
Organoboron Compounds: Effective Antibacterial and Antiparasitic Agents

 ,
, 


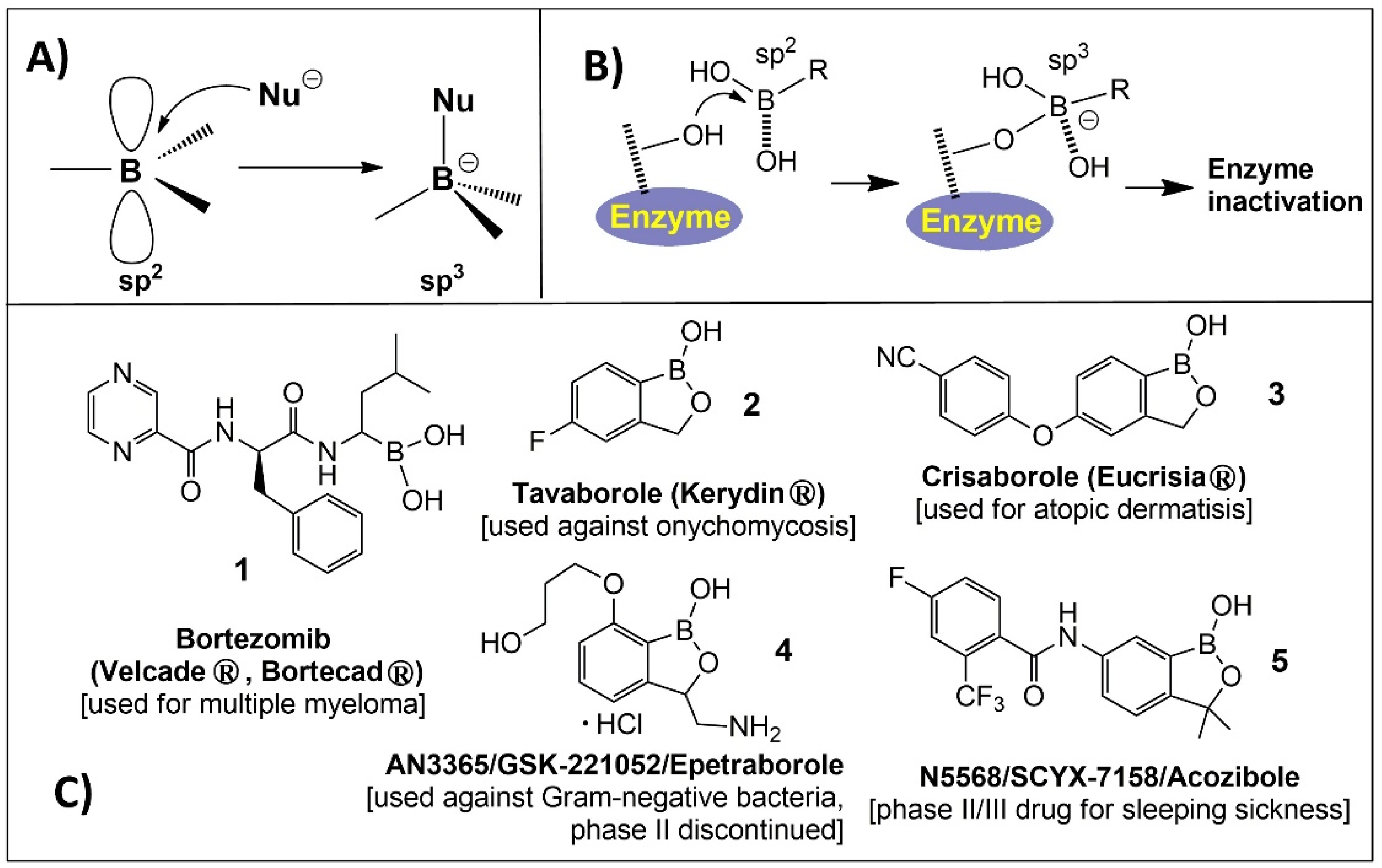
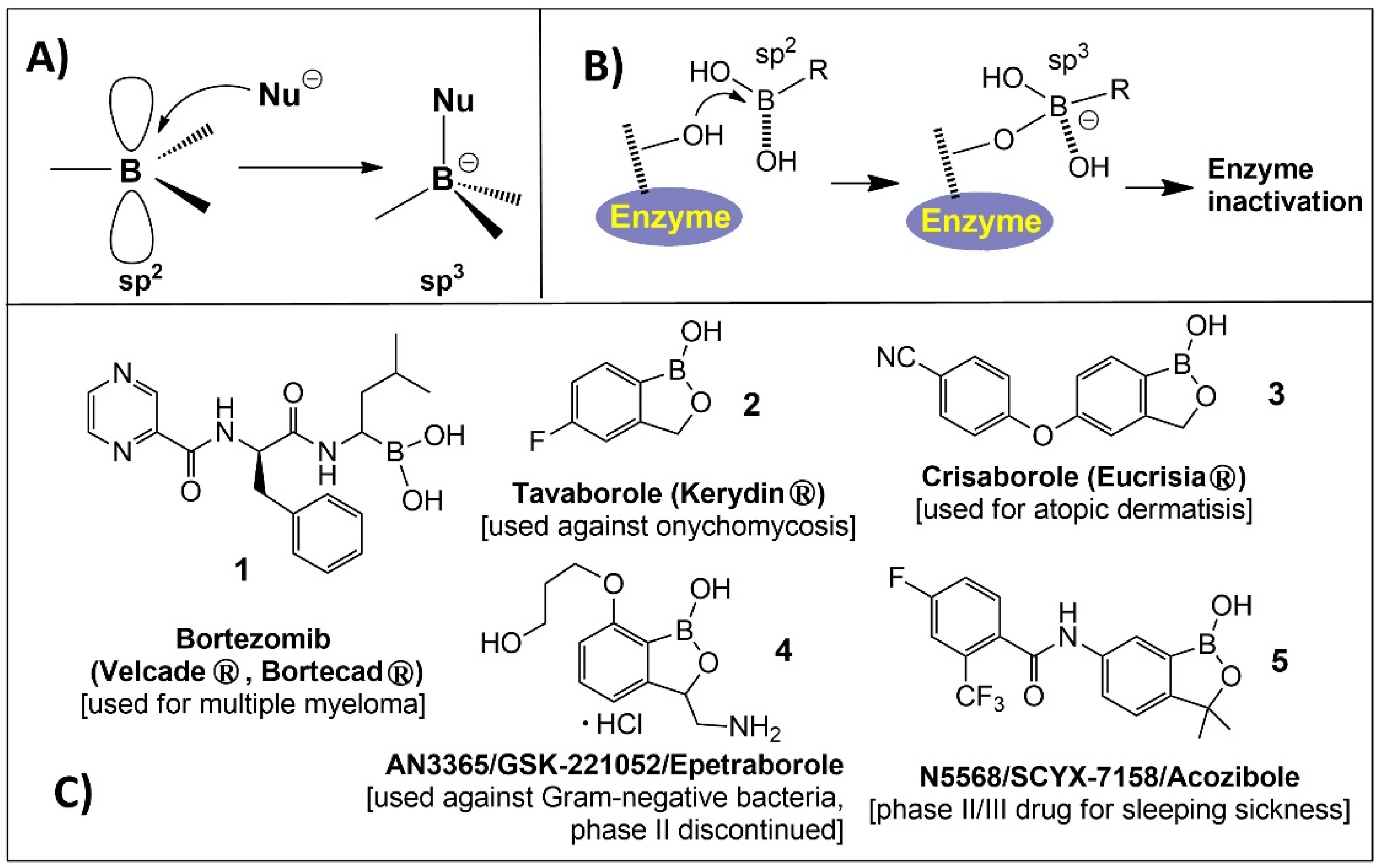{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Tuberculosis and Antifungal Activity
2.1. Benzoxaboroles
2.2. Peptidyl Boronates/Boronic Acids
2.3. Other Small Compounds of Boron (Diazoborines, Antibiotic)
3. Malaria
4. Neglected Tropical Diseases (NTD)
4.1. Trypanosomiasis
4.2. Leishmaniasis
4.3. Onchocerciasis (River Blindness) and Lymphatic Filariasis (Elephantiasis)
5. Cryptosporidiosis and Toxoplasmosis
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Mycobacterium tuberculosis | Mtb |
| H. proteasome | Human proteasome |
| MIC | Minimum inhibitory concentration |
| ACT | Artemisinin-based combination therapies |
| aaRSAmino | Acyl tRNA synthetase |
| CQ | Chloroquine |
| ADME | Absorption, distribution, metabolism, and excretion |
| SsrA-tagged protein | Caseinolytic-protease-specific degradation protein |
| RLU | Relative luminescence |
| WT cell lines | Wild-type cell lines |
| aaRS | Aminoacyl-tRNA synthetase |
| ClpP | Caseinolytic proteases |
| TB | Tuberculosis |
| Chloromethyl ketones | CMKs |
| enoyl- reductase | ENR |
| Enoyl-[acyl-carrier-protein] reductase [NADH] | InhA |
| Nicotinamide adenine dinucleotide oxidized | NAD+ |
| Nicotinamide adenine dinucleotide reduced | NADH |
| artemisinin-based combination therapies | ACT |
| Plasmodium Falciparum | P. Falciparum |
| Plasmodium falciparum 3D7 | CQ-sensitive 3D7 |
| structure−activity relationship studies | SARs |
| 3D7 | Chloroquine (CQ) sensitive P. falciparum strain |
| W2 | Chloroquine (CQ) resistant P. falciparum strain |
| D2d | Chloroquine (CQ) resistant P. falciparum strain |
| half-life | t1/2 |
| availability | F |
| WT cells | Wild-type cells |

References
- Yuan, T.; Sampson, N.S. Hit Generation in TB Drug Discovery: From Genome to Granuloma. Chem. Rev. 2018, 118, 1887–1916. [Google Scholar] [CrossRef]
- Delves, M.J.; Migue-Blanco, C.; Matthews, H.; Molina, I.; Yahiya, S.; Straschil, U.; Abraham, M.; Leon, M.L.; Fischer, O.J.; Rueda-Zubiaurre, A.; et al. A high throughtput screen for next-generation leads targeting malaria parasite transmission. Nat. Commun. 2018, 9, 3805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization (WHO). Why Are Some Tropical Diseases Called “Neglected”? Available online: http://www.who.int/features/qa/58/en/ (accessed on 10 January 2021).
- Hotez, P.J.; Aksoy, S.; Brindley, P.J.; Kamhawi, S. What constitutes a neglected tropical disease? PLoS Negl. Trop. Dis. 2020, 14, e0008001. [Google Scholar] [CrossRef] [Green Version]
- Lanata, C.F.; Fischer-Walker, C.L.; Olascoaga, A.C.; Torres, C.X.; Aryee, M.J.; Black, R.E. Global causes of diarrheal disease mortality in children <5 years of age: A systematic review. PLoS ONE 2013, 8, e72788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, B.C.; Thapa, P.; Karki, R.; Schinke, C.; Das, S.; Kambhampati, S.; Banerjee, S.K.; Veldhuizen, V.P.; Verma, A.; Weiss, L.M.; et al. Boron chemicals in diagnosis, therapeutics. Future Med. Chem. 2013, 5, 653–676. [Google Scholar] [CrossRef] [Green Version]
- Smedskjaer, M.M.; Mauro, J.C.; Youngman, R.E.; Hogue, C.L.; Potuzak, M.; Yue, Y. Topological principles of borosilicate glass chemistry. J. Phys. Chem. B. 2011, 115, 12930–12946. [Google Scholar] [CrossRef] [PubMed]
- Hosmane, N.S.; Maguire, J.A.; Zhu, Y.; Takagaki, M. Boron and Gadolinium Neutron Capture Therapy for Cancer Treatment; World Scientific Publishing Co. Pte. Ltd.: Singapore, 2012. [Google Scholar]
- Wisniak, J. Borax, boric acid, boron—From exotic to commodity. Indian J. Chem. Technol. 2005, 12, 488–500. [Google Scholar]
- Zhu, Y.; Lin, X.; Xie, H.; Li, J.; Hosmane, N.S.; Zhang, Y. The current status and perspectives of delivery strategy for boronbased drugs. Curr. Med. Chem. 2019, 26, 5019–5035. [Google Scholar]
- Scholz, M.; Hey-Hawkins, E. Carbaboranes as pharmacophores: Properties, synthesis, application strategies. Chem. Rev. 2011, 111, 7035–7062. [Google Scholar] [CrossRef]
- Baker, S.J.; Tomsho, J.W.; Benkovic, S.J. Boron-containing inhibitors of synthetases. Chem. Soc. Rev. 2011, 40, 4279–4285. [Google Scholar] [CrossRef]
- Baker, S.J.; Ding, C.Z.; Akama, T.; Zhang, Y.-K.; Xia, Y.; Hernandez, V. Therapeutic potential of boron containing compounds. Future Med. Chem. 2009, 1, 1275–1288. [Google Scholar] [CrossRef] [Green Version]
- Adams, J.; Behnke, M.; Chen, S.W.; Cruickshank, A.A.; Dick, L.R.; Grenier, L.; Klunder, J.M.; Ma, Y.T.; Plamondon, L.; Stein, R.L. Potent, selective inhibitors of the proteasome: Dipeptidyl boronic acids. Bioorg. Med. Chem. Lett. 1998, 8, 333–338. [Google Scholar] [CrossRef]
- Adamczyk-Woźniak, A.; Borys, K.M.; Sporzyński, A. Recent developments in the chemistry and biological applications of benzoxaboroles. Chem. Rev. 2015, 115, 5224–5247. [Google Scholar] [CrossRef]
- Anacor Pharmaceuticals. FDA Approves Anacor Pharmaceuticals’ KERYDIN® (Tavaborole) Topical Solution, 5% for the Treatment of Onychomycosis of the Toenails. Press Release, 8 July 2014. [Google Scholar]
- Hoy, S.M. Crisaborole ointment 2%: A review in mild to moderate atopic dermatitis. Am. J. Clin. Dermatol. 2017, 18, 837–843. [Google Scholar] [CrossRef] [PubMed]
- Scifinder, Version 2019; Chemical Abstracts Service: Columbus, OH, USA, 2019; (accessed on 15 March 2019).
- Zhang, P.; Ma, S. Recent development of leucyl-tRNA synthetase inhibitors as antimicrobial agents. Med. Chem. Commun. 2019, 10, 1329–1341. [Google Scholar] [CrossRef]
- Yang, F.; Zhu, M.; Zhang, J.; Zhou, H. Synthesis of biologically active boron-containing compounds. Med. Chem. Comm. 2018, 9, 201. [Google Scholar] [CrossRef] [PubMed]
- Dembitsky, V.M.; Quntar, A.A.A.; Srebnik, M. Natural and Synthetic Small Boron-Containing Molecules as Potential Inhibitors of Bacterial and Fungal Quorum Sensing. Chem. Rev. 2011, 111, 209–237. [Google Scholar] [CrossRef]
- Silva, M.P.; Saraiva, L.; Pinto, M.; Sousa, M.E. Boronic Acids and Their Derivatives in Medicinal Chemistry: Synthesis and Biological Applications. Molecules 2020, 25, 4323. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://apps.who.int/iris/bitstream/handle/10665/336069/9789240013131-eng.pdf (accessed on 11 February 2021).
- Ernst, J.D. The immunological life cycle of tuberculosis. Nat. Rev. Immunol. 2012, 12, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Vjecha, M.J.; Tiberi, S.; Zumla, A. Accelerating the development of therapeutic strategies for drug-resistant tuberculosis. Nat. Rev. Drug. Discov. 2018, 17, 607–608. [Google Scholar] [CrossRef] [PubMed]
- Chetty, S.; Ramesh, M.; Singh-Pillay, A.; Soliman, M.E. Recent advancements in the development of anti-tuberculosis drugs. Bioorg. Med. Chem. Lett. 2017, 27, 370–386. [Google Scholar] [CrossRef]
- Torssell, K. Bromination of tolylboronic acids according to Wohl-Ziegler. Ark. Kemi. 1957, 10, 507–511. [Google Scholar]
- Adamczyk-Wozniak, A.; Cyranski, M.K.; Zubrowska, A.; Sporzynski, A. Benzoxaboroles—Old compounds with new applications. J. Organomet. Chem. 2009, 694, 3533–3541. [Google Scholar] [CrossRef]
- Mereddy, G.R.; Chakradhar, A.; Rutkoski, R.M.; Jonnalagadda, S.C. Benzoboroxoles: Synthesis and applications in medicinal chemistry. J. Organomet. Chem. 2018, 865, 12–22. [Google Scholar] [CrossRef]
- Nocentini, A.; Supuran, C.T.; Winum, J.Y. Benzoxaborole compounds for therapeutic uses: A patent review (2010–2018). Expert. Opin. Ther. Pat. 2018, 28, 493–504. [Google Scholar] [CrossRef]
- Zhang, J.; Zhu, M.; Lin, Y.; Zhou, H. The synthesis of benzoxaboroles and their applications in medicinal chemistry. Sci. China Chem. 2013, 56, 1372–1381. [Google Scholar] [CrossRef]
- Rock, F.L.; Mao, W.; Yaremchuk, A.; Tukalo, M.; Crepin, T.; Zhou, H.; Zhang, Y.K.; Hernandez, V.; Akama, T.; Baker, S.J.; et al. An antifungal agent inhibits an aminoacyl-tRNA synthetase by trapping tRNA in the editing site. Science 2007, 316, 1759–1761. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; Simpson, F.C. New therapeutic options for onychomycosis. Expert. Opin. Pharmacother. 2012, 13, 1131–1142. [Google Scholar] [CrossRef]
- Sharma, N.; Sharma, D. An upcoming drug for onychomycosis: Tavaborole. J. Pharmacol. Pharmacother. 2015, 6, 236–239. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.Y.; Kim, S.; Kim, M.H. Aminoacyl-tRNA synthetases, therapeutic targets for infectious diseases. Biochem. Pharmacol. 2018, 154, 424–434. [Google Scholar] [CrossRef]
- Seiradake, E.; Mao, W.; Hernandez, V.; Baker, S.J.; Plattner, J.J.; Alley, M.R.; Cusack, S. Crystal structures of the human and fungal cytosolic Leucyl-tRNA synthetase editing domains: A structural basis for the rational design of antifungal benzoxaboroles. J. Mol. Biol. 2009, 390, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.A.; Arora, K.; Gurrapu, S.; Jonnalagadda, S.K.; Nelson, G.L.; Kiprof, P.; Jonnalagadda, S.C.; Mereddy, V.R. Synthesis and evaluation of functionalized benzoboroxoles as potential anti-tuberculosis agents. Tetrahedron 2016, 72, 3795–3801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palencia, A.; Li, X.; Bu, W.; Choi, W.; Ding, C.Z.; Easom, E.E.; Feng, L.; Hernandez, V.; Houston, P.; Liu, L.; et al. Discovery of Novel Oral Protein Synthesis Inhibitors of Mycobacterium Tuberculosis That Target Leucyl-tRNA Synthetase. Antimicrob. Agents Chemother. 2016, 60, 6271–6280. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Liao, J.; Zhu, B.; Wang, E.D.; Ding, J. Crystal structures of the editing domain of Escherichia coli leucyl-tRNA synthetase and its complexes with Met and Ile reveal a lock-and-key mechanism for amino acid discrimination. Biochem. J. 2006, 394, 399–407. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Hernandez, V.; Rock, F.L.; Choi, W.; Mak, Y.S.L.; Mohan, M.; Mao, W.; Zhou, Y.; Easom, E.E.; Plattner, J.J.; et al. Discovery of a Potent and Specific M. tuberculosis Leucyl-tRNA Synthetase Inhibitor: (S)-3-(Aminomethyl)-4-chloro-7-(2-hydroxyethoxy)benzo[c][1,2]oxaborol-1(3H)-ol (GSK656). J. Med. Chem. 2017, 60, 8011–8026. [Google Scholar] [CrossRef]
- Vshyvenko, S.; Clapson, M.L.; Suzuki, I.; Hall, D.G. Characterization of the dynamic equilibrium between closed and open forms of the benzoxaborole pharmacophore. ACS Med. Chem. Lett. 2016, 7, 1097–1101. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://clinicaltrials.gov/ct2/show/NCT03075410 (accessed on 10 March 2021).
- Patel, N.; O’Malley, T.; Zhang, Y.K.; Xia, Y.; Sunde, B.; Flint, L.; Korkegian, A.; Loerger, T.R.; Sacchettini, J.; Alley, M.R.K.; et al. A Novel 6-Benzyl Ether Benzoxaborole Is Active against Mycobacterium tuberculosis in vitro. Antimicrob. Agents Chemother. 2017, 61, pii: e01205-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korkegian, A.; O’Malley, T.; Xia, Y.; Zhou, Y.; Carter, D.S.; Sunde, B.; Flint, L.; Thompson, D.; Ioerger, T.R.; Sacchettini, J.; et al. The 7-phenyl benzoxaborole series is active against Mycobacterium tuberculosis. Tuberculosis 2018, 108, 96–98. [Google Scholar] [CrossRef] [PubMed]
- Yano, T.; Rahimian, M.; Aneja, K.K.; Schechter, N.M.; Rubin, H.; Scott, C.P. Mycobacterium tuberculosis type II NADH-menaquinone oxidoreductase catalyzes electron transfer through a two-site ping-pong mechanism and has two quinone-binding sites. Biochemistry 2014, 53, 1179–1190. [Google Scholar] [CrossRef]
- Lee, A.S.; Teo, A.S.; Wong, S.Y. Novel mutations in ndh in isoniazid-resistant Mycobacterium tuberculosis isolates. Antimicrob. Agents Chemother. 2001, 45, 2157–2159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sena, F.V.; Batista, A.P.; Catarino, T.; Brito, J.A.; Archer, M.; Viertler, M.; Madl, T.; Cabrita, E.J.; Pereira, M.M. Type-II NADH:quinone oxidoreductase from Staphylococcus aureus has two distinct binding sites and is rate limited by quinone reduction. Mol. Microbiol. 2015, 98, 272–288. [Google Scholar] [CrossRef] [Green Version]
- Guy, C.S.; Murray, K.; Gibson, M.I.; Fullam, E. Dimeric benzoboroxoles for targeted activity against Mycobacterium tuberculosis. Org. Biomol. Chem. 2019, 17, 9524–9528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Wu, G.; Zhu, X.; Ma, Y.; Zhao, X.; Li, Y.; Yuan, Y.; Yang, J.; Yu, S.; Shao, F.; et al. Synthesis, in vitro and in vivo Biological Evaluation, and Comprehensive Understanding of Structure—Activity Relationships of Dipeptidyl Boronic Acid Proteasome Inhibitors Constructed from β-Amino Acids. J. Med. Chem. 2010, 53, 8619–8626. [Google Scholar] [CrossRef]
- Han, L.; Wen, Y.; Li, R.; Xu, B.; Ge, Z.; Wang, X.; Cheng, T.; Cui, J.; Li, R. Synthesis and biological activity of peptide proline-boronic acids as proteasome inhibitors. Bioorg. Med. Chem. 2017, 25, 4031–4044. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Frezza, M.; Schmitt, S.; Kanwar, J.; Dou, Q.P. Bortezomib as the first proteasome inhibitor anticancer drug: Current status and future perspectives. Curr. Cancer Drug Targets 2011, 11, 239–253. [Google Scholar] [CrossRef] [Green Version]
- Groll, M.; Berkers, C.R.; Ploegh, H.L.; Ovaa, H. Crystal structure of the boronic acid-based proteasome inhibitor bortezomib in complex with the yeast 20S proteasome. Structure 2006, 14, 451–456. [Google Scholar] [CrossRef] [Green Version]
- Bonvini, P.; Zorzi, E.; Basso, G.; Rosolen, A. Bortezomib-mediated 26S proteasome inhibition causes cell-cycle arrest and induces apoptosis in CD-30 anaplastic large cell lymphoma. Leukemia 2007, 21, 838–842. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, D.; Tian, Z.; Zhou, B.; Kuhn, D.; Orlowski, R.; Raje, N.; Richardson, P.; Anderson, K.C. In vitro and in vivo selective antitumor activity of a novel orally bioavailable proteasome inhibitor MLN9708 against multiple myeloma cells. Clin. Cancer. Res. 2011, 17, 5311–5321. [Google Scholar] [CrossRef] [Green Version]
- Brötz-Oesterhelt, H.; Sass, P. Bacterial caseinolytic proteases as novel targets for antibacterial treatment. Int. J. Med. Microbiol. 2014, 304, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Raju, R.M.; Jedrychowski, M.P.; Wei, J.; Pinkham, J.T.; Park, A.S.; O’Brien, K.; Rehren, G.; Schnappinger, D.; Gygi, S.P.; Rubin, E.J. Post-translational regulation via Clp protease is critical for survival of Mycobacterium tuberculosis. PLoS Pathog. 2014, 10, e1003994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keiler, K.C. Biology of trans-translation. Annu. Rev. Microbiol. 2008, 62, 133–151. [Google Scholar] [CrossRef]
- Raju, R.M.; Unnikrishnan, M.; Rubin, D.H.; Krishnamoorthy, V.; Kandror, O.; Akopian, T.N.; Goldberg, A.L.; Rubin, E.J. Mycobacterium tuberculosis ClpP1 and ClpP2 function together in protein degradation and are required for viability in vitro and during infection. PLoS Pathog. 2012, 8, e1002511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreira, W.; Ngan, G.J.; Low, J.L.; Poulsen, A.; Chia, B.C.; Ang, M.J.; Yap, A.; Fulwood, J.; Lakshmanan, U.; Lim, J.; et al. Target mechanism-based whole-cell screening identifies bortezomib as an inhibitor of caseinolytic protease in mycobacteria. mBio 2015, 6, e00253-15. [Google Scholar] [CrossRef] [Green Version]
- Bogyo, M.; Wang, E.W. Proteasome inhibitors: Complex tools for a complex enzyme. Curr. Top. Microbiol. Immunol. 2002, 268, 185–208. [Google Scholar] [PubMed]
- Moreira, W.; Santhanakrishnan, S.; Dymock, B.W.; Dick, T. Bortezomib Warhead-Switch Confers Dual Activity against Mycobacterial Caseinolytic Protease and Proteasome and Selectivity against Human Proteasome. Front. Microbiol. 2017, 27, 746. [Google Scholar] [CrossRef] [PubMed]
- Moreira, W.; Santhanakrishnan, S.; Ngan, G.J.Y.; Low, C.B.; Sangthongpitag, K.; Poulsen, A.; Dymock, B.W.; Dick, T. Towards Selective Mycobacterial ClpP1P2 Inhibitors with Reduced Activity against the Human Proteasome. Antimicrob. Agents Chemother. 2017, 61, e02307-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug. Deliv. Rev. 1997, 23, 3–26. [Google Scholar] [CrossRef]
- Fernandes, G.F.S.; Denny, W.A.; Dos Santos, J.L. Boron in drug design: Recent advances in the development of new therapeutic agents. Eur. J. Med. Chem. 2019, 179, 791–804. [Google Scholar] [CrossRef]
- Grassberger, M.A.; Turnowsky, F.; Hildebrandt, J. Preparation and antibacterial activities of new 1,2,3-diazaborine derivatives and analogs. J. Med. Chem. 1984, 27, 947–953. [Google Scholar] [CrossRef]
- Baldock, C.; Rafferty, J.B.; Sedelnikova, S.E.; Baker, P.J.; Stuitje, A.R.; Slabas, A.R.; Hawkes, T.R.; Rice, D.W. Mechanism of Drug Action Revealed by Structural Studies of Enoyl Reductase. Science 1996, 274, 2107–2110. [Google Scholar] [CrossRef] [Green Version]
- De Boer, G.J.; Pielage, G.J.; Nijkamp, H.J.; Slabas, A.R.; Rafferty, J.B.; Baldock, C.; Rice, D.W.; Stuitje, A.R. Molecular genetic analysis of enoyl-acyl carrier protein reductase inhibition by diazaborine. Mol. Microbiol. 1999, 31, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Zhou, Y.; Carter, D.S.; McNeil, M.B.; Choi, W.; Halladay, J.; Berry, P.W.; Mao, W.; Hernandez, V.; O’Malley, T.; et al. Discovery of a cofactor-independent inhibitor of Mycobacterium tuberculosis InhA. Life Sci. Alliance 2018, 1, e201800025. [Google Scholar] [CrossRef] [Green Version]
- Takayama, K.; Wang, C.; Besra, G.S. Pathway to synthesis and processing of mycolic acids in Mycobacterium tuberculosis. Clin. Microbiol. Rev. 2005, 18, 81–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, M.C.; Franzblau, S.G.; Martin, A.R. Syntheses and evaluation of benzodiazaborine compounds against M. tuberculosis H37Rv in vitro. Bioorg. Med. Chem. Lett. 1998, 8, 843–846. [Google Scholar] [CrossRef]
- Kanichar, D.; Roppiyakuda, L.; Kosmowska, E.; Faust, M.A.; Tran, K.P.; Chow, F.; Buglo, E.; Groziak, M.P.; Sarina, E.A.; Olmstead, M.M. Synthesis, Characterization, and Antibacterial Activity of Structurally Complex 2-Acylated 2,3,1-Benzodiazaborines and Related Compounds. Chem. Biodivers. 2014, 11, 1381–1397. [Google Scholar] [CrossRef] [PubMed]
- Hicks, J.W.; Kyle, C.B.; Vogels, C.M.; Wheaton, S.L.; Baerlocher, F.J.; Decken, A.; Westcott, S.A. Synthesis, characterization, and antifungal activity of boron-containing thiosemicarbazones. Chem. Biodivers. 2008, 5, 2415–2422. [Google Scholar] [CrossRef]
- Rajan, R.; Budihal, V.; Renold, P.; Stierli, D. Preparation of Boron Containing Fungicides and Their Use in Compns. and Methods for the Control and/or Prevention of Microbial Infection in Plants. MedChemComm 2018, 9, 201–211. [Google Scholar]
- Moreira, W.; Aziz, D.B.; Dick, T. Boromycin Kills Mycobacterial Persisters without Detectable Resistance. Front. Microbiol. 2016, 7, 199. [Google Scholar] [CrossRef]
- Available online: https://www.who.int/publications/i/item/9789240015791 (accessed on 27 April 2021).
- Ouji, M.; Augereau, J.M.; Paloque, L.; Benoit-Vical, F. Plasmodium falciparum resistance to artemisinin-based combination therapies: A sword of Damocles in the path toward malaria elimination. Parasite 2018, 25, 24. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://www.cdc.gov/malaria/about/biology/index.html (accessed on 10 February 2021).
- Artesunate. Available online: http://apps.who.int/medicinedocs/en/d/Jh2922e/2.5.11.html (accessed on 10 February 2021).
- Sibley, C.H. Understanding artemisinin resistance. Science 2015, 347, 373–374. [Google Scholar] [CrossRef]
- Heller, L.E.; Roepe, P.D. Artemisinin-Based Antimalarial Drug Therapy: Molecular Pharmacology and Evolving Resistance. Trop. Med. Infect. Dis. 2019, 4, 89. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, R.T.; Plattner, J.J.; Keenan, M. Boron-based drugs as antiprotozoals. Curr. Opin. Infect. Dis. 2011, 24, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-K.; Plattner, J.J.; Freund, Y.R.; Easom, E.E.; Zhou, Y.; Gut, J.; Rosenthal, P.J.; Waterson, D.; Gamo, F.; Angulo-Barturen, I.; et al. Synthesis and structure-activity relationships of novel benzoxaboroles as a new class of antimalarial agents. Bioorg. Med. Chem. Lett. 2011, 21, 644–651. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-K.; Plattner, J.J.; Easom, E.E.; Waterson, D.; Ge, M.; Li, Z.; Li, L.; Jian, Y. An efficient synthesis for a new class antimalarial agent, 7-(2-carboxyethyl)-1,3-dihydro-1-hydroxy-2,1-benzoxaborole. Tetrahedron Lett. 2011, 52, 3909–3911. [Google Scholar] [CrossRef]
- Zhang, Y.-K.; Plattner, J.J.; Freund, Y.R.; Easom, E.E.; Zhou, Y.; Huchen, L.Y.; Zhou, H.; Waterson, D.; Gamo, F.; Sanz, L.M.; et al. Benzoxaborole antimalarial agents. Part 2: Discovery of fluoro-substituted 7-(2-carboxyethyl)-1,3-dihydro-1-hydroxy-2,1-benzoxaboroles. Bioorg. Med. Chem. Lett. 2012, 22, 1299–1307. [Google Scholar] [CrossRef]
- Zhang, Y.-K.; Plattner, J.J.; Easom, E.E.; Jacobs, R.T.; Guo, D.; Sanders, V.; Freund, Y.R.; Campo, B.; Rosenthal, P.J.; Bu, W.; et al. Benzoxaborole antimalarial agents. Part 4. Discovery of potent 6-(2-(alkoxycarbonyl)pyrazinyl-5-oxy)-1,3-dihydro-1-hydroxy-2,1-benzoxaboroles. J. Med. Chem. 2015, 58, 5344–5354. [Google Scholar] [CrossRef]
- Zhang, Y.-K.; Plattner, J.J.; Easom, E.E.; Jacobs, R.T.; Guo, D.; Freund, Y.R.; Berry, P.; Ciaravino, V.; Erve, J.C.L.; Rosenthal, P.J.; et al. Benzoxaborole Antimalarial Agents. Part 5. Lead Optimization of Novel Amide Pyrazinyloxy Benzoxaboroles and Identification of a Preclinical Candidate. J. Med. Chem. 2017, 60, 5889–5908. [Google Scholar] [CrossRef]
- Hu, Q.H.; Liu, R.J.; Fang, Z.P.; Zhang, J.; Ding, Y.Y.; Tan, M.; Wang, M.; Pan, W.; Zhou, H.C.; Wang, E.D. Discovery of a potent benzoxaborole-based anti-pneumococcal agent targeting leucyl-tRNA synthetase. Sci. Rep. 2013, 3, 2475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonoiki, E.; Palencia, A.; Guo, D.; Ahyong, V.; Dong, C.; Li, X.; Hernandez, V.S.; Zhang, Y.K.; Choi, W.; Gut, J.; et al. Antimalarial Benzoxaboroles Target Plasmodium falciparum Leucyl-tRNA Synthetase. Antimicrob. Agents Chemother. 2016, 60, 4886–4895. [Google Scholar] [CrossRef] [Green Version]
- Hernandez, V.S.; Ding, C.; Plattner, J.J.; Alley, M.R.K.; Rock, F.; Zhang, S.; Easom, E.; Li, X.; Zhou, D. Benzoxaborole Derivatives for Treating Bacterial Infections. WO2012033858A2, 15 March 2012. [Google Scholar]
- Sonoiki, E.; Ng, C.L.; Lee, M.C.; Guo, D.; Zhang, Y.K.; Zhou, Y.; Alley, M.R.K.; Ahyong, V.; Sanz, L.M.; Lafuente-Monasterio, M.J.; et al. A potent antimalarial benzoxaborole targets a Plasmodium falciparum cleavage and polyadenylation specificity factor homologue. Nat. Commun. 2017, 8, 14574. [Google Scholar] [CrossRef]
- Mandel, C.R.; Kaneko, S.; Zhang, H.; Gebauer, D.; Vethantham, V.; Manley, J.L.; Tong, L. Polyadenylation factor CPSF-73 is the pre-mRNA 3′-end-processing endonuclease. Nature 2006, 444, 953–956. [Google Scholar] [CrossRef]
- The WHO List of 17 Neglected Tropical Diseases. Available online: https://www.who.int/teams/control-of-neglected-tropical-diseases/overview/progress-dashboard-2011-2020 (accessed on 28 April 2021).
- Peacock, L.; Cook, S.; Ferris, V.; Bailey, M.; Gibson, W. The life cycle of Trypanosoma (Nannomonas) congolense in the tsetse fly. Parasites Vectors 2012, 5, 109. [Google Scholar] [CrossRef] [Green Version]
- Ding, D.; Zhao, Y.; Meng, Q.; Xie, D.; Nare, B.; Chen, D.; Bacchi, C.J.; Yarlett, N.; Zhang, Y.-K.; Hernandez, V.; et al. Discovery of Novel Benzoxaborole-Based Potent Antitrypanosomal Agents. ACS Med. Chem. Lett. 2010, 1, 165–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nare, B.; Wring, S.; Bacchi, C.; Beaudet, B.; Bowling, T.; Brun, R.; Chen, D.; Ding, C.; Freund, Y.; Gaukel, E.; et al. Discovery of Novel Orally Bioavailable Oxaborole 6-Carboxamides That Demonstrate Cure in a Murine Model of Late-Stage Central Nervous System African Trypanosomiasis. Antimicrobic. Agents Chemother. 2010, 54, 4379–4388. [Google Scholar] [CrossRef] [Green Version]
- Doan, K.M.; Wring, S.A.; Shampine, L.J.; Jordan, K.H.; Bishop, J.P.; Kratz, J.; Yang, E.; Serabjit-Singh, C.J.; Adkison, K.K.; Polli, J.W. Steady-state brain concentrations of antihistamines in rats: Interplay of membrane permeability, P-glycoprotein efflux and plasma protein binding. Pharmacology 2004, 72, 92–98. [Google Scholar] [CrossRef]
- Jacobs, R.T.; Plattner, J.J.; Nare, B.; Wring, S.A.; Chen, D.; Freund, Y.; Gaukel, E.G.; Orr, M.D.; Perales, J.B.; Jenks, M.; et al. Benzoxaboroles: A new class of potential drugs for human African trypanosomiasis. Future Med. Chem. 2011, 3, 1259–1278. [Google Scholar] [CrossRef]
- Jacobs, R.T.; Nare, B.; Wring, S.A.; Orr, M.D.; Chen, D.; Sligar, J.M.; Jenks, M.X.; Noe, R.A.; Bowling, T.S.; Mercer, L.T.; et al. SCYX-7158, an orally-active benzoxaborole for the treatment of stage 2 human African trypanosomiasis. PLoS Negl. Trop. Dis. 2011, 5, e1151. [Google Scholar] [CrossRef] [Green Version]
- Drugs for Neglected Diseases Initiative (Dndi). Dndi Announces Successful Completion of SCYX-7158 Phase I Study for Treatment of Sleeping Sicknes. 2015. Available online: https://www.news-medical.net/news/20150909/DNDi-announces-successful-completion-of-SCYX-7158-Phase-I-study-for-treatment-of-sleeping-sickness.aspx (accessed on 11 March 2021).
- Available online: https://clinicaltrials.gov/ct2/show/NCT03087955 (accessed on 20 April 2021).
- Aponte, J.C.; Verastegui, M.; Malaga, E.; Zimic, M.; Quiliano, M.; Vaisberg, A.J.; Gilman, R.H.; Hammond, G.B. Synthesis, cytotoxicity, and anti-Trypanosoma cruzi activity of new chalcones. J. Med. Chem. 2008, 51, 6230–6234. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Z.; Wang, Q.; Zhang, F.; Wang, Z.; Bowling, T.; Nare, B.; Jacobs, R.T.; Zhang, J.; Ding, D.; Liu, Y.; et al. Chalcone–Benzoxaborole Hybrid Molecules as Potent Antitrypanosomal Agents. J. Med. Chem. 2012, 55, 3553–3557. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Zhang, J.; Meng, Q.; Nare, B.; Jacobs, R.T.; Zhou, H. Novel pyrrolobenzoxaboroles: Design, synthesis, and biological evaluation against Trypanosoma brucei. Eur. J. Med. Chem. 2014, 81, 59–75. [Google Scholar] [CrossRef] [PubMed]
- Gumbo, M.; Beteck, R.M.; Mandizvo, T.; Seldon, R.; Warner, D.F.; Hoppe, H.C.; Isaacs, M.; Laming, D.; Tam, C.C.; Cheng, L.W.; et al. Cinnamoyl-Oxaborole Amides: Synthesis and Their in Vitro Biological Activity. Molecules. 2018, 23, 2038. [Google Scholar] [CrossRef] [Green Version]
- Ding, D.; Meng, Q.; Gao, G.; Zhao, Y.; Wang, Q.; Nare, B.; Jacobs, R.T.; Rock, F.; Alley, M.R.; Plattner, J.J.; et al. Design, synthesis, and structure-activity relationship of Trypanosoma brucei leucyl-tRNA synthetase inhibitors as antitrypanosomal agents. J. Med. Chem. 2011, 54, 1276–1287. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.who.int/news-room/fact-sheets/detail/leishmaniasis (accessed on 29 April 2021).
- Torres-Guerrero, E.; Quintanilla-Cedillo, M.R.; Ruiz-Esmenjaud, J.; Arenas, R. Leishmaniasis: A review. F1000Res. 2017, 6, 750. [Google Scholar] [CrossRef] [PubMed]
- Sundar, S.; Pandey, K.; Thakur, C.P.; Jha, T.K.; Das, V.N.R.; Verma, N.; Lal, C.S.; Verma, D.; Alam, S.; Das, P. Efficacy and safety of amphotericin B emulsion versus liposomal formulation in Indian patients with visceral leishmaniasis: A randomized, open-label study. PLoS Negl. Trop. Dis. 2014, 8, e3169. [Google Scholar] [CrossRef]
- Tiwari, N.; Gedda, M.R.; Tiwari, V.K.; Singh, S.P.; Singh, R.K. Limitations of current therapeutic options, possible drug targets and scope of natural products in control of leishmaniasis. Mini. Rev. Med. Chem. 2018, 18, 26–41. [Google Scholar] [CrossRef]
- Pandey, B.D.; Pandey, K.; Kaneko, O.; Yanagi, T.; Hirayama, K. Relapse of visceral leishmaniasis after miltefosine treatment in a Nepalese patient. Am. J. Trop. Med. Hyg. 2009, 80, 580–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WHO. Accelerating work to overcome the global impact of neglected tropical diseases: A roadmap for implementation. WHO/HTM/NTD/2012. World Health Organization, Geneva, Switzerland. 2012. Available online: http://www.who.int/neglected_diseases/NTD_RoadMap_2012_Fullversion.pdf (accessed on 22 February 2021).
- Manhas, R.; Tandon, S.; Sen, S.S.; Tiwari, N.; Munde, M.; Madhubala, R. Leishmania donovani Parasites Are Inhibited by the Benzoxaborole AN2690 Targeting Leucyl-tRNA Synthetase. Antimicrob. Agents Chemother. 2018, 62, e00079-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermelho, A.B.; Capaci, G.R.; Rodrigues, I.A.; Cardoso, V.S.; MariaMazotto, A.; Supuran, C.T. Carbonic anhydrases from Trypanosoma and Leishmania as anti-protozoan drug targets. Bioorg. Med. Chem. 2017, 25, 1543–1555. [Google Scholar] [CrossRef] [PubMed]
- Capasso, C.; Supuran, C.T. Bacterial, fungal and protozoan carbonic anhydrases as drug targets. Expert Opin. Ther. Targets 2015, 19, 1689–1704. [Google Scholar] [CrossRef] [PubMed]
- Nocentini, A.; Cadoni, R.; Dumy, P.; Supuran, C.T.; Winum, J.-Y. Carbonic anhydrases from Trypanosoma cruzi and Leishmania donovani chagasi are inhibited by benzoxaboroles. Enzyme Inhib. Med. Chem. 2018, 33, 286–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Bocxlaer, K.; Gaukel, E.; Hauser, D.; Park, S.H.; Schock, S.; Yardley, V.; Randolph, R.; Plattner, J.J.; Merchant, T.; Croft, S.L.; et al. Topical Treatment for Cutaneous Leishmaniasis: Dermato-Pharmacokinetic Lead Optimization of Benzoxaboroles. Antimicrob. Agents Chemother. 2018, 62, e02419-17. [Google Scholar] [CrossRef] [Green Version]
- Available online: https://www.who.int/blindness/partnerships/onchocerciasis_disease_information/en/ (accessed on 21 April 2021).
- Available online: https://www.who.int/lymphatic_filariasis/en/ (accessed on 21 April 2021).
- Kavanagh, F.; Hervey, A.; Robbins, W.J. Antibiotic substances from basidiomycetes: IX. Drosophila subtarata. (batsch ex fr.) quel. Proc. Natl. Acad. Sci. USA 1952, 38, 555–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlunzen, F.; Pyetan, E.; Fucini, P.; Yonath, A.; Harms, J.M. Inhibition of peptide bond formation by pleuromutilins: The structure of the 50S ribosomal subunit from Deinococcus radiodurans in complex with tiamulin. Mol. Microbiol. 2004, 54, 1287–1294. [Google Scholar] [CrossRef]
- Jacobs, R.T.; Lunde, C.S.; Freund, Y.R.; Hernandez, V.; Li, X.; Xia, Y.; Carter, D.S.; Berry, P.W.; Halladay, J.; Rock, F.; et al. Boron-Pleuromutilins as Anti-Wolbachia Agents with Potential for Treatment of Onchocerciasis and Lymphatic Filariasis. J. Med. Chem. 2019, 62, 2521–2540. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.-K.; Zhou, H.; Ding, C.; Plattner, J.J.; Freund, Y. Boron-Containing Small Molecules as Antihelminth Agents. WO201106, 26 May 2011. [Google Scholar]
- Available online: https://en.wikipedia.org/wiki/Cryptosporidiosis (accessed on 29 April 2021).
- WHO. World Health Organization. 2018. Available online: https://www.who.int/water_sanitation_health/gdwqrevision/cryptodraft2.pdf (accessed on 1 March 2021).
- Dubey, J.P. Chapter 1—The History and Life Cycle of Toxoplasma Gondii in Toxoplasma Gondii, 2nd ed.; Weiss, L.M., Ed.; Academic Press: Cambridge, MA, USA, 2014; pp. 1–17. [Google Scholar]
- Lv, P.-C.; Zhu, H.-L. Aminoacyl-tRNA synthetase inhibitors as potent antibacterials. Curr. Med. Chem. 2012, 19, 3550–3563. [Google Scholar] [CrossRef] [PubMed]
- Palencia, A.; Liu, R.J.; Lukarska, M.; Gut, J.; Bougdour, A.; Touquet, B.; Wang, E.D.; Li, X.; Alley, M.R.; Freund, Y.R.; et al. Cryptosporidium and Toxoplasma Parasites Are Inhibited by a Benzoxaborole Targeting Leucyl-tRNA Synthetase. Antimicrob. Agents Chemother. 2016, 60, 5817–5827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prudêncio, M.; Costa, J.C. Research funding after COVID-19. Nat. Microbiol. 2020, 5, 986. [Google Scholar] [CrossRef]
- Burki, T.K. Cuts in cancer research funding due to COVID-19. Lancet Oncol. 2021, 22, E6. [Google Scholar] [CrossRef]













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coghi, P.S.; Zhu, Y.; Xie, H.; Hosmane, N.S.; Zhang, Y. Organoboron Compounds: Effective Antibacterial and Antiparasitic Agents. Molecules 2021, 26, 3309. https://doi.org/10.3390/molecules26113309
Coghi PS, Zhu Y, Xie H, Hosmane NS, Zhang Y. Organoboron Compounds: Effective Antibacterial and Antiparasitic Agents. Molecules. 2021; 26(11):3309. https://doi.org/10.3390/molecules26113309
Chicago/Turabian StyleCoghi, Paolo Saul, Yinghuai Zhu, Hongming Xie, Narayan S. Hosmane, and Yingjun Zhang. 2021. "Organoboron Compounds: Effective Antibacterial and Antiparasitic Agents" Molecules 26, no. 11: 3309. https://doi.org/10.3390/molecules26113309