrophosphonates to imines.
Next, the reduction and hydrolysis of α-nitrophosphonate 144 (Ar = p-PhOC6H4) under acidic conditions results in the formation of enantioenriched α,β-diaminophosphonate 145 in 84% yield with retention of the absolute configuration. According to the authors, the chiral Brönsted acid catalyst activates both the imine and nitro phosphonate substrates through hydrogen bonding. Since both nitro and phosphoryl groups may be activated by acid catalysts, a bulky phosphonate is selected in order to minimize the activation of the phosphoryl group enhancing the steric repulsion. The nitro group is, in this way, located close to the catalyst and the 144-anti product is favored through transition state TS10, while the high bulkiness of the phosphonate moiety prevents the formation of TS11 and 144-syn isomer is obtained as the minor product.
As a continuation of this work, Namboothiri and colleagues reported in 2012 the conjugate addition of α-nitrophosphonates 146 to α,β-unsaturated ketones 147 uing in this case cinchona alkaloid-derived thiourea VI as a bifunctional catalyst (Scheme 27) [62]. For this transformation, a tentative model of addition is proposed by the authors, where both acidic protons of the thiourea moiety activate ketones by means of hydrogen bonding, while the basic nitrogen of the quinuclidine unit stabilizes the nitrophosphonate anion (TS12-13). Although the reaction affords optically active α-nitrophosphonates 148 in high yields (70–97%), the enantioselectivity of the reaction is found to be strongly dependent on the substituent of the ketone. Thus, when aromatic groups bearing electron-donating substituents are used, enantioselectivities ranging from 70% to 94% are obtained. In contrast, the use of some electron poor aromatic groups, such as 4-nitrophenyl, heteroaryl substituents such as 2-furyl or 2-thienyl, and aliphatic cyclohexyl substituents results in a drastic decrease into the enantioselectivity (35–44%).
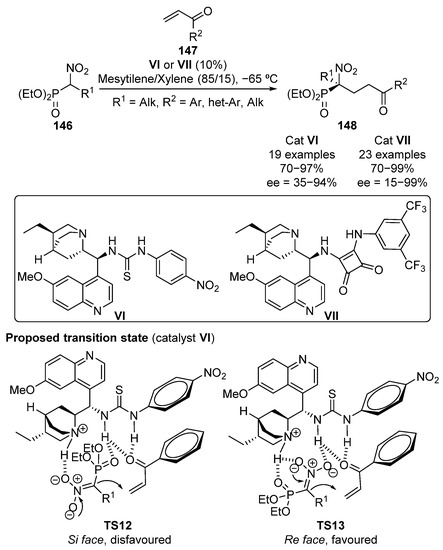
Scheme 27.
Enantioselective conjugate addition of α-nitrophosphonates to enones.
Following their interest in this reaction, a few years later, the same authors proposed that the use of stronger acidic squaramide catalyst VII in the reaction improves the poor enantioselectivity when electron-withdrawing groups are used [63] (Scheme 27). A relevant improvement not only on the stereocontrol but also on the reactivity was reported. Thus, yields above 90% and enantioselectivities ranging from 85% to 99% for all the aromatic and heteroaromatic ketones except for 2-substituted aryl groups such as 2-Cl-C6H4 (97%, 74% ee) and 1-naphthyl (98%, 15% ee) are obtained. In contrast, cyclohexyl substituted enone only affords moderate yield and enantioselectivity (70%, 51% ee).
In addition, the authors reported several useful transformations of the obtained optically active α-nitrophosphonates into α-aminophosphonates (Scheme 28). For instance, the reduction of the nitro group in the presence of zinc and ammonium chloride results in the unprotected amino group that spontaneously leads to the formation of cyclic imine 149 in 86% yield.
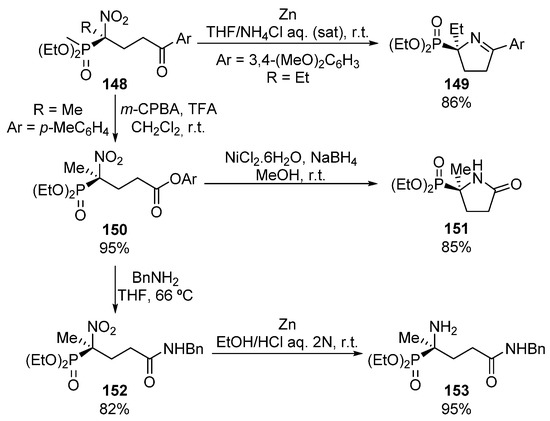
Scheme 28.
Synthetic applications of α-nitrophosphonates 148.
On the other hand, the Baeyer–Villiger oxidation allows obtaining of the corresponding ester 150 in almost quantitative yield. This ester 150 can be subsequently used for the synthesis of cyclic lactam 151 after reduction of the nitro group and subsequent intramolecular lactamization reaction (Scheme 28). Moreover, the reaction of ester 150 with a primary amine to yield the acyclic amide 152, followed by the Clemmensen reduction of the nitro group affords the acyclic α-aminophosphonate derivative 153.
Following the same line, in 2013, Jászay and colleagues reported the addition of α-nitrophosphonates 154 to aryl acrylates 155 using also an squaramide organocatalyst VIII (Scheme 29) [64]. Even though some bulky phosphonates were tested in the reaction with phenyl acrylate, the use of iso-propyl and butyl phosphonates does not result in a further improvement on the reaction yield or enantiocontrol (82–85%, 52–64% ee) and the best enantiomeric excesses are obtained with ethyl phosphonates (93%, 76% ee).
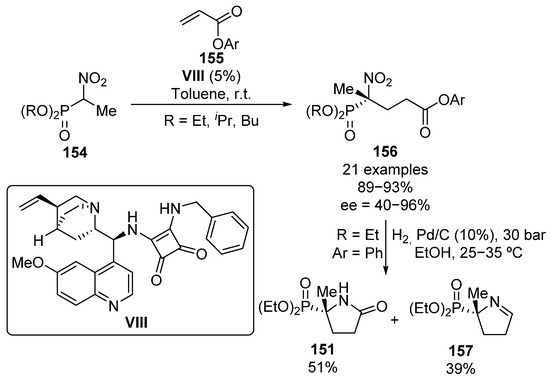
Scheme 29.
Enantioselective addition of α-nitrophosphonates to conjugated esters.
Concerning aryl acrylate substrates, the best enantioselectivities are obtained for electron-donating aryl groups (e.g., 2,6-(OMe)2C6H3, 92%, 96% ee). In contrast, although no relevant effect is observed on the reaction yield, the use of strongly electron-withdrawing aromatic rings such as 2-NO2C6H4 results in a lower stereocontrol (90%, 40% ee). Besides, the reduction of the nitro group results in a mixture of phosphorus substituted γ-lactam 151 and cyclic α-iminophosphonate 157 in variable ratios depending on the reaction pressure and the aryl groups present on the ester moiety.
Other Michael acceptors different than conjugated carbonyl compounds have been used in enantioselective reactions with α-nitrophosphonates. Specifically, in 2012 Mukherjee’s group reported the use of thiourea–alkaloid bifunctional catalyst IX for the addition of α-nitrophosphonates 146 to conjugated nitroalkenes 158 to provide tetrasubstituted α-nitrophosphonates 159, in yields ranging from 64% to 82% when using both, aryl and alkyl groups on the nitroalkene (Scheme 30) [65]. Curiously, when 2-naphthyl nitroalkene is used, the corresponding α-nitrophosphonate 159 is formed in only 38% yield. Nevertheless, diastereomeric ratios ranging from 83:17 to 95:5 and enantiomeric excesses up to >99% are obtained in all cases. The concomitant reduction of both nitro groups results in an intramolecular cyclization reaction, which leads to chiral pirazolidine 160 in 60% yield with no loss of the optical purity.
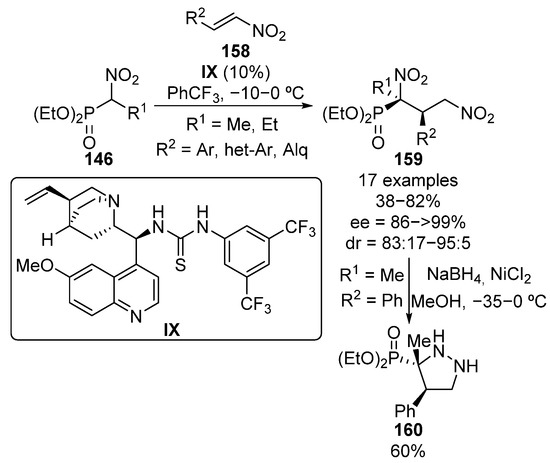
Scheme 30.
Enantioselective addition of α-nitrophosphonates to nitroalkenes.
In 2013, the use of vinyl sulfones as Michael acceptors in the addition of α-nitrophosphonates 146 was simultaneously reported by Namboothiri and Lu (Scheme 31) [66,67].
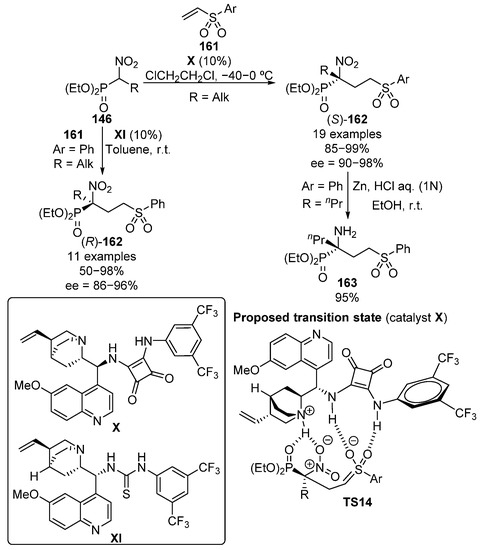
Scheme 31.
Enantioselective addition of α-nitrophosphonates to vinyl sulfones.
Namboothiri proposes an enhancement of the electrophilicity of vinyl sulfone substrates through the establishment of two hydrogen bonds between the squaramide catalyst X acidic protons and the sulfone oxygen atoms, while the basic nitrogen of the alkaloid moiety activates the α-nitrophosphonate as a nucleophile (Scheme 31, TS14). The reaction products are obtained in this case in excellent yield and enantiocontrol when aryl sulfones 161 are used (85–99%, 90–98% ee). In contrast, the use of tetrazole-derived sulfone results in a decrease in the enantiomeric excesses (74–79% ee). Moreover, the reduction of the nitro group affords α-aminophosphonate 163 in almost quantitative yield (95%). Slightly lower yields but similar enantiomeric excesses were obtained by Lu and colleagues by using thiourea catalyst XI (50–98%, 86–95% ee), obtaining, in this case, the opposite enantiomer (S)-162.
Analogously to α-nitrophosphonates, α-isothiocyanatophosphonates 164 possess a significantly acidic proton in the alpha position, which makes them suitable to be used as nucleophiles. In this regard, Yuan and colleagues reported in 2013 the enantioselective addition of α-isothiocyanatophosphonates 164 to aldehydes 165 catalyzed by bifunctional thiourea catalyst IX (Scheme 32) [68]. In this case, the initial nucleophilic addition to aldehydes 165 proceeds in a similar manner as in the case of α-nitrophosphonates, through the activation of aldehyde electrophile165 by the thiourea acidic protons and a simultaneous deprotonation of α-isothiocyanatophosphonates 164 by the quinuclidine basic unit of the catalyst (TS15). Due to the electrophilic character of the central carbon in isothiocyanates, a subsequent intramolecular addition of the alcohol occurs (TS16), leading to the formation of cyclic α-aminophosphonates 166 in moderate to good yield and diastereocontrol (36–93%, 84:16 to >99:1 dr). However, only moderate enantiomeric excesses are obtained (68–81% ee) if aromatic aldehydes are used. Besides, the use of acetaldehyde as electrophile results in a dramatic drop into the enantioselectivity (66%, 85:15 dr, 55% ee).
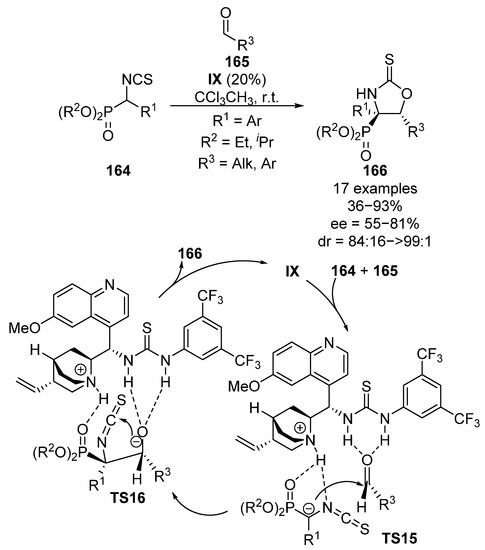
Scheme 32.
Enantioselective nucleophilic addition of α-isocyanatophosphonates to aldehydes.
One year later, Wang and colleagues reported an improvement in Yuan’s work using in this case squaramide catalyst XII (Scheme 33) [69]. In this reaction, they obtain cyclic α-aminophosphonates 169 in excellent yield and stereocontrol using aldehydes 165 bearing not only electron-donating and electron-withdrawing groups, but also heteroaryl (2-furyl, 2-thienyl) aldehydes or conjugated cinnamaldehyde (86–99%, 94:6 to >95:5 dr, 87 ≥ 99% ee). Moreover, they also extended this methodology to N-tosyl aldimines 168, cyclic thioureas 170 with similar results (80–99%, 86:14 to 92:8 dr, 92 ≥ 99% ee).
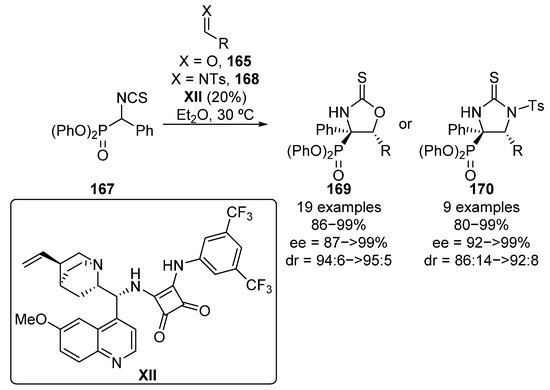
Scheme 33.
Enantioselective nucleophilic addition of α-isocyanatophosphonates to aldehydes and imines.
More recently, Albrecht and colleagues described the synthesis of tetrasubstituted spirocyclic chiral α-aminoesters and α-aminophosphonates 173 through a conjugate addition of α-isocyanates 171 to conjugated barbiturates 172 in the presence of squaramide catalyst X (Scheme 34) [70]. Although the scope is limited, spirocyclic α-aminophosphonates 173 are obtained in high yields and stereocontrol (60–99%, 88:12 to >95:5 dr, 92–98% ee).
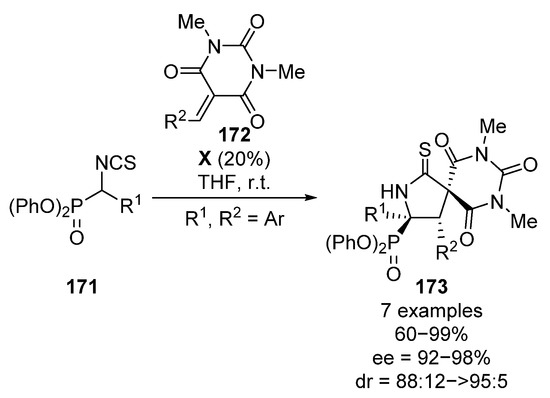
Scheme 34.
Enantioselective synthesis of spirocyclic α-aminophosphonates.
So far, the reactions described in this chapter, giving an account of reactions where the key step implies the formation of a C-C bond, entail the addition of an α-aminophosphonate equivalent onto an electrophile (Figure 4a; vide supra). A complementary general method involving the generation of C-C bonds that also leads to tetrasubstituted α-aminophosphonates consists of the addition of carbon nucleophiles to α-iminophosphonates (Figure 4b; vide supra) [71].
The synthesis of activated α-ketiminophosphonate substrates is known to be a challenging task, mainly due to the low reactivity found in the typical amine-carbonyl condensation reactions, where deactivated amide substrates are required, and the intrinsic tendency of α-ketophosphonates to eliminate the phosphonate substituent, which leads to acylation reactions [72]. Moreover, the high moisture sensitivity of such substrates entails additional obstacles for the purification of the imines which very often have to be prepared in situ. For this reason, it was not until 2012 when our research group reported an efficient synthesis of α-ketiminophosphonates 174 and the enantioselective addition of nucleophiles to such substrates (Scheme 35) [73]. In this reaction, the cinchonidine (XIII)-catalyzed nucleophilic addition of cyanide to α-phosphorated ketimines 174 provides optically active α-cyano α-aminophosphonates 176 in high yield (75–80%) and enantioselectivities ranging from 73% to 92%. The presence of bulky isopropyl groups was found to be crucial in order to obtain high enantiocontrol if compared with other alkyl and aryl phosphonates. The fact that in alcoholic solvents, the reaction proceeds fast but with no enantiocontrol might indicate a crucial role for the hydroxyl group of cinchona alkaloid in the transition state TS17, which may activate the substrate via hydrogen bonding with the iminic nitrogen. In addition, optically active α-aminophosphonic acid 177 was obtained in 80% yield without racemization by the hydrolysis reaction of the cyano group under strong acidic conditions.
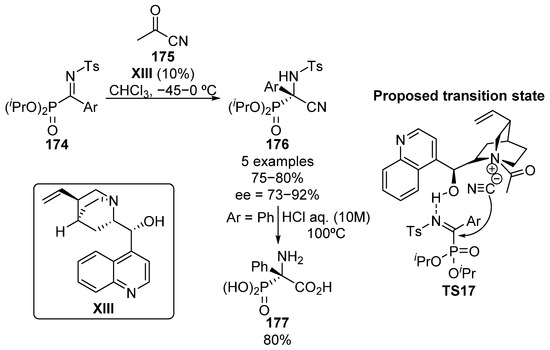
Scheme 35.
First synthesis of tetrasubstituted α-aminophosphonic acids through a nucleophilic addition to α-iminophosphonates.
As a continuation of our research on the enantioselective nucleophilic addition reactions to α-ketiminophosphonates, a few years later, we reported the asymmetric aza-Henry reaction with ketimines 178 (Scheme 36) [74,75,76], using bifunctional thiourea-alkaloid a catalyst XIV. The reaction allows the use of electron-donating and electron-withdrawing aromatic groups at the imine substrates with no relevant differences in the yield or the enantioselectivity of the obtained α-amino-β-nitrophosphonates 180 (82–87%, 80–84% ee). In addition, the reduction of the nitro group is also reported, leading to α,β-diaminophosphonate 181 in almost quantitative yield.
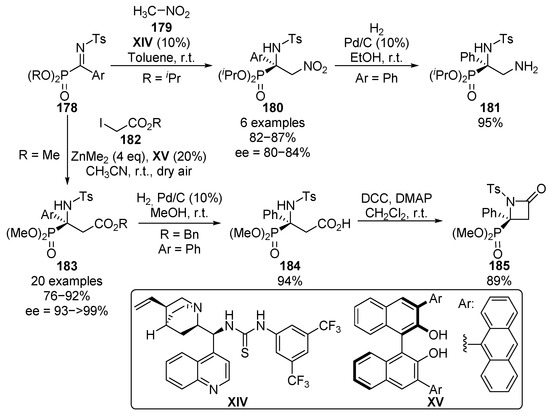
Scheme 36.
Further catalytic asymmetric additions to α-iminophosphonates.
In the same context, more recently, we described the first example of an enantioselective aza-Reformatsky reaction with non-cyclic ketimines 178, using dialkyl zinc reagents and BINOL-derived chiral ligand XV. The presence of molecular oxygen is crucial in this case in order to obtain a high yield, since other byproducts are observed when an inert atmosphere is used. The reaction can be successfully generalized to several aryl and heteroaryl ketimines 178 and alkyl iodoacetates 182, affording tetrasubstituted α-aminophosphonates 183 in excellent yield and enantiocontrol (76–92%, 93–>99% ee). Furthermore, the synthesis of β-lactam 185 containing a tetrasubstituted α-aminophosphonate is also described by a selective deprotection of the ester group and subsequent lactamization reaction.
Although enantioselective nucleophilic additions to α-alkyl iminophosphonates remains almost unexplored, in 2014, a particular case using α-trifluoromethyl α-iminophosphonates was reported by Onys’ko and colleagues (Scheme 37) [77]. In particular, proline (I)-catalyzed nucleophilic addition of acetone (33) to N-unprotected α-iminophosphonate 186 yields tetrasubstituted α-iminophosphonate 187 in high yield and enantiocontrol (80%, 90% ee). Moreover, some further transformations of α-aminophosphonate 187 are reported by the authors. For instance, the reaction of substrate 187 with 2,5-dimethoxyfuran in acid media leads to N-heterocyclic derivative 189 in 84% yield through an aldol reaction of the in situ generated pyrrole 188. On the other hand, the reaction with aryl isocyanates leads to pyrimidine 191 via urea intermediate 190. As in the previous case, an intramolecular condensation involving the ketone moiety affords substrate 191 in 89% yield without racemization.
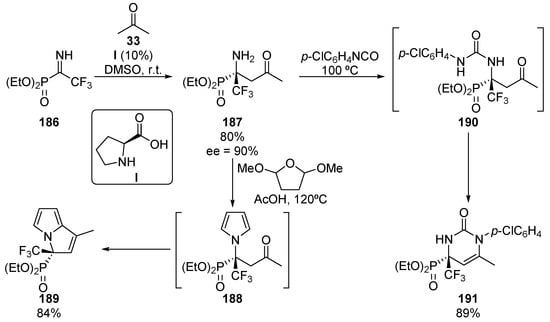
Scheme 37.
Enantioselective preparation of α-aminophosphonate 187 and its synthetic applications.
Following a similar approach, Ohshima and colleagues reported some examples of a rhodium complex XVI-catalyzed enantioselective alkynylation of α-CF3 α-iminophosphonates 192 (Scheme 38) [78]. Once the alkyne 193 is inserted into the catalyst by displacement of TMS substituted alkyne (195), an enantioselective alkynylation of imine 192 takes place leading to amide–rhodium complex 196, where a new chiral center is formed. Then, a new insertion occurs by the introduction of a second unit of alkyne 193, leading to the formation of rhodium complex 197. Finally, amide deprotonation of the terminal alkyne ends with the formation of α-alkynyl α-iminophosphonates 194 and the consequent regeneration of the active catalyst 195. The use of aryl and cyclopropyl provides α-aminophosphonates 194 in excellent yields and enantioselectivity (86–99%, 80–93% ee).
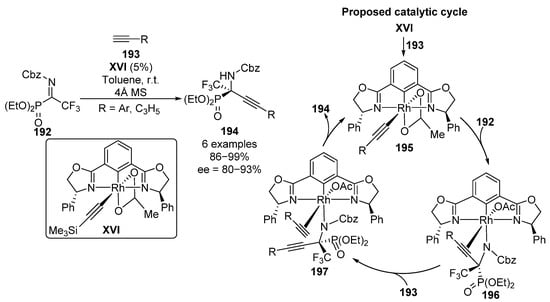
Scheme 38.
Rhodium-catalyzed enantioselective synthesis of α-alkynyl α-aminophosphonates 194.
Following a similar approach, in 2012, Che’s group reported the use of chiral rhodium catalysts XVII and XVIII on the enantioselective multicomponent reaction of diazophosphonates 198, anilines 199 and aromatic aldehydes 165 (Scheme 39) [79]. Here, in the first reaction step, the rhodium catalyst reacts with diazo compound 198 to form rhodium carbene species 201 with the release of nitrogen gas. The subsequent insertion of aniline moiety leads to an ionic intermediate 202, which easily evolves to zwiterionic species 203. At this point, the rhodium-phosphonate undergoes an addition reaction to the corresponding aldehyde substrate 165, leading to optically active tetrasubstituted α-amino-β-hydroxy phosphonates 200 while the catalyst unit is released for a new catalytic cycle. In this reaction, tetrasubstituted α-aminophosphonates 200 are obtained in moderate to excellent yield and stereocontrol (56–86%, 76:24 to 94:6 dr, 60–98% ee).
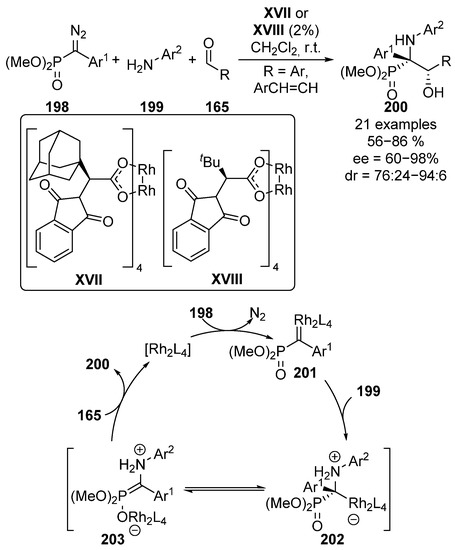
Scheme 39.
Rhodium-catalyzed enantioselective multicomponent reaction.
During the last lustrum, a new family of cyclic α-ketiminophosphonates 204 has been used as electrophilic sources in enantioselective nucleophilic addition reactions. In particular, the palladium–XIX-catalyzed enantioselective arylation reaction of α-iminophosphonates 204 was reported in 2016 by Zhou’s group (Scheme 40) [80].
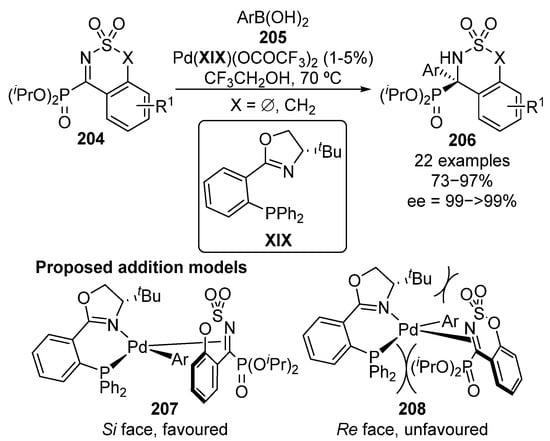
Scheme 40.
Palladium-catalyzed enantioselective arylation of cyclic imines 204.
The reaction can be generalized to several α-iminophosphonate substrates 204 and boronic acids 205, bearing electron-donating and electron-withdrawing aryl groups. In consistence with other reported examples, the use of bulky iso-propyl phosphonates results in higher enantiocontrol, providing cyclic α-aminophosphonates 206 in excellent yields (73–97%) and enantioselectivities above 99%. The high steric bulkiness of the phosphonate moiety induces the coordination of the catalyst to the imine group through Si-face (207) since the coordination through Re-face (208) implies steric repulsions not only between phosphine and phosphonate moieties but also between tert-butyl group at the oxazoline ring and sulfonyl protecting group at the imine group.
One year later, the enantioselective Friedel–Crafts reaction of indoles 210 to five-membered imines 209 was also reported (Scheme 41) [81]. In this case, phosphoric acid XX was selected as the optimal catalyst, affording optically active α-aminophosphonate functionalized indoles 211. The reaction can be successfully generalized to several indole substrates bearing electron-donating and electron-withdrawing groups with excellent yields and enantiocontrol (85–98%, 87–98% ee). However, the use of 2-methylindole results in a drastic drop in the enantioselectivity (91%, 59% ee). In addition, the addition of simple pyrrole to imines 209, leads to the formation of the analogous α-aminophosphonate substituted pyrroles in 98% yield with 84% enantiomeric excess.
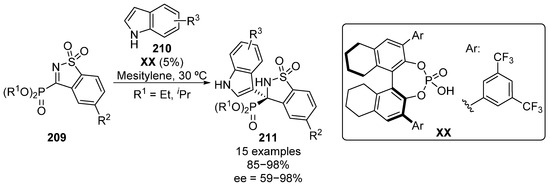
Scheme 41.
Enantioselective addition of indoles to cyclic α-iminophosphonates.
In the same context, Zhang and colleagues described in 2018 a single example on the enantioselective Mannich-type addition of glycine Schiff bases 213 to five-membered iminophosphonates 212 (Scheme 42), providing tetrasubstituted α-aminophosphonate 214 in moderate yield and stereocontrol (48%, 80:20 dr, 83% ee) [82].
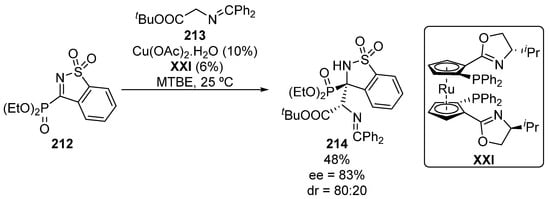
Scheme 42.
Enantioselective addition of Schiff base 213 to α-iminophosphonate 212.
In the same year, Ma and colleagues reported the enantioselective decarboxylative addition of β-keto acids 215 to cyclic α-iminophosphonates 204 (Scheme 43) [83]. The reaction affords α-amino-β-ketophosphonates 216 when five- or six-membered cyclic imines 204 are used as electrophile substrates. In addition, several alkyl and (hetero)aryl keto acids were tested, obtaining in all cases excellent yields and enantiocontrol (77–93%, 90–99% ee), and allowing a decrease in the catalyst loading down to 1% without any lose in the enantioselectivity. Likewise, the reduction of the ketone to obtain chiral alcohol 217 (88%, 94:6 dr) and the synthesis of aziridine 218 in presence of tert-butyl hydroperoxide (61%, single diastereoisomer) are described with high yields and diastereocontrol.
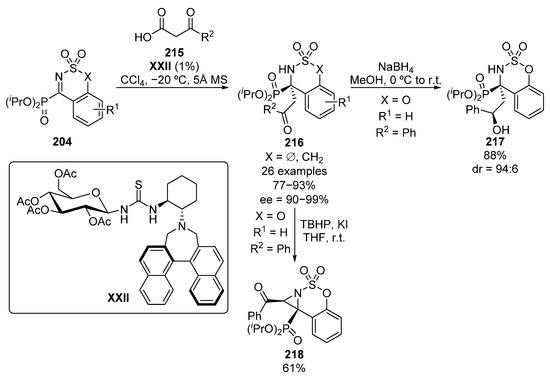
Scheme 43.
Enantioselective decarboxylative Mannich addition to imines 204.
Besides reactions that imply C-C bond formation, through the functionalization of trisubstituted α-aminophosphonate derivatives with electrophiles or by the addition of nucleophiles to α-iminophosphonates, cycloaddition reactions are also efficient protocols leading to the formation of optically active tetrasubstituted α-aminophosphonate derivatives. The first example of such reaction was published in 2011 by Kobayashi (Scheme 44) [84]. In particular, they reported a [3+2] reaction between Schiff bases 219 and tert-butyl acrylate 220, using silver hexamethyldisilazane as a catalyst and chiral bisphosphine ligand XXIII. The reaction can be successfully generalized to several chiral pyrrolidines 221 showing a tetrasubstituted α-aminophosphonate moiety in excellent yields and stereocontrol (72–81%, >99:<1 dr, 90–98% ee). According to the authors, the in situ formed silver–XXIII complex catalyzes the enolization of the phosphoryl group, leading to the active reagent for the exo-[3+2] cyclization through transition state TS18 (Scheme 44).
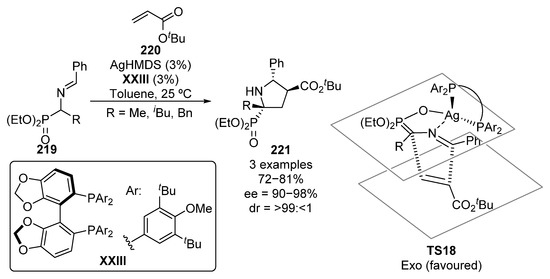
Scheme 44.
Enantioselective [3+2] cycloaddition with α-aminophosphonates 219.
More recently, an example of a dipolar cycloaddition was reported by Peng and colleagues (Scheme 45) [85]. The reaction between diazophosphonates 222 and acryloyl oxazolidones 223 in presence of Mg–XXIV complex afforded enantioenriched pyrazolines 224 in high yield and enantiocontrol.
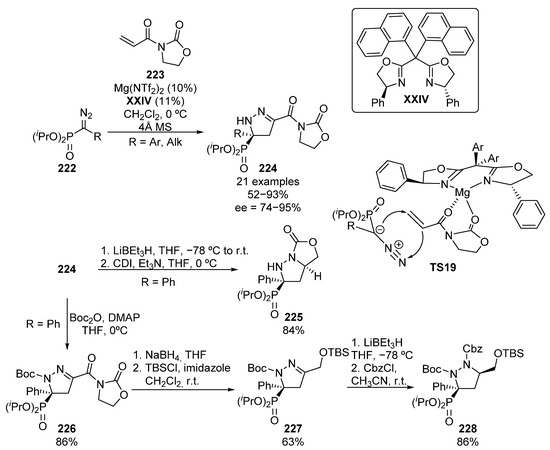
Scheme 45.
Enantioselective dipolar cycloaddition for the synthesis of chiral α-aminophosphonates.
For this reaction, the authors propose an activation of the electrophile through a double coordination of the chiral Mg-complex to both carbonyl groups of acryloyl oxazolidones 223, inducing the preliminary addition of the nucleophile from the Re-face in TS19. The subsequent trapping of the in situ formed enolate by the nitrogen atom of the diazo compound leads to cyclic α-aminophosphonates 224 in moderate to excellent yield and enantiocontrol (52–93%, 74–95% ee).
In addition, the double reduction of the pyrazoline and oxazolidone moieties, followed by treatment with carbonyl di-imidazole (CDI), leads to bicyclic pyrazolidine 225 in 84% yield. On the other hand, Boc-protected pyrazoline 226 can be also obtained in high yield (86%) by treatment with terc-butyl dicarbonate. Next, the selective deprotection of oxazolidone moiety by a reduction reaction with sodium borohydride, and the subsequent protection of the resulting alcohol affords pyrazoline 227 in 63% yield. Finally, the reduction of pyrazoline, and the in situ Cbz-protection of the newly formed NH group leads to pyrazolidine 228 in 86% yield.
Moreover, although not strictly a cycloaddition reaction, in 2018, Chi’s group reported the use of five- and six-membered cyclic α-iminophosphonates 229 in a N-heterocyclic carbene XXV-catalyzed formal [4+2] cycloaddition with α,β-unsaturated aldehydes 230 (Scheme 46) [86,87].
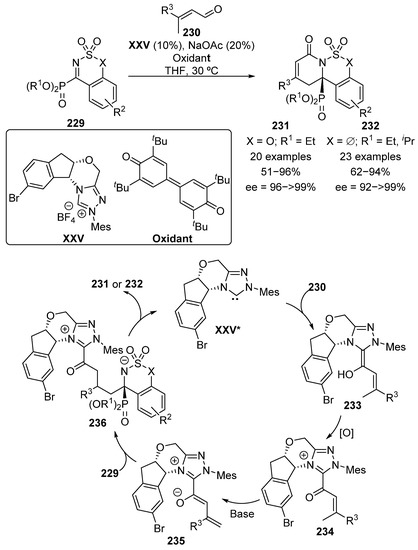
Scheme 46.
NHC-catalyzed formal [4+2] cycloaddition. XXV* represents the active carbene derived form of the catalyst XXV.
In the catalytic cycle proposed by the authors, after the activation of carbene catalyst XXV*, the reaction starts with an addition of the carbine to aldehyde substrate to generate intermediate 233. Then, a stoichiometric amount of an oxidant reagent is needed to re-generate the carbonyl group in species 234. Next, a basic source generates enolate 235 and the enantioselective vinylogous addition to the imine takes place giving rise to adduct 236. The cycle ends with an intramolecular addition of the nitrogen atom to the carbonyl moiety, which leads to cyclic α-aminophosphonates 231 or 232, after releasing the active carbene catalyst XXV*. The reaction products were obtained in moderate to excellent yield (51–96%) and enantioselectivities above 92%. Remarkably, simultaneously to Chi’s work, Ye and colleagues reported the same transformation in similar reaction conditions with a different NHC catalyst, resulting in the formation of the opposite enantiomer of α-aminophosphonates 231 and 232 [88].
3.2. C-P Bond Formation
One of the simplest strategies for the preparation of α-aminophosphonates is the addition of phosphorus nucleophiles to imines. Although the first catalytic asymmetric hydrophosphonylation of aldimines was described by Shibasaki in 1995 [89], it is easy to understand the further complication of developing a catalytic system that works on ketimines, due to the more difficult discrimination between both faces in the prochiral species if compared to aldimines [90,91,92]. Therefore, it was not until 2009 when Nakamura’s group described the first nucleophilic addition of diphenyl phosphite to N-sulfonyl ketimines 237 catalyzed by cinchona alkaloids XXVI and XXVII (Scheme 47) [93]. In the presence of a base and 2% of alkaloid XXVI, tetrasubstituted α-aminophosphonates 238 in excellent yields (93–99%) are obtained, with moderate to excellent enantiomeric excesses (55–97% ee). The use of hydroquinidine XXVII epimer as organocatalyst results equally in the formation of α-aminophosphonates 238 with the opposite configuration, in yields up to 99% with good stereocontrol (52–95% ee). Since it was evidenced that the reaction does not work without the presence of a base, the transition state TS20 proposed by the authors might consist in a coordination between the alkaloid nitrogen with the sodium cation that would improve the nucleophilic character of the phosphite reagent. Furthermore, the use of an alkaloid with a protected alcohol group results in a drop in the enantioselectivity, which may indicate a dual activation mode of the alkaloid in which the hydroxyl group activates the imine 237 by hydrogen bonding.
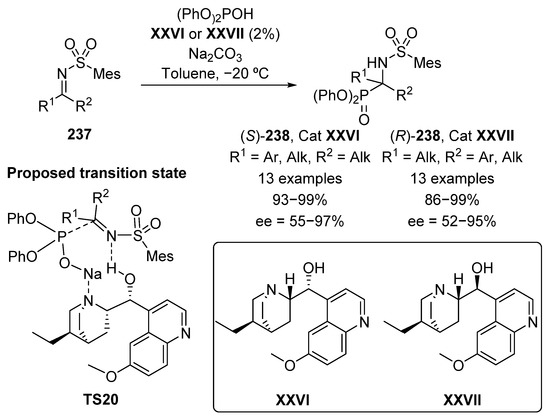
Scheme 47.
First enantioselective hydrophosphorylation of ketimines.
A few years later, Shibasaki reported the hydrophosphorylation reaction of N-thiophosphinyl imines 239 with various phosphites, using copper complexes and chiral bis-phosphine-based ligands as catalysts, thus obtaining tetrasubstituted α-aminophosphonates 240 with excellent enantioselectivities ranging between 86% and 97% (Scheme 48) [94,95]. The reaction works very efficiently even using only 0.5–2% copper–XXVIII catalyst, which can also be reused.
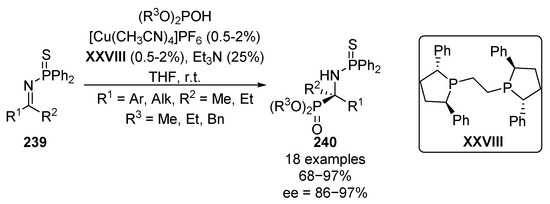
Scheme 48.
Catalytic asymmetric hydrophosphorylation of N-thiophosphinyl ketimines 239.
Another approach that can be used for the hydrophosphorylation reaction of α-phosphorated ketimines consists in the use of N-phosphinyl imines 241 in the presence of a bifunctional thiourea–iminophosphorane catalyst XXIX (Scheme 49) [96]. This type of catalyst has a superbase iminophosphorane acceptor and a classical thiourea donor unit. As described by the authors, the proposed transition state TS21 may involve the initial deprotonation of the phosphite reagent by the superbase unit, while the thiourea donor activates the imine electrophile 226 by hydrogen bonding. The resulting tetrasubstituted α-aminophosphonates 242 are obtained in excellent yields (78–99%) and with moderate to good enantiomeric excesses (46–71% ee).
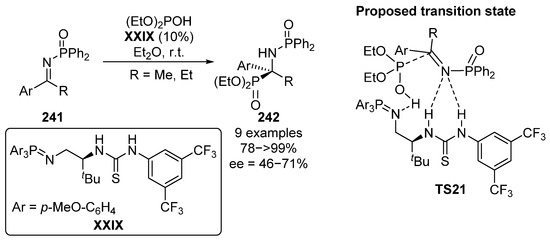
Scheme 49.
Organocatalyzed asymmetric phospha–Mannich reaction.
The first example of a hydrophosphonylation reaction of cyclic ketimines was described in 2013 [97]. In this case, trifluoromethylimines 243 derived from quinazolinone react with methyl, ethyl, benzyl or phenyl phosphites in the presence of a bifunctional alkaloid-thiourea catalyst IX (Scheme 50). The reaction is highly dependent on the medium and chloroform, dichloromethane or hexane: dichloromethane (5:1) mixtures were used depending on the phosphite reagent. In this reaction, α-aminophosphonates 244 were obtained in high yields (75–91%) and excellent stereocontrol (81–93% ee). In the proposed transition state TS22, the two hydrogens are in a gauche conformation with respect to the bulkiest groups, in order to minimize steric interactions. The basic amine and the donor thiourea adopt also a gauche conformation, so that they can simultaneously activate the nucleophilic phosphite and the electrophilic imine 243, through an acid–base interaction and a double hydrogen bond of the thiourea moiety with nitrogen and carbonyl, respectively. This rigid conformation directs the nucleophilic attack from the Re-face and supports the stereochemical outcome of the addition adduct with the R configuration.
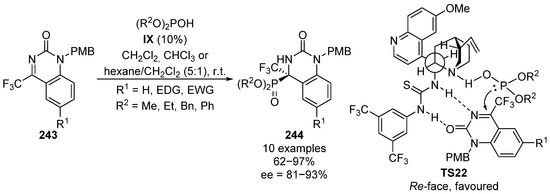
Scheme 50.
Enantioselective hydrophosphorylation of cyclic imines 243.
Later on, the addition of diphenyl phosphite to isatin-derived ketimines 245 using a bifunctional organocatalyst derived from squaramide XXX was described with high yields (85–96%) and excellent stereocontrol (83–97% ee) (Scheme 51) [98].
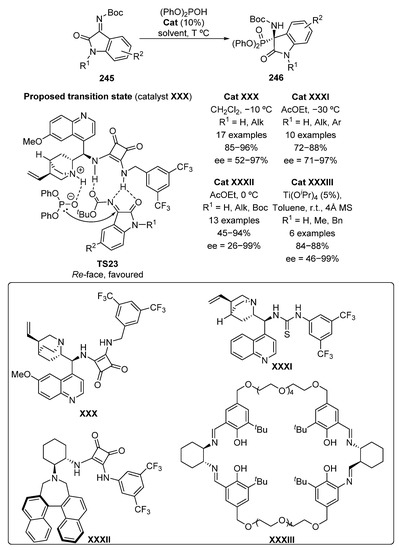
Scheme 51.
Organocatalyzed nucleophilic addition of diphenyl phosphite to isatin ketimines 245.
The use of 4-Br or 5-NO2 as substituents into the phenyl ring of the isatin derivatives decreases notably the enantioselectivities in this reaction, with excesses between 52% and 68%. Taking into account the poor results obtained for the addition of phosphites to the analogous ketones, the authors figure out an essential role of the iminic protecting group and propose transition state TS23, where the squaramide unit binds with isatin-derived imine while quinuclidine moiety deprotonates the phosphite reagent, in order to facilitate the reaction. A preferential Re-face attack of the phosphite to ketimine 245 affords the (R)-isomer of α-aminophosphonates 246.
In the same year, a similar approach was described by Chimni’s group, that is, the addition of diphenyl phosphite to ketimines 245, mediated by Chinchona-derived catalyst XXXI, to provide similar yields (72–88%) and enantiomeric excesses (71–97% ee) (Scheme 51) [99].
Another example regarding the addition of phosphites to isatin-derived imines 245 was described by Kim in 2016, using a bifunctional squaramide XXXII originated from binaphthyl (Scheme 51) [100]. The catalytic system is very efficient for unsubstituted isatins or those substituted with alkyl groups, obtaining yields up to 94% and enantioselectivities ranging from 73% to 99%. It should be noted that the use of an allyl or benzyl protecting group at the nitrogen of the amide moiety is strongly relevant for this process, since the presence of a carbamate group (R1 = Boc) in that position results in a strong drop in the yield (45%) and almost a total loss of stereocontrol (26% ee). Following the line of the previous examples, the proposed transition state is expected to be similar to TS23, showing a bifunctional activation of the phosphite and the ketimine 245. In this conformation, the phosphite nucleophile may attack preferentially the Re-face of the imine.
Some enantioselective methods for the addition of phosphites to imines derived from isatins 245, making use of metallic catalysts instead of organocatalysis have been also developed. In 2016, titanium–Salen complex Ti-XXXIII was used as catalyst, in the hydrophosphorylation reaction of ketimines 245 to provide tetrasubstituted α-aminophosphonates 246 with high yields (84–88%) and good enantiomeric excesses (63–99%) (Scheme 51) [101]. It should be noted that the amide group of the isatin moiety must be substituted, since the presence of an unprotected nitrogen resulted in a significant decrease in enantiomeric excess (46% ee), although without affecting the yield (88%).
A useful strategy for the in situ generation of cyclic ketimines and their subsequent activation for the nucleophilin addition of a phosphite reagent was reported by Singh in 2017 (Scheme 52) [102]. In this report, ketimines are produced from α-hydroxyamines 247 and then activated by the presence of phosphoric acid catalyst XXXIV as in TS24, leading to the formation of tetrasubstituted isoindolinones 248, with yields up to 98% and high enantioselectivities (72–97%).
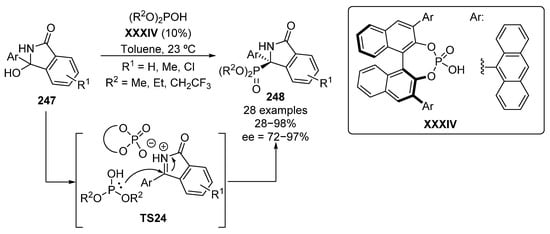
Scheme 52.
Enantioselective hydrophosphorylation of in situ generated N-acyl ketimines catalyzed by BINOL-derived phosphoric acid.
Furthermore, the addition of diphenyl phosphite to azirines 249 was described a few years ago, using zinc complexes with chiral bisimidazolines XXXV, with yields up to 99% and enantiomeric excesses between 80% and 96% (Scheme 53) [103]. The resulting phosphorus-substituted aziridines 250 were converted to oxazolines 253 through an initial acylation of the nitrogen with 3,5-dinitrobenzoyl chloride (251), followed by treatment with boron trifluoride. Additionally, the ring opening of aziridines 250 with hydrobromic acid under ultrasound treatment, leads to the corresponding β-bromo α-aminophosphonates 254. The halogen atom can be also removed by a radical reaction to give α-aminophosphonate 255 without any loss of enantiomeric purity (92% ee).
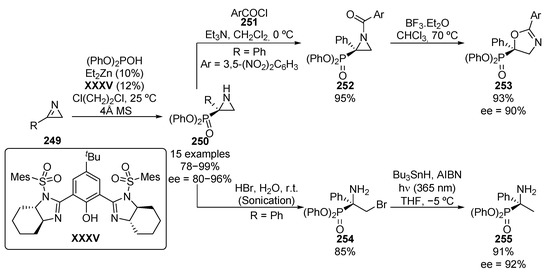
Scheme 53.
Enantioselective synthesis of aziridines 250 and their applications.
The proposed pathway for this transformation may consist of an initial formation of a Zn(II)–XXXV complex 256 in which the metal coordinates with the oxygen and only one of the imidazoline moieties of the ligand, leaving the other nitrogen atom free. The zinc atom then coordinates with azirine 249 in a tetrahedral mode (TS25), and the phosphite moiety establishes a hydrogen bond with the second free imidazoline unit in TS26. Azirine is therefore positioned with its substituent facing outwards, favoring a Re-face attack of the nucleophile. After the addition and formation of the corresponding phosphorus aziridines 250 the Zn catalyst is released, in order to be able to return to the catalytic cycle (Scheme 54).
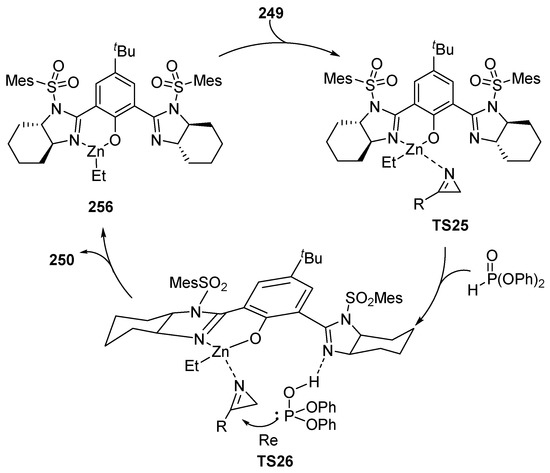
Scheme 54.
Proposed mechanism for the hydrophosphorylation of azirines 249.
3.3. C-N Bond Formation
Although there are only a few reports in the literature, the electrophilic amination reaction of trisubstituted phosphonates can also be an effective method for the preparation of tetrasubstituted α-aminophosphonates.
In 2005, Jørgensen and Kim described, almost simultaneously, the electrophilic amination of β-ketophophosphonates with azodicarboxylates. Jørgensen’s method makes use of a zinc–oxazolidine complex XXXVI, in order to generate an enolate from β-ketophosphonate 257, which undergoes an enantioselective nucleophilic addition to the nitrogen electrophile 258, providing the corresponding α-aminophosphonates 259 with yields up to 98% and excellent stereocontrol (85–98% ee) (Scheme 55) [104].
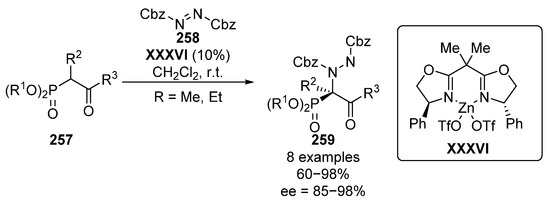
Scheme 55.
Enantioselective amination of β-ketophosphonates 257 catalyzed by zinc–oxazolidine complex.
On the other hand, Kim described a similar reaction using a chiral palladium-complex XXXVII in order to catalyze the addition of β-ketophosphonates 260 to diethyl azocarboxylates 261 with similar yields (68–92%) and enantiomeric excesses (99% ee) (Scheme 56) [105].
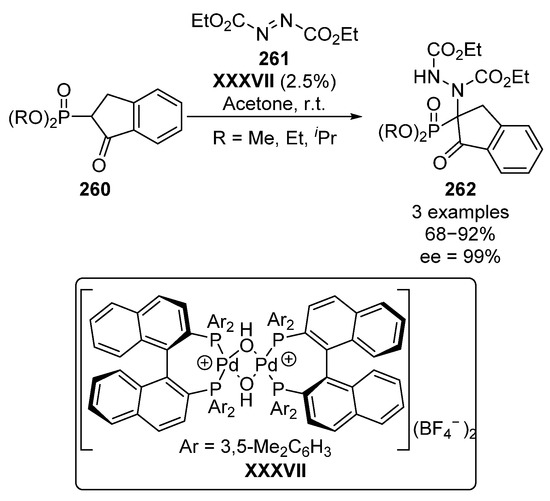
Scheme 56.
Chiral palladium complexes-catalyzed amination of phosphonates 260.
Finally, although only one example is reported with an acceptable yield (72%) and moderate enantioselectivity (50% ee), phosphonate-substituted aziridine 265 is obtained from α, β-unsaturated β-ketophosphonates 263 using a bifunctional catalyst derived from thiourea XXXVIII (Scheme 57) [106]. The transition state TS27 might consist of a double activation of the ketophosphonate 263 by the thiourea unit while the basic unit deprotonates the nitrogen of the hydroxylamine derivative 264. Then, the amphiphilic oxycarbamate reacts with the olefin through a conjugate addition reaction, while the enolate attacks the nitrogen atom of the amine, with concomitant release of the tosyl group.
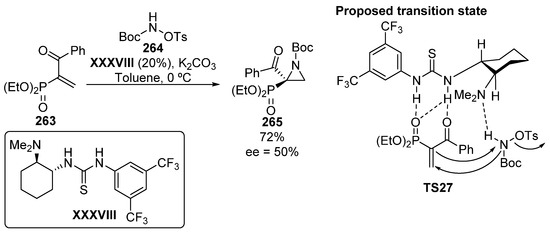
Scheme 57.
Organocatalyzed asymmetric synthesis of tetrasubstituted aziridines 265.
4. Final Remarks
Even though some examples of stereocontrolled synthesis of tetrasubstituted α-aminophosphonates have been reported, they are still rather limited if compared with the homologous reactions for the preparation of trisubstituted α-aminophosphonates. In particular, in recent years, the main efforts have been focused on the enantioselective transformations, which are known to be more attractive than diastereoselective ones. It should be noted that most of the enantioselective reactions summarized in this review were published during the past decade and thus, more articles regarding this approach are expected in the following years.
One of the most promising topics is related to nucleophilic additions to α-phosphorylated ketimines, which experienced an important growth during the last lustrum. Another promising topic is the enantioselective addition of phosphorus nucleophiles to ketimines. It has been slightly explored, with just a few examples reported to date, but due to the vast number of synthetic protocols for the preparation of imines known in the literature, the development of new enantioselective protocols for this transformation would constitute a relevant improvement in order to expand the structural diversity of tetrasubstituted α-aminophosphonates.
Author Contributions
Conceptualization, A.L.-F., X.d.C., A.M., E.M.d.M., F.P. and J.V.; methodology, A.L.-F. and X.d.C.; software, A.L.-F., A.M. and X.d.C.; validation, E.M.d.M. and J.V.; formal analysis, A.L.-F. and X.d.C.; investigation, A.L.-F. and X.d.C.; resources, E.M.d.M., F.P. and J.V.; data curation, A.L.-F. and X.d.C.; writing—original draft preparation, A.L.-F., A.M., X.d.C. and J.V.; writing—review and editing, A.L.-F., X.d.C., A.M., E.M.d.M., F.P. and J.V.; visualization, E.M.d.M., F.P. and J.V.; supervision, E.M.d.M. and J.V.; project administration, E.M.d.M. and J.V.; funding acquisition, E.M.d.M., F.P. and J.V. All authors have read and agreed to the published version of the manuscript.
Funding
Financial support by Ministerio de Economía, Industria y Competividad (MINECO, CTQ-2015-67871R) and Gobierno Vasco (GV, IT 992-16) is gratefully acknowledged. X.d.C. and A.L.-F. thank the Basque Country Government for a predoctoral grant.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Duthaler, R.O. Recent developments in the stereoselective synthesis of α-aminoacids. Tetrahedron 1994, 50, 1539–1650. [Google Scholar] [CrossRef]
- Nájera, C.; Sansano, J.M. Catalytic asymmetric synthesis of α-amino acids. Chem. Rev. 2007, 107, 4584–4671. [Google Scholar] [CrossRef]
- O’Donnell, M.J. The enantioselective synthesis of α-amino acids by phase-transfer catalysis with achiral Schiff base esters. Acc. Chem. Res. 2004, 37, 506–517. [Google Scholar] [CrossRef]
- Berlicki, L.; Kafarski, P. Computer-Aided Analysis and Design of Phosphonic and Phosphinic Enzyme Inhibitors as Potential Drugs and Agrochemicals. Curr. Org. Chem. 2005, 9, 1829–1850. [Google Scholar] [CrossRef]
- Sieńczyk, M.; Winiarski, Ł.; Kasperkiewicz, P.; Psurski, M.; Wietrzyk, J.; Oleksyszyn, J. Simple phosphonic inhibitors of human neutrophil elastase. Bioorg. Med. Chem. Lett. 2011, 21, 1310–1314. [Google Scholar] [CrossRef]
- Vassiliou, S.; Węglarz-Tomczak, E.; Berlicki, Ł.; Pawełczak, M.; Nocek, B.; Mulligan, R.; Joachimiak, A.; Mucha, A. Structure-guided, single-point modifications in the phosphinic dipeptide structure yield highly potent and selective inhibitors of neutral aminopeptidases. J. Med. Chem. 2014, 57, 8140–8151. [Google Scholar] [CrossRef] [PubMed]
- Arya, T.; Reddi, R.; Kishor, C.; Ganji, R.J.; Bhukya, S.; Gumpena, R.; McGowan, S.; Drag, M.; Addlagatta, A. Identification of the molecular basis of inhibitor selectivity between the human and streptococcal type i methionine aminopeptidases. J. Med. Chem. 2015, 58, 2350–2357. [Google Scholar] [CrossRef]
- Veera Narayana Reddy, M.; Balakrishna, A.; Anil Kumar, M.; Chandra Sekhar Reddy, G.; Uma Ravi Sankar, A.; Suresh Reddy, C.; Murali Krishna, T. One-step synthesis and bioassay of N-phosphoramidophosphonates. Chem. Pharm. Bull. 2009, 57, 1391–1395. [Google Scholar] [CrossRef] [PubMed]
- Che, J.Y.; Xu, X.Y.; Tang, Z.L.; Gu, Y.C.; Shi, D.Q. Synthesis and herbicidal activity evaluation of novel α-amino phosphonate derivatives containing a uracil moiety. Bioorg. Med. Chem. Lett. 2016, 26, 1310–1313. [Google Scholar] [CrossRef]
- Narayana Reddy, M.V.; Siva Kumar, B.; Balakrishna, A.; Reddy, C.S.; Nayak, S.K.; Reddya, C.D. One-pot synthesis of novel α-amino phosphonates using tetramethylguanidine as a catalyst. Arkivoc 2007, 2007, 246–254. [Google Scholar] [CrossRef]
- Dake, S.A.; Raut, D.S.; Kharat, K.R.; Mhaske, R.S.; Deshmukh, S.U.; Pawar, R.P. Ionic liquid promoted synthesis, antibacterial and in vitro antiproliferative activity of novel α-aminophosphonate derivatives. Bioorg. Med. Chem. Lett. 2011, 21, 2527–2532. [Google Scholar] [CrossRef]
- Sivala, M.R.; Devineni, S.R.; Golla, M.; Medarametla, V.; Pothuru, G.K.; Chamarthi, N.R. A heterogeneous catalyst, SiO2-ZnBr2: An efficient neat access for α-aminophosphonates and antimicrobial activity evaluation. J. Chem. Sci. 2016, 128, 1303–1313. [Google Scholar] [CrossRef]
- Rao, K.U.M.; Swapna, S.; Manidhar, D.M.; Reddy, K.M.K.; Reddy, C.S. Efficient synthesis of α-aminophosphonates and evaluation of significance of PO group towards antioxidant activity. Phosphorus Sulfur Silicon Relat. Elem. 2015, 190, 232–239. [Google Scholar] [CrossRef]
- Cassien, M.; Petrocchi, C.; Thétiot-Laurent, S.; Robin, M.; Ricquebourg, E.; Kandouli, C.; Asteian, A.; Rockenbauer, A.; Mercier, A.; Culcasi, M.; et al. On the vasoprotective mechanisms underlying novel β-phosphorylated nitrones: Focus on free radical characterization, scavenging and NO-donation in a biological model of oxidative stress. Eur. J. Med. Chem. 2016, 119, 197–217. [Google Scholar] [CrossRef] [PubMed]
- Damiche, R.; Chafaa, S. Synthesis of new bioactive aminophosphonates and study of their antioxidant, anti-inflammatory and antibacterial activities as well the assessment of their toxicological activity. J. Mol. Struct. 2017, 1130, 1009–1017. [Google Scholar] [CrossRef]
- Li, Y.J.; Wang, C.Y.; Ye, M.Y.; Yao, G.Y.; Wang, H.S. Novel coumarin-containing aminophosphonatesas antitumor agent: Synthesis, cytotoxicity, DNA-Binding and apoptosis evaluation. Molecules 2015, 20, 14791–14809. [Google Scholar] [CrossRef]
- Huang, R.Z.; Wang, C.Y.; Li, J.F.; Yao, G.Y.; Pan, Y.M.; Ye, M.Y.; Wang, H.S.; Zhang, Y. Synthesis, antiproliferative and apoptosis-inducing effects of novel asiatic acid derivatives containing α-aminophosphonates. RSC Adv. 2016, 6, 62890–62906. [Google Scholar] [CrossRef]
- Wang, Q.; Yang, L.; Ding, H.; Chen, X.; Wang, H.; Tang, X. Synthesis, X-ray crystal structure, DNA/protein binding and cytotoxicity studies of five α-aminophosphonate N-derivatives. Bioorg. Chem. 2016, 69, 132–139. [Google Scholar] [CrossRef]
- Stamper, C.; Bennett, B.; Edwards, T.; Holz, R.C.; Ringe, D.; Petsko, G. Inhibition of the aminopeptidase from Aeromonas proteolytica by l-leucinephosphonic acid. Spectroscopic and crystallographic characterization of the transition state of peptide hydrolysis. Biochemistry 2001, 40, 7035–7046. [Google Scholar] [CrossRef]
- Kafarski, P.; Lejczak, B.; Szewczyk, J. Optically active 1-aminoalkanephosphonic acids. Dibenzoyl-l-tartaric anhydride as an effective agent for the resolution of racemic diphenyl 1-aminoalkanephosphonates. Can. J. Chem. 1983, 61, 2425–2430. [Google Scholar] [CrossRef]
- Solodenko, V.A.; Kukhar, V.P. Stereoselective papain-catalyzed Synthesis of Alafosfalin. Tetrahedron Lett. 1989, 30, 6917–6918. [Google Scholar] [CrossRef]
- Poliak, M.S. Antibiotics of the phosphonic acid group. Antibiot. Med Biotechnol. 1987, 32, 66–75. [Google Scholar]
- Ordóñez, M.; Sayago, F.J.; Cativiela, C. Synthesis of quaternary α-aminophosphonic acids. Tetrahedron 2012, 68, 6369–6412. [Google Scholar] [CrossRef]
- Bera, K.; Namboothiri, I.N.N. Asymmetric Synthesis of Quaternary α-Amino Acids and Their Phosphonate Analogues. Asian J. Org. Chem. 2014, 3, 1234–1260. [Google Scholar] [CrossRef]
- Shimizu, M. Construction of asymmetric quaternary carbon centers with high enantioselectivity. Angew. Chem. Int. Ed. 2011, 50, 5998–6000. [Google Scholar] [CrossRef]
- Hawner, C.; Alexakis, A. Metal-catalyzed asymmetric conjugate addition reaction: Formation of quaternary stereocenters. Chem. Commun. 2010, 46, 7295–7306. [Google Scholar] [CrossRef] [PubMed]
- Christoffers, J.; Baro, A. Quaternary Stereocenters: Challenges and Solutions for Organic Synthesis; John Wiley & Sons: Hoboken, NJ, USA, 2006; ISBN 3527311076. [Google Scholar]
- Riant, O.; Hannedouche, J. Asymmetric catalysis for the construction of quaternary carbon centres: Nucleophilic addition on ketones and ketimines. Org. Biomol. Chem. 2007, 5, 873–888. [Google Scholar] [CrossRef] [PubMed]
- Studer, A.; Seebach, D. Preparation of either enantiomer of 1,2-diaminoalkane-2-phosphonic acid derivatives. Heterocycles 1995, 40, 357–378. [Google Scholar] [CrossRef]
- Fitzi, R.; Seebach, D. Resolution and use in α-amino acid synthesis of imidazolidinone glycine derivatives. Tetrahedron 1988, 44, 5277–5292. [Google Scholar] [CrossRef]
- Schickll, C.P.; Seebach, D. Preparation of Chiral Electrophilic Glycine and (E)-2, 3-Dehydroamino Acid Derivatives from tert.-Butyl 2-tert.-Butyl-3-methyl-4-oxo-1-imidazolidinecarboxylate (Boc-BMI). ChemInform 1991, 22, 655–668. [Google Scholar] [CrossRef]
- Davis, F.A.; Wu, Y.; Xu, H.; Zhang, J. Asymmetric synthesis of cis-5-substituted pyrrolidine 2-phosphonates using metal carbenoid NH insertion and δ-amino β-ketophosphonates. Org. Lett. 2004, 6, 4523–4525. [Google Scholar] [CrossRef]
- Davis, F.A.; Xu, H.; Wu, Y.; Zhang, J. Asymmetric synthesis of polyfunctionalized pyrrolidines from sulfinimine-derived pyrrolidine 2-phosphonates. Synthesis of pyrrolidine 225C. Org. Lett. 2006, 8, 2273–2276. [Google Scholar] [CrossRef] [PubMed]
- Katritzky, A.R.; Cui, X.L.; Yang, B.; Steel, P.J. Asymmetric syntheses of 2-substituted and 2,5-disubstituted pyrrolidines from (3S,5R,7aR)-5-(benzotriazol-1-yl)-3-phenyl[2,1-b]oxazolopyrrolidine. J. Org. Chem. 1999, 64, 1979–1985. [Google Scholar] [CrossRef] [PubMed]
- Amedjkouh, M.; Westerlund, K. Self-reproduction of chirality on α-aminophosphonates: Asymmetric synthesis of α-alkylated diethyl pyrrolidin-2-yl-phosphonate. Tetrahedron Lett. 2004, 45, 5175–5177. [Google Scholar] [CrossRef]
- Vicario, J.; Ortiz, P.; Palacios, F. Synthesis of tetrasubstituted α-aminophosphonic acid derivatives from trisubstituted α-aminophosphonates. Eur. J. Org. Chem. 2013, 7095–7100. [Google Scholar] [CrossRef]
- Rassukana, Y.V.; Stanko, O.V.; Onys’ko, P.P. Enantiomeric O,O-dimenthyl α-iminotrifluoroethylphosphonates: Novel chiral building blocks in asymmetric synthesis of α-trifluoromethylated α-aminophosphonic acid derivatives. J. Fluor. Chem. 2019, 219, 123–128. [Google Scholar] [CrossRef]
- Rassukana, Y.V.; Bezgubenko, L.V.; Stanko, O.V.; Rusanov, E.B.; Kulik, I.B.; Onys’ko, P.P. Diastereoselective cycloaddition of (S)-N-(1-phenylethylimino)trifluoropropionate and trifluoroethylphosphonate with diazomethane. Tetrahedron Asymmetry 2017, 28, 555–560. [Google Scholar] [CrossRef]
- Davis, F.A.; Lee, S.; Yan, H.; Titus, D.D. Asymmetric Synthesis of Quaternary α-Amino Phosphonates Using Sulfinimines. Org. Lett. 2001, 3, 1757–1760. [Google Scholar] [CrossRef]
- Davis, F.A.; Lee, S.H.; Xu, H. Asymmetric synthesis of cyclic α-amino phosphonates using masked oxo sulfinimines (N-sulfinyl imines). J. Org. Chem. 2004, 69, 3774–3781. [Google Scholar] [CrossRef]
- Chen, Q.; Yuan, C. A new and convenient asymmetric synthesis of α-amino- and α-alkyl-α-aminophosphonic acids using N-tert-butylsulfinyl imines as chiral auxiliaries. Synthesis 2007, 24, 3779–3786. [Google Scholar] [CrossRef]
- Chen, Q.; Li, J.; Yuan, C. Sulfinimine-mediated asymmetric synthesis of acyclic and cyclic α-aminophosphonates. Synthesis 2008, 18, 2986–2990. [Google Scholar] [CrossRef]
- Khan, H.A.; Ellman, J.A. Asymmetric synthesis of α-aminophosphonate esters by the addition of dialkyl phosphites to tert-butanesulfinyl imines. Synthesis 2013, 45, 3147–3150. [Google Scholar] [CrossRef]
- Li, P.; Jiang, M.; Liu, J.T. Asymmetric Pudovik Reaction of Chiral Fluoroalkyl α,β-Unsaturated Ketimines and Diphenyl Phosphite. Chin. J. Chem. 2014, 32, 1003–1006. [Google Scholar] [CrossRef]
- Pedroni, J.; Cramer, N. Enantioselective C–H Functionalization—Addition Sequence Delivers Densely Substituted 3-Azabicyclo[3.1.0]hexanes. J. Am. Chem. Soc. 2017, 139, 12398–12401. [Google Scholar] [CrossRef] [PubMed]
- Fadel, A.; Tesson, N. Synthesis of enantiomerically pure (1S,2S)-1-aminocyclopropanephosphonic acids from (2S)-methylcyclopropanone acetal. Eur. J. Org. Chem. 2000, 2153–2159. [Google Scholar] [CrossRef]
- Tesson, N.; Dorigneux, B.; Fadel, A. Synthesis of (1S,2S)- and (1R,2R)-1-amino-2-methylcyclopropane-phosphonic acids from racemic methylcyclopropanone acetal. Tetrahedron Asymmetry 2002, 13, 2267–2276. [Google Scholar] [CrossRef]
- Fadel, A.; Tesson, N. Preparation of enantiomerically pure (1S,2S)-1-aminocyclopropanephosphonic acid from methylcyclopropanone acetal via spirophosphonate intermediates. Tetrahedron Asymmetry 2000, 11, 2023–2031. [Google Scholar] [CrossRef]
- Louaisil, N.; Rabasso, N.; Fadel, A. Asymmetric synthesis of (R)- and (S)-α-amino-3-piperidinylphosphonic acids via phosphite addition to iminium ions. Synthesis 2007, 2, 289–293. [Google Scholar] [CrossRef]
- Louaisil, N.; Rabasso, N.; Fadel, A. Highly stereoselective synthesis of new α-amino-β-hydroxy six-membered heterocyclic phosphonic acids, serine analogues. Tetrahedron 2009, 65, 8587–8595. [Google Scholar] [CrossRef]
- Iwanejko, J.; Brol, A.; Szyja, B.M.; Daszkiewicz, M.; Wojaczyńska, E.; Olszewski, T.K. Aminophosphonates and aminophosphonic acids with tetrasubstituted stereogenic center: Diastereoselective synthesis from cyclic ketimines. Org. Biomol. Chem. 2019, 17, 7352–7359. [Google Scholar] [CrossRef]
- Iwanejko, J.; Sowiński, M.; Wojaczyńska, E.; Olszewski, T.K.; Górecki, M. An approach to new chiral bicyclic imines and amines: Via Horner-Wadsworth-Emmons reaction. RSC Adv. 2020, 10, 14618–14629. [Google Scholar] [CrossRef]
- Fang, Z.; Yang, H.; Miao, Z.; Chen, R. Asymmetric Mannich-type synthesis of N-phosphinyl α-aminophosphonic acid monoesters. Helv. Chim. Acta 2011, 94, 1586–1593. [Google Scholar] [CrossRef]
- Kuliszewska, E.; Hanbauer, M.; Hammerschmidt, F. Preparation of α-aminobenzylphosphonic acids with a stereogenic quaternary carbon atom via microscopically configurationally stable α-aminobenzyllithiums. Chem. Eur. J. 2008, 14, 8603–8614. [Google Scholar] [CrossRef] [PubMed]
- Qian, R.; Roller, A.; Hammerschmidt, F. Phosphonate-phosphinate rearrangement. J. Org. Chem. 2015, 80, 1082–1091. [Google Scholar] [CrossRef] [PubMed]
- Davis, F.A.; McCoull, W.; Titus, D.D. Asymmetric synthesis of α-methylphosphophenylalanine derivatives using sulfinimine-derived enantiopure aziridine-2-phosphonates. Org. Lett. 1999, 1, 1053–1055. [Google Scholar] [CrossRef] [PubMed]
- Hercouet, A.; Le Corre, M.; Carboni, B. Asymmetric synthesis of a phosphonic analogue of (−)-allo-norcoronamic acid. Tetrahedron Lett. 2000, 41, 197–199. [Google Scholar] [CrossRef]
- Sawamura, M.; Hamashima, H.; Ito, Y. Rhodium-catalyzed enantioselective Michael addition of (1-cyanoethyl) phosphonate: Synthesis of optically active phosphonic acid derivatives with phosphorus-substituted quaternary asymmetric carbon center. Bull. Chem. Soc. Jpn. 2000, 73, 2559–2562. [Google Scholar] [CrossRef]
- Wąsek, K.; Kędzia, J.; Krawczyk, H. An efficient approach to the synthesis of enantiomerically pure trans-1-amino-2-(hydroxymethyl)cyclopropanephosphonic acids. Tetrahedron Asymmetry 2010, 21, 2081–2086. [Google Scholar] [CrossRef]
- Kuwano, R.; Nishio, R.; Ito, Y. Enantioselective construction of quaternary α-carbon centers on α-amino phosphonates via catalytic asymmetric allylation. Org. Lett. 1999, 1, 837–839. [Google Scholar] [CrossRef]
- Wilt, J.C.; Pink, M.; Johnston, J.N. A diastereo- and enantioselective synthesis of α-substituted anti-α,β-diaminophosphonic acid derivatives. Chem. Commun. 2008, 4177–4179. [Google Scholar] [CrossRef]
- Bera, K.; Namboothiri, I.N.N. Enantioselective synthesis of quaternary α-aminophosphonates via conjugate addition of α-nitrophosphonates to enones. Org. Lett. 2012, 14, 980–983. [Google Scholar] [CrossRef]
- Bera, K.; Namboothiri, I.N.N. Quinine-derived thiourea and squaramide catalyzed conjugate addition of α-nitrophosphonates to enones: Asymmetric synthesis of quaternary α-aminophosphonates. J. Org. Chem. 2015, 80, 1402–1413. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.S.; Gönczi, K.; Kardos, G.; Süle, K.; Hegedus, L.; Kállay, M.; Kubinyi, M.; Szabó, P.; Petneházy, I.; Toke, L.; et al. Cinchona based squaramide catalysed enantioselective Michael addition of α-nitrophosphonates to aryl acrylates: Enantioselective synthesis of quaternary α-aminophosphonates. Tetrahedron Asymmetry 2013, 24, 1605–1614. [Google Scholar] [CrossRef]
- Tripathi, C.B.; Kayal, S.; Mukherjee, S. Catalytic asymmetric synthesis of α,β-disubstituted α,γ-diaminophosphonic acid precursors by michael addition of α-substituted nitrophosphonates to nitroolefins. Org. Lett. 2012, 14, 3296–3299. [Google Scholar] [CrossRef]
- Bera, K.; Namboothiri, I.N.N. Enantioselective synthesis of α-amino-γ-sulfonyl phosphonates with a tetrasubstituted chiral α-carbon via quinine-squaramide-catalyzed michael addition of nitrophosphonates to vinyl sulfones. Adv. Synth. Catal. 2013, 355, 1265–1270. [Google Scholar] [CrossRef]
- Chen, G.Y.; Lu, Y. Highly enantioselective Michael addition of α-substituted nitrophosphonates to a vinyl sulfone. Synthesis 2013, 45, 1654–1658. [Google Scholar] [CrossRef]
- Han, W.Y.; Zhao, J.Q.; Wu, Z.J.; Zhang, X.M.; Yuan, W.C. Asymmetric synthesis of β-hydroxy-α-amino phosphonic acid derivatives via organocatalytic direct aldol reaction of α-isothiocyanato phosphonates with aldehydes. J. Org. Chem. 2013, 78, 10541–10547. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.M.; Shen, F.F.; Zhang, F.T.; Zhang, J.L.; Wang, R. Catalytic asymmetric 1,2-addition of α-isothiocyanato phosphonates: Synthesis of chiral β-hydroxy- or β-amino-substituted α-amino phosphonic acid derivatives. Angew. Chem. Int. Ed. 2014, 53, 1862–1866. [Google Scholar] [CrossRef]
- Frankowski, S.; Gajda, T.; Albrecht, Ł. Isothiocyanate Strategy for the Synthesis of Quaternary α-Amino Acids Bearing a Spirocyclic Ring System. Adv. Synth. Catal. 2018, 360, 1822–1832. [Google Scholar] [CrossRef]
- Maestro, A.; de Marigorta, E.M.; Palacios, F.; Vicario, J. α-Iminophosphonates: Useful Intermediates for Enantioselective Synthesis of α-Aminophosphonates. Asian J. Org. Chem. 2020, 9, 538–548. [Google Scholar] [CrossRef]
- Sekine, M.; Kume, A.; Hata, T. Dialkyl acylphosphonates: A new acylating agent of alcohols. Tetrahedron Lett. 1981, 22, 3617–3620. [Google Scholar] [CrossRef]
- Vicario, J.; Ezpeleta, J.M.; Palacios, F. Asymmetric cyanation of a-ketiminophosphonates catalyzed by Cinchona alkaloids: Enantioselective synthesis of tetrasubstituted a-aminophosphonic acid derivatives from trisubstituted a-aminophosphonates. Adv. Synth. Catal. 2012, 354, 2641–2647. [Google Scholar] [CrossRef]
- Vicario, J.; Ortiz, P.; Ezpeleta, J.M.; Palacios, F. Asymmetric synthesis of functionalized tetrasubstituted α-aminophosphonates through enantioselective aza-Henry reaction of phosphorylated ketimines. J. Org. Chem. 2015, 80, 156–164. [Google Scholar] [CrossRef]
- Maestro, A.; del Corte, X.; Martinez de Marigorta, E.; Palacios, F.; Vicario, J. Enantioselective synthesis of functionalized αα-aminophosphonic acid derivatives. Phosphorus Sulfur Silicon Relat. Elem. 2019, 194, 287–291. [Google Scholar] [CrossRef]
- Maestro, A.; Martinez de Marigorta, E.; Palacios, F.; Vicario, J. Enantioselective Aza-Reformatsky Reaction with Ketimines. Org. Lett. 2019, 21, 9473–9477. [Google Scholar] [CrossRef]
- Rassukana, Y.V.; Yelenich, I.P.; Vlasenko, Y.G.; Onys’ko, P.P. Asymmetric synthesis of phosphonotrifluoroalanine derivatives via proline-catalyzed direct enantioselective CC bond formation reactions of NH trifluoroacetimidoyl phosphonate. Tetrahedron Asymmetry 2014, 25, 1234–1238. [Google Scholar] [CrossRef]
- Morisaki, K.; Sawa, M.; Yonesaki, R.; Morimoto, H.; Mashima, K.; Ohshima, T. Mechanistic Studies and Expansion of the Substrate Scope of Direct Enantioselective Alkynylation of α-Ketiminoesters Catalyzed by Adaptable (Phebox)Rhodium(III) Complexes. J. Am. Chem. Soc. 2016, 138, 6194–6203. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.Y.; Wang, J.C.; Wei, J.; Xu, Z.J.; Guo, Z.; Low, K.H.; Che, C.M. Dirhodium carboxylates catalyzed enantioselective coupling reactions of α-diazophosphonates, anilines, and electron-deficient aldehydes. Angew. Chem. Int. Ed. 2012, 51, 11376–11380. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Wu, B.; Gao, X.; Zhou, Y.G. Enantioselective synthesis of quaternary α-aminophosphonates by Pd-catalyzed arylation of cyclic α-ketiminophosphonates with arylboronic acids. Chem. Commun. 2016, 52, 10882–10885. [Google Scholar] [CrossRef]
- Yan, Z.; Gao, X.; Zhou, Y.G. Enantioselective synthesis of quaternary α-aminophosphonates by organocatalytic Friedel-Crafts reactions of indoles with cyclic α-ketiminophosphonates. Chin. J. Catal. 2017, 38, 784–791. [Google Scholar] [CrossRef]
- Shao, Q.; Wu, L.; Chen, J.; Gridnev, I.D.; Yang, G.; Xie, F.; Zhang, W. Copper (II)/RuPHOX-Catalyzed Enantioselective Mannich-Type Reaction of Glycine Schiff Bases with Cyclic Ketimines. Adv. Synth. Catal. 2018, 360, 4625–4633. [Google Scholar] [CrossRef]
- Liu, Y.J.; Li, J.S.; Nie, J.; Ma, J.A. Organocatalytic Asymmetric Decarboxylative Mannich Reaction of β-Keto Acids with Cyclic α-Ketiminophosphonates: Access to Quaternary α-Aminophosphonates. Org. Lett. 2018, 20, 3643–3646. [Google Scholar] [CrossRef]
- Yamashita, Y.; Imaizumi, T.; Guo, X.X.; Kobayashi, S. Chiral silver amides as effective catalysts for enantioselective [3+2] cycloaddition reactions. Chem. Asian J. 2011, 6, 2550–2559. [Google Scholar] [CrossRef]
- Huang, N.; Tong, X.; Zhou, S.; Guo, Q.; Peng, Y. Asymmetric [3+2] Cycloaddition Reactions of α-Substituted Diazophosphonates with 3-Acryloyl-2-oxazolidinone to Access Chiral Pyrazoline Derivatives with Phosphonyl at a Tetrasubstituted Stereogenic Center. Adv. Synth. Catal. 2019, 361, 4805–4810. [Google Scholar] [CrossRef]
- Sun, J.; Mou, C.; Liu, C.; Huang, R.; Zhang, S.; Zheng, P.; Chi, Y.R. Enantioselective access to multi-cyclic α-amino phosphonates: Via carbene-catalyzed cycloaddition reactions between enals and six-membered cyclic imines. Org. Chem. Front. 2018, 5, 2992–2996. [Google Scholar] [CrossRef]
- Sun, J.; Mou, C.; Wang, Z.; He, F.; Wu, J.; Chi, Y.R. Carbene-Catalyzed [4+2] Cycloadditions of Vinyl Enolate and (in Situ Generated) Imines for Enantioselective Synthesis of Quaternary α-Amino Phosphonates. Org. Lett. 2018, 20, 5969–5972. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Gao, Z.H.; Zhang, C.L.; Chen, K.Q.; Ye, S. Enantioselective Synthesis of Cyclic α-Aminophosphonates through N-Heterocyclic Carbene-Catalyzed [4+2] Annulation of Enals with α-Iminophosphonates. Asian J. Org. Chem. 2018, 7, 2452–2455. [Google Scholar] [CrossRef]
- Sasai, H.; Arai, S.; Tahara, Y.; Shibasaki, M. Catalytic Asymmetric Synthesis of a-Amino Phosphonates Using Lanthanoid-Potassium-BINOL Complexes. J. Org. Chem. 1995, 60, 6656–6657. [Google Scholar] [CrossRef]
- Merino, P.; Marqués-López, E.; Herrera, R.P. Catalytic enantioselective hydrophosphonylation of aldehydes and imines. Adv. Synth. Catal. 2008, 350, 1195–1208. [Google Scholar] [CrossRef]
- Bhadury, P.S.; Li, H. Organocatalytic asymmetric hydrophosphonylation/Mannich reactions using thiourea, cinchona and brnsted acid catalysts. Synlett 2012, 23, 1108–1131. [Google Scholar] [CrossRef]
- Spilling, C.D.; Malla, R.K. Synthesis of Non-Racemic α-Hydroxyphosphonates via Asymmetric Phospho-Aldol Reaction; Springer: Berlin/Heidelberg, Germany, 2015; Volume 361, ISBN 978-3-540-73346-1. [Google Scholar]
- Nakamura, S.; Hayashi, M.; Hiramatsu, Y.; Shibata, N.; Funahashi, Y.; Toru, T. Catalytic enantioselective hydrophosphonylation of ketimines using cinchona alkaloids. J. Am. Chem. Soc. 2009, 131, 18240–18241. [Google Scholar] [CrossRef]
- Yin, L.; Bao, Y.; Kumagai, N.; Shibasaki, M. Catalytic asymmetric hydrophosphonylation of ketimines. J. Am. Chem. Soc. 2013, 135, 10338–10341. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Otsuka, Y.; Yin, L.; Kumagai, N.; Shibasaki, M. Catalytic Enantioselective Addition of Diethyl Phosphite to N-Thiophosphinoyl Ketimines: Preparation of (R)-Diethyl (1-Amino-1-phenylethyl)phosphonate. Org. Synth. 2017, 94, 313–331. [Google Scholar] [CrossRef]
- Robertson, G.P.; Farley, A.J.M.; Dixon, D.J. Bifunctional Iminophosphorane Catalyzed Enantioselective Ketimine Phospha-Mannich Reaction. Synlett 2016, 27, 21–24. [Google Scholar] [CrossRef][Green Version]
- Xie, H.; Song, A.; Zhang, X.; Chen, X.; Li, H.; Sheng, C.; Wang, W. Quinine-thiourea catalyzed enantioselective hydrophosphonylation of trifluoromethyl 2(1H)-quinazolinones. Chem. Commun. 2013, 49, 928–930. [Google Scholar] [CrossRef] [PubMed]
- George, J.; Sridhar, B.; Reddy, B.V.S. First example of quinine-squaramide catalyzed enantioselective addition of diphenyl phosphite to ketimines derived from isatins. Org. Biomol. Chem. 2014, 12, 1595–1602. [Google Scholar] [CrossRef]
- Kumar, A.; Sharma, V.; Kaur, J.; Kumar, V.; Mahajan, S.; Kumar, N.; Chimni, S.S. Cinchona-derived thiourea catalyzed hydrophosphonylation of ketiminesdan enantioselective synthesis of α-amino phosphonates. Tetrahedron 2014, 70, 7044–7049. [Google Scholar] [CrossRef]
- Jang, H.S.; Kim, Y.; Kim, D.Y. Enantioselective addition of diphenyl phosphonate to ketimines derived from isatins catalyzed by binaphthyl-modified organocatalysts. Beilstein J. Org. Chem. 2016, 12, 1551–1556. [Google Scholar] [CrossRef]
- Nazish, M.; Jakhar, A.; Khan, N.U.H.; Verma, S.; Kureshy, R.I.; Abdi, S.H.R.; Bajaj, H.C. Enantioselective hydrophosphonylation of N-benzyl imines, isatin derived ketimines and isatins catalyzed by in-situ generated Ti(IV) macrocyclic salen complexes. Appl. Catal. A Gen. 2016, 515, 116–125. [Google Scholar] [CrossRef]
- Suneja, A.; Unhale, R.A.; Singh, V.K. Enantioselective Hydrophosphonylation of in Situ Generated N-Acyl Ketimines Catalyzed by BINOL-Derived Phosphoric Acid. Org. Lett. 2017, 19, 476–479. [Google Scholar] [CrossRef]
- Nakamura, S.; Hayama, D. Enantioselective Reaction of 2H-Azirines with Phosphite Using Chiral Bis(imidazoline)/Zinc(II) Catalysts. Angew. Chem. Int. Ed. 2017, 56, 8785–8789. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, L.; Zhuang, W.; Jørgensen, K.A. An easy approach to optically active α-amino phosphonic acid derivatives by chiral Zn(II)-catalyzed enantioselective amination of phosphonates. J. Am. Chem. Soc. 2005, 127, 5772–5773. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Kim, H.R.; Kim, Y.K. Catalytic enantioselective fluorination and amination of β-keto phosphonates catalyzed by chiral palladium complexes. Org. Lett. 2005, 7, 2309–2311. [Google Scholar] [CrossRef] [PubMed]
- de Fusco, C.; Fuoco, T.; Croce, G.; Lattanzi, A. Noncovalent organocatalytic synthesis of enantioenriched terminal aziridines with a quaternary stereogenic center. Org. Lett. 2012, 14, 4078–4081. [Google Scholar] [CrossRef] [PubMed]

































































Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).