
Photoredox Catalyzed Dealkylative Aromatic Halogen Substitution with Tertiary Amines

Abstract
:1. Introduction
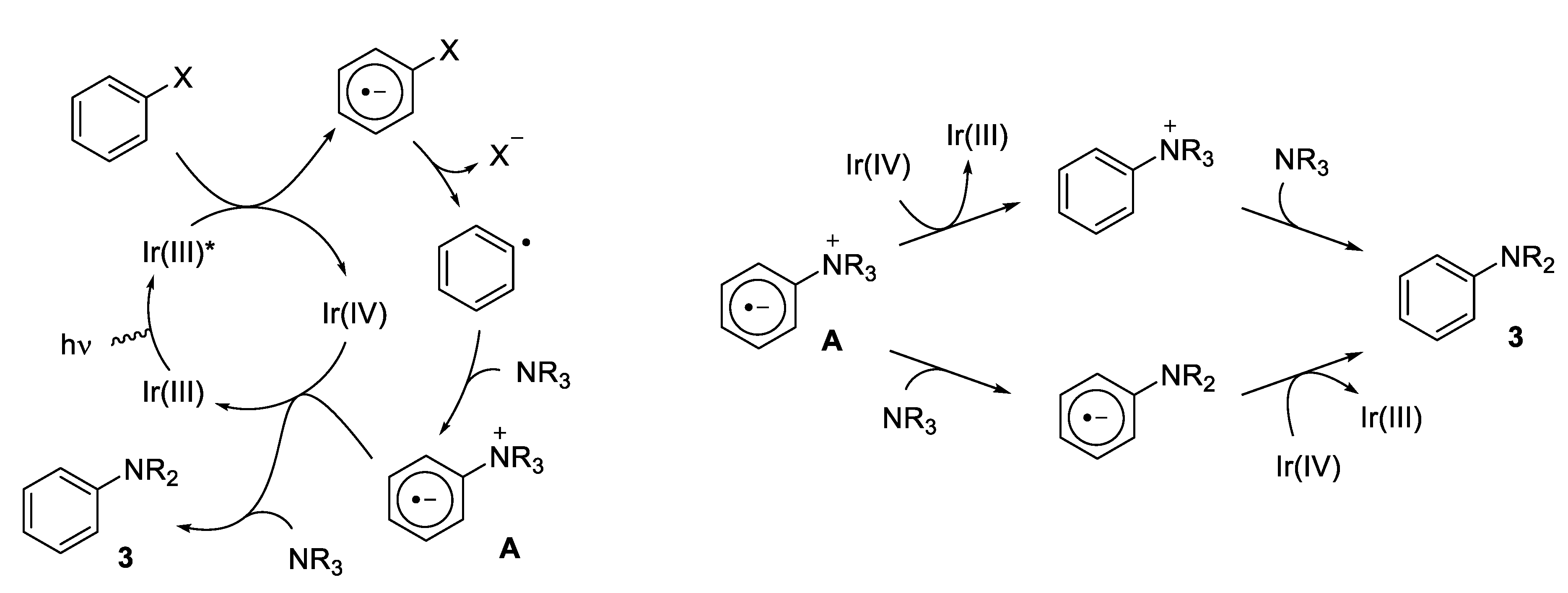
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Reactions of Aryl Halides with Amines
3.2.1. Reaction of 2,4-Dinitrofluorobenzene with Tertiary Amines. Synthesis of Amines 3. General Procedure 1 (GP 1)
3.2.2. Reactions of Aryl Halides with Tertiary Amines. Synthesis of Amines 4. General Procedure 2 (GP 2)
3.2.3. Mechanistic Experiment
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Terrier, F. Modern Nucleophilic Aromatic Substitution; Wiley-VCH: Weinheim, Germany, 2013. [Google Scholar]
- Caron, S.; McInturff, E. Nucleophilic aromatic substitution. In Practical Synthetic Organic Chemistry; Caron, S., Ed.; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2020; pp. 231–246. [Google Scholar]
- Sandford, G. Perfluoroheteroaromatic chemistry: Multifunctional systems from perfluorinated heterocycles by nucleophilic aromatic substitution processes. Top. Heterocycl. Chem. 2012, 27, 1–32. [Google Scholar]
- Langlois, B.; Gilbert, L.; Forat, G.; Jean-Roger, D.; Serge, R. Fluorination of aromatic compounds by halogen exchange with fluoride anions (‘‘halex’’ reaction). In Industrial Chemistry Library; Desmurs, J.-R., Ratton, S., Eds.; Elsevier: Amsterdam, The Netherlands, 1996; Volume 8, pp. 244–292. [Google Scholar]
- Hartwig, J.F. Transition metal catalyzed synthesis of arylamines and aryl ethers from aryl halides and triflates: Scope and mechanism. Angew. Chem. Int. Ed. 1998, 37, 2047–2067. [Google Scholar] [CrossRef]
- Yang, B.H.; Buchwald, S.L. Palladium-catalyzed amination of aryl halides and sulfonates. J. Organomet. Chem. 1999, 576, 125–146. [Google Scholar] [CrossRef]
- Buchwald, S.L.; Mauger, C.; Mignani, G.; Scholz, U. Industrial-scale palladium-catalyzed coupling of aryl halides and amines—A personal account. Adv. Synth. Catal. 2006, 348, 23–39. [Google Scholar] [CrossRef]
- Sambiagio, C.; Marsden, S.P.; Blacker, A.J.; McGowan, P.C. Copper catalysed Ullmann type chemistry: From mechanistic aspects to modern development. Chem. Soc. Rev. 2014, 43, 3525–3550. [Google Scholar] [CrossRef] [PubMed]
- Walsh, K.; Sneddon, H.F.; Moody, C.J. Amination of heteroaryl chlorides: Palladium catalysis or SNAr in green solvents? ChemSusChem 2013, 6, 1455–1460. [Google Scholar] [CrossRef] [Green Version]
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W.C. Visible light photoredox catalysis with transition metal complexes: Applications in organic synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marzo, L.; Pagire, S.K.; Reiser, O.; König, B. Visible-light photocatalysis: Does it make a difference in organic synthesis? Angew. Chem. Int. Ed. 2018, 57, 10034–10072. [Google Scholar] [CrossRef]
- Majek, M.; Jacobi von Wangelin, A. Mechanistic perspectives on organic photoredox catalysis for aromatic substitutions. Acc. Chem. Res. 2016, 49, 2316–2327. [Google Scholar] [CrossRef]
- Tay, N.E.S.; Nicewicz, D.A. Cation radical accelerated nucleophilic aromatic substitution via organic photoredox catalysis. J. Am. Chem. Soc. 2017, 139, 16100–16104. [Google Scholar] [CrossRef]
- Pistritto, V.A.; Schutzbach-Horton, M.E.; Nicewicz, D.A. Nucleophilic aromatic substitution of unactivated fluoroarenes enabled by organic photoredox catalysis. J. Am. Chem. Soc. 2020, 142, 17187–17194. [Google Scholar] [CrossRef] [PubMed]
- Venditto, N.J.; Nicewicz, D.A. Cation radical-accelerated nucleophilic aromatic substitution for amination of alkoxyarenes. Org. Lett. 2020, 22, 4817–4822. [Google Scholar] [CrossRef] [PubMed]
- Holmberg-Douglas, N.; Nicewicz, D.A. Arene cyanation via cation-radical accelerated-nucleophilic aromatic substitution. Org. Lett. 2019, 21, 7114–7118. [Google Scholar] [CrossRef]
- Tay, N.E.S.; Chen, W.; Levens, A.; Pistritto, V.A.; Huang, Z.; Wu, Z.; Li, Z.; Nicewicz, D.A. 19F- and 18F-arene deoxyfluorination via organic photoredox-catalysed polarity-reversed nucleophilic aromatic substitution. Nat. Catal. 2020, 3, 734–742. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Li, J.; Huang, C.-Y.; Li, C.-J. Aromatic chemistry in the excited state: Facilitating metal-free substitutions and cross-couplings. Angew. Chem. Int. Ed. 2020, 59, 1786–1796. [Google Scholar] [CrossRef] [PubMed]
- Cowper, N.G.W.; Chernowsky, C.P.; Williams, O.P.; Wickens, Z.K. Potent reductants via electron-primed photoredox catalysis: Unlocking aryl chlorides for radical coupling. J. Am. Chem. Soc. 2020, 142, 2093–2099. [Google Scholar] [CrossRef]
- Corcoran, E.B.; Pirnot, M.T.; Lin, S.; Dreher, S.D.; DiRocco, D.A.; Davies, I.W.; Buchwald, S.L.; MacMillan, D.W.C. Aryl amination using ligand-free Ni(II) salts and photoredox catalysis. Science 2016, 353, 279–283. [Google Scholar] [CrossRef] [Green Version]
- Zuo, Z.; Cong, H.; Li, W.; Choi, J.; Fu, G.C.; MacMillan, D.W.C. Enantioselective decarboxylative arylation of o±-amino acids via the merger of photoredox and nickel catalysis. J. Am. Chem. Soc. 2016, 138, 1832–1835. [Google Scholar] [CrossRef] [Green Version]
- Lavagnino, M.N.; Liang, T.; MacMillan, D.W.C. HARC as an open-shell strategy to bypass oxidative addition in Ullmann–Goldberg couplings. Proc. Natl. Acad. Sci. USA 2020, 117, 21058–21064. [Google Scholar] [CrossRef]
- Kim, K.; Hong, S.H. Photoinduced Copper(I)-catalyzed cyanation of aromatic halides at room temperature. Adv. Synth. Catal. 2017, 359, 2345–2351. [Google Scholar] [CrossRef]
- Arora, A.; Weaver, J.D. Visible Light photocatalysis for the generation and use of reactive azolyl and polyfluoroaryl intermediates. Acc. Chem. Res. 2016, 49, 2273–2283. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Fennell, C.J.; Weaver, J.D. Photocatalyst size controls electron and energy transfer: Selectable E/Z isomer synthesis via C–F alkenylation. Chem. Sci. 2016, 7, 6796–6802. [Google Scholar] [CrossRef] [Green Version]
- Meyer, A.U.; Slanina, T.; Yao, C.-J.; Koenig, B. Metal-free perfluoroarylation by visible light photoredox catalysis. ACS Catal. 2016, 6, 369–375. [Google Scholar] [CrossRef]
- Senaweera, S.; Weaver, J.D. Dual C–F, C–H Functionalization via photocatalysis: Access to multifluorinated biaryls. J. Am. Chem. Soc. 2016, 138, 2520–2523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, W.; Zou, R.; Han, M.; Yu, L.; Su, C. Tailorable carbazolyl cyanobenzene-based photocatalysts for visible light-induced reduction of aryl halides. Chin. Chem. Lett. 2020, 31, 1899–1902. [Google Scholar] [CrossRef]
- Hamilton, G.L.; Backes, B.J. Dealkylative functionalization of tertiary amines with electron deficient heteroaryl chlorides. Tetrahedron Lett. 2006, 47, 2229–2231. [Google Scholar] [CrossRef]
- Khalaf, A.I.; Alvarez, R.G.; Suckling, C.J.; Waigh, R.D. Unexpected dealkylation during nucleophilic substitution: Synthesis of 2-N,N-dialkylamino benzoxazoles and benzothiazoles. Tetrahedron 2000, 56, 8567–8571. [Google Scholar] [CrossRef]
- Kolesinska, B.; Kaminski, Z.J. The umpolung of substituent effect in nucleophilic aromatic substitution. A new approach to the synthesis of N,N-disubstituted melamines (triazine triskelions) under mild reaction conditions. Tetrahedron 2009, 65, 3573–3576. [Google Scholar] [CrossRef]
- Lee, M.; Rucil, T.; Hesek, D.; Oliver, A.G.; Fisher, J.F.; Mobashery, S. Regioselective control of the SNAr amination of 5-substituted-2,4-dichloropyrimidines using tertiary amine nucleophiles. J. Org. Chem. 2015, 80, 7757–7763. [Google Scholar] [CrossRef]
- Zubkov, M.O.; Kosobokov, M.D.; Levin, V.V.; Kokorekin, V.A.; Korlyukov, A.A.; Hu, J.; Dilman, A.D. A novel photoredox-active group for the generation of fluorinated radicals from difluorostyrenes. Chem. Sci. 2020, 11, 737–741. [Google Scholar] [CrossRef] [Green Version]
- Panferova, L.I.; Zubkov, M.O.; Kokorekin, V.A.; Levin, V.V.; Dilman, A.D. Using the thiyl radical for aliphatic hydrogen-atom transfer: Thiolation of unactivated C–H bonds. Angew. Chem. Int. Ed. 2021, 60, 2849–2854. [Google Scholar] [CrossRef] [PubMed]
- Ormazabal-Toledo, R.; Richter, S.; Robles-Navarro, A.; Maulen, B.; Matute, R.A.; Gallardo-Fuentes, S. Meisenheimer complexes as hidden intermediates in the aza-SNAr mechanism. Org. Biomol. Chem. 2020, 18, 4238–4247. [Google Scholar] [CrossRef]
- Christopher, J.A.; Brophy, L.; Lynn, S.M.; Miller, D.D.; Sloan, L.A.; Sandford, G. Synthetic utility of 4-bromo-2,3,5,6-tetrafluoropyridine. J. Fluorine Chem. 2008, 129, 447–454. [Google Scholar] [CrossRef]
- Grossi, L.; Strazzari, S. Aromatic radical anions as possible intermediates in the nucleophilic aromatic substitution (SNAr): An EPR study. J. Chem. Soc. Perkin Trans. 2 1999, 2141–2146. [Google Scholar] [CrossRef]
- Imoto, M.; Matsui, Y.; Takeda, M.; Tamaki, A.; Taniguchi, H.; Mizuno, K.; Ikeda, H. A probable hydrogen-bonded meisenheimer complex: An unusually high SNAr reactivity of nitroaniline derivatives with hydroxide ion in aqueous media. J. Org. Chem. 2011, 76, 6356–6361. [Google Scholar] [CrossRef]
- Kuehne, M.E. The arylation of enamines. J. Am. Chem. Soc. 1962, 84, 837–847. [Google Scholar] [CrossRef]
- Panahi, F.; Daneshgar, F.; Haghighi, F.; Khalafi-Nezhad, A. Immobilized Pd nanoparticles on silica-starch substrate (PNP-SSS): Efficient heterogeneous catalyst in Buchwald–Hartwig C–N cross coupling reaction. J. Organomet. Chem. 2017, 851, 210–217. [Google Scholar] [CrossRef]
- Al-Rawi, J.M.A.; Khuthier, A.-H.; Hanna, S.Y. Carbon-13 nuclear magnetic resonance spectra of some N-(2,4-dinitrophenyl) Amines, The Corresponding N-Oxides, and Their Thermal Rearrangement Products. Spectrosc. Lett. 1988, 21, 249–259. [Google Scholar] [CrossRef]
- Feng, Y.-S.; Mao, L.; Bu, X.-S.; Dai, J.-J.; Xu, H.-J. Pd(OAc)2-catalyzed dinitration reaction of aromatic amines. Tetrahedron 2015, 71, 3827–3832. [Google Scholar] [CrossRef]
- Bellucci, G. Synthesis and determination of the configuration of the diastereoisomeric 4,N-dimethyl-N-phenylcyclohexylamines. Gazz. Chim. Ital. 1969, 99, 1208–1216. [Google Scholar]
- Graymore, J. 175. The reduction products of certain cyclic methyleneamines. Part II. J. Chem. Soc. 1932, 1353–1357. [Google Scholar] [CrossRef]
- Kleinschmidt, R.F.; Cope, A.C. Rearrangement of allyl groups in dyad systems. amine oxides. J. Am. Chem. Soc. 1944, 66, 1929–1933. [Google Scholar] [CrossRef]
- McIntire, F.C.; Clements, L.M.; Sproull, M. 1-Fluoro-2,4-dinitrobenzene as quantitative reagent for primary and secondary amines. Anal. Chem. 1953, 25, 1757–1758. [Google Scholar] [CrossRef]
- Matsuda, N.; Hirano, K.; Satoh, T.; Miura, M. Copper-catalyzed direct amination of polyfluoroarenes and azoles with hydroxylamines and its application to the synthesis of 3-aminobenzoheteroles. Synthesis 2012, 44, 1792–1797. [Google Scholar] [CrossRef]
- Grube, H.; Suhr, H. Nucleophile substitution, XI. reaktionen von chlorsubstituierten heteroaromaten mit aminen. Chem. Ber. 1969, 102, 1570–1579. [Google Scholar] [CrossRef]
- Qiu, Y.; Struwe, J.; Meyer, T.H.; Oliveira, J.C.A.; Ackermann, L. Catalyst- and reagent-free electrochemical azole C–H amination. Chem. Eur. J. 2018, 24, 12784–12789. [Google Scholar] [CrossRef] [PubMed]
- Yonemoto, K.; Shibuya, I. Reaction of 1,4,2-dithiazolium salts with amino compounds. Bull. Chem. Soc. Jpn. 1988, 61, 4043–4049. [Google Scholar] [CrossRef] [Green Version]
- Jo, J.; Kim, S.H.; Kim, H.; Jeong, M.; Kwak, J.-H.; Taek Han, Y.; Jeong, J.-Y.; Jung, Y.-S.; Yun, H. Discovery and SAR studies of novel 2-anilinopyrimidine-based selective inhibitors against triple-negative breast cancer cell line MDA-MB-468. Bioorg. Med. Chem. Lett. 2019, 29, 62–65. [Google Scholar] [CrossRef] [PubMed]
- Su, D.; Duan, H.; Wei, Z.; Cao, J.; Liang, D.; Lin, Y. Condensation of Vilsmeier salts, derived from tetraalkylureas, with amidoximes: A novel approach to access N,N-dialkyl-1,2,4-oxadiazol-5-amines. Tetrahedron Lett. 2013, 54, 6959–6963. [Google Scholar] [CrossRef]
- Joardar, S.; Bhattacharyya, A.; Das, S. A Palladium on carbon catalyzed one-pot synthesis of substituted benzimidazoles. Synthesis 2014, 46, 3121–3132. [Google Scholar] [CrossRef]
- Adamson, A.J.; Jondi, W.J.; Tipping, A.E. Reaction of metal diethylnitroxides with pentafluoropyridine, pentafluorobenzene, octafluorotoluene and 2-chloro-3- or 5-nitropyridine. J. Fluorine Chem. 1996, 76, 67–78. [Google Scholar] [CrossRef]
- Nandi, D.; Islam, R.U.; Devi, N.; Siwal, S.; Mallick, K. A palladium nanoparticle-catalyzed aryl–amine coupling reaction: High performance of aryl and pyridyl chlorides as the coupling partner. New J. Chem. 2018, 42, 812–816. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | PC 1 | Solv. | Ratio 1a:3a |
|---|---|---|---|
| 1 2 | – | DMSO | 100:0 |
| 2 2,3 | – | DMSO | 95:5 |
| 3 | – | DMSO | 90:10 |
| 4 | Ru(phen)3(PF6)2 (1%) | DMSO | 68:32 |
| 5 | 4CzIPN (2%) | DMSO | 33:67 |
| 6 | 3DPA2FBN (2%) | DMSO | 28:72 |
| 7 | Ir(ppy)2dtbbpy PF6 (0.5%) | DMSO | 29:71 |
| 8 | Ir(ppy)3 (0.3%) | DMSO | 15:85 (84) 4 |
| 9 | Ir(ppy)3 (0.3%), 1.1 equiv of NEt3 | DMSO | 61:39 |
| 10 | Ir(ppy)3 (0.3%) | DMF | 35:65 |
| 11 | Ir(ppy)3 (0.3%) | MeCN | 57:43 |
| 12 | Ir(ppy)3 (0.3%) | CH2Cl2 | 85:15 |

| Amine | Product | Y., % 1 | Amine | Major Product | Y., % 1 | Minor Product | Y., % 1 | |||
|---|---|---|---|---|---|---|---|---|---|---|
 |  | 3a | 84 |  |  | 3h | 86 |  | 3g | 1 |
 |  | 3b | 68 |  |  | 3i | 64 |  | 3g | 3 |
 |  | 3c | 71 |  |  | 3j | 83 |  | 3g | 9 |
 |  | 3d | 87 |  |  | 3k | 76 |  | 3g | 10 |
 |  | 3e | 86 |  |  | 3l | 70 |  | 3g | 19 |
 |  | 3m | 70 |  | 3g | 20 | ||||
 |  | 3f | 88 |  |  | 3n | 55 |  | 3g | 32 |
 |  | 3a | 50 |  | 3o | 41 | ||||
 |  | 3g | 85 |  |  | 3p | 59 |  | 3g | 30 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lipilin, D.L.; Frumkin, A.E.; Tyurin, A.Y.; Levin, V.V.; Dilman, A.D. Photoredox Catalyzed Dealkylative Aromatic Halogen Substitution with Tertiary Amines. Molecules 2021, 26, 3323. https://doi.org/10.3390/molecules26113323
Lipilin DL, Frumkin AE, Tyurin AY, Levin VV, Dilman AD. Photoredox Catalyzed Dealkylative Aromatic Halogen Substitution with Tertiary Amines. Molecules. 2021; 26(11):3323. https://doi.org/10.3390/molecules26113323
Chicago/Turabian StyleLipilin, Dmitry L., Alexander E. Frumkin, Alexey Y. Tyurin, Vitalij V. Levin, and Alexander D. Dilman. 2021. "Photoredox Catalyzed Dealkylative Aromatic Halogen Substitution with Tertiary Amines" Molecules 26, no. 11: 3323. https://doi.org/10.3390/molecules26113323