
Copper(II) 2,2-Bis(Hydroxymethyl)Propionate Coordination Compounds with Hexamethylenetetramine: From Mononuclear Complex to One-Dimensional Coordination Polymer


Abstract
:
1. Introduction
2. Results and Discussion
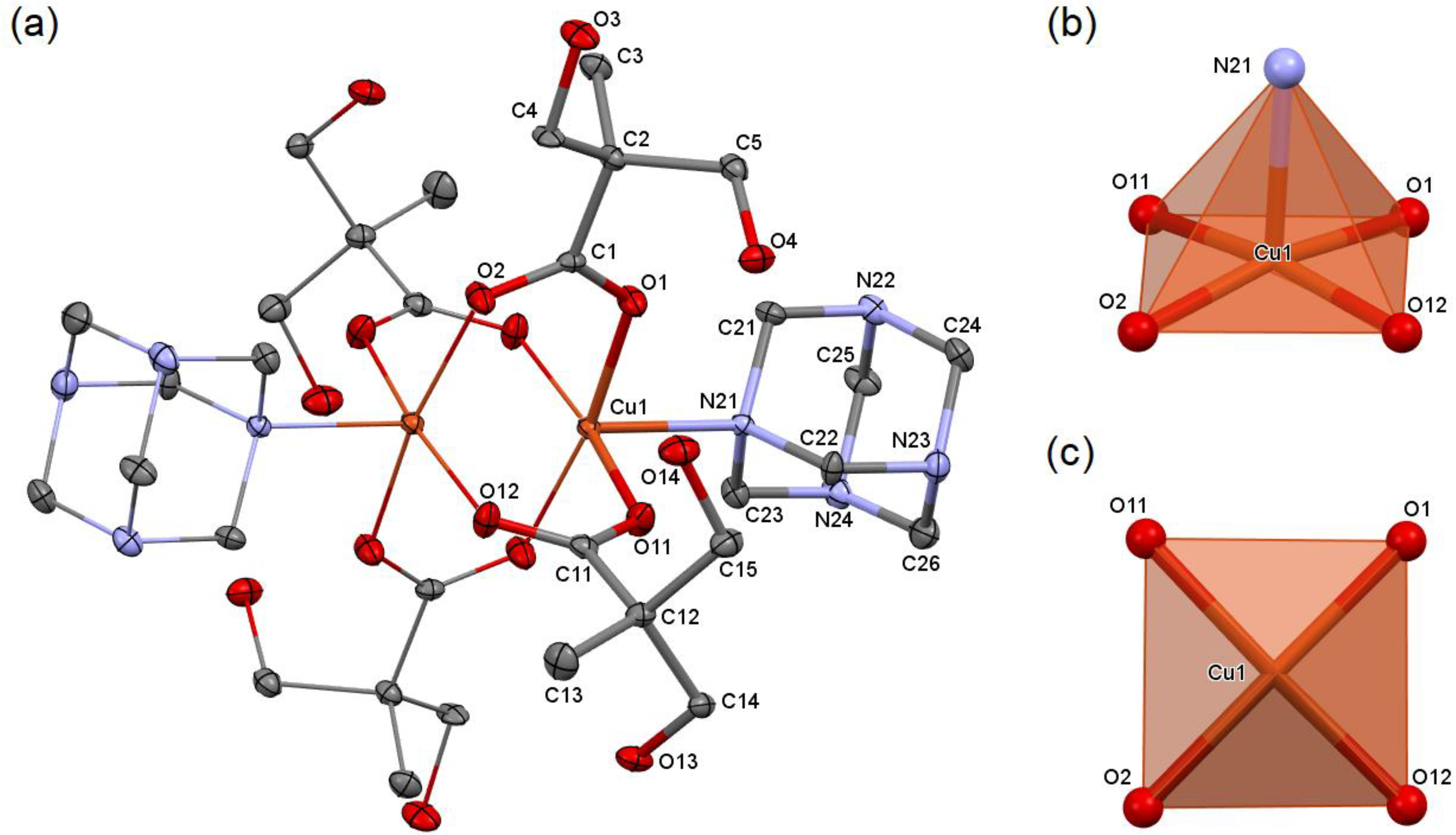
2.1. Synthesis and Structural Analysis of Copper Coordination Compounds
2.2. IR Spectroscopy Analysis
2.3. UV-Vis Spectroscopy Analysis
2.4. Thermal Analysis
3. Materials and Methods
3.1. Synthesis of Copper Coordination Compounds
3.2. Crystal Structure Determination
3.3. Physicochemical Measurements
3.4. Quantum-Mechanical Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Desiraju, G.R.; Vittal, J.J.; Ramanan, A. Crystal Engineering. A Textbook; World Scientific Publishing: Singapore, 2011. [Google Scholar]
- Biradha, K. Crystal engineering: From weak hydrogen bonds to co-ordination bonds. CrystEngComm 2003, 5, 374–384. [Google Scholar] [CrossRef]
- Danilescu, O.; Bourosh, P.N.; Petuhov, O.; Kulikova, O.V.; Bulhac, I.; Chumakov, Y.M.; Croitor, L. Crystal Engineering of Schiff Base Zn(II) and Cd(II) Homo and Zn(II)M(II) (M = Mn or Cd) Heterometallic Coordination Polymers and Their Ability to Accommodate Solvent Guest Molecules. Molecules 2021, 26, 2317. [Google Scholar] [CrossRef]
- Dey, C.; Kundu, T.; Biswal, B.P.; Mallick, A.; Banerjeea, R. Crystalline metal-organic frameworks (MOFs): Synthesis, structure and function. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2014, 70, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, K.; Sun, Y.; Lollar, C.T.; Li, J.; Zhou, H.-C. Recent advances in gas storage and separation using metal–organic frameworks. Mater. Today 2018, 21, 108–121. [Google Scholar] [CrossRef]
- Mukherjee, S.; Sensharma, D.; Chen, K.-J.; Zaworotko, M.J. Crystal engineering of porous coordination networks to enable separation of C2 hydrocarbons. Chem. Commun. 2020, 56, 10419–10441. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Zaworotko, M.J. Crystal Engineering of Hybrid Coordination Networks: From Form to Function. Trends Chem. 2020, 2, 506–518. [Google Scholar] [CrossRef]
- Konnertha, H.; Matsagara, B.M.; Chena, S.S.; Prechtl, M.H.G.; Shiehb, F.-K.; Wu, K.C.-W. Metal-organic framework (MOF)-derived catalysts for fine chemical production. Coord. Chem. Rev. 2020, 416, 213319. [Google Scholar] [CrossRef]
- Alhumaimess, M.S. Metal-organic frameworks and their catalytic applications. J. Saudi Chem. Soc. 2020, 24, 461–473. [Google Scholar] [CrossRef]
- Goetjen, T.A.; Liu, J.; Wu, Y.; Sui, J.; Zhang, X.; Hupp, J.T.; Farha, O.K. Metal–organic framework (MOF) materials as polymerization catalysts: A review and recent advances. Chem. Commun. 2020, 56, 10409. [Google Scholar] [CrossRef] [PubMed]
- Kirillov, A.M. Hexamethylenetetramine: An old new building block for design of coordination polymers. Coord. Chem. Rev. 2011, 255, 1603–1622. [Google Scholar] [CrossRef]
- Qiao, X.; Ge, Y.; Li, Y.; Niu, Y.; Wu, B. Preparation and Analyses of the Multifunctional Properties of 2D and 3D MOFs Constructed from Copper(I) Halides and Hexamethylenetetramine. ACS Omega 2019, 4, 12402–12409. [Google Scholar] [CrossRef] [Green Version]
- Kaihua, J.; Shuhong, B. Coordination compounds of hexamethylenetetramine with metal salts: A review. Johns. Matthey Technol. Rev. 2018, 62, 4–31. [Google Scholar] [CrossRef]
- Halder, P.; Zangrando, E.; Paine, T.K. Copper(II) α-hydroxycarboxylate complexes of bis(2-pyridylcarbonyl)amine ligand: From mononuclear complex to one-dimensional coordination polymer. Polyhedron 2010, 29, 434–440. [Google Scholar] [CrossRef]
- Halder, P.; Chakraborty, B.; Banerjee, P.R.; Zangrando, E.; Paine, T.K. Role of α-hydroxycarboxylic acids in the construction of supramolecular assemblies of nickel(II) complexes with nitrogen donor coligands. CrystEngComm 2009, 11, 2650–2659. [Google Scholar] [CrossRef]
- Carballo, R.; Covelo, B.; Fernández-Hermida, N.; García-Martinez, E.; Lago, A.B.; Vázquez-López, E.M. Structural Versatility in Copper(II) α-Hydroxycarboxylate Complexes with the Twisted Ligands 4,4′-Dipyridyldisulfide and Bis(4-pyridylthio)methane: From Molecular Compounds to Two-Dimensional Coordination Polymers. Cryst. Growth Des. 2008, 8, 995–1004. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. Cambridge Structural Database, CSD v 5.39, August 2018. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Kruszynski, R.; Sierański, T.; Świątkowski, M.; Zielak, M.; Wojciechowski, J.; Dzierżawska, M.; Lewiński, B. On the coordination behavior of the hmta toward zinc and cadmium cations in presence of sulfate(VI) and nitrate(V) anions. J. Coord. Chem. 2014, 67, 1332–1352. [Google Scholar] [CrossRef]
- Kruszynski, R.; Sieranski, T.; Swiatkowski, M.; Zielak, M.; Wojciechowski, J.; Dzierzawska, M.; Czubacka, E. On the Coordination Behaviour of the hmta Toward Alkali Metal Cations in Presence of Perchlorate Anions. J. Chem. Crystallogr. 2015, 45, 484–494. [Google Scholar] [CrossRef] [Green Version]
- Swiatkowski, M.; Kruszynski, R. Structurally diverse coordination compounds of zinc as effective precursors of zinc oxide nanoparticles with various morphologies. Appl. Organomet. Chem. 2019, 33, e4812. [Google Scholar] [CrossRef]
- Kepert, D.L. Aspects of the Stereochemistry of Seven-Coordination. In Progress in Inorganic Chemistry; Lippard, S.J., Ed.; John Wiley: New York, NY, USA, 1979; Volume 25, pp. 41–144. [Google Scholar]
- Zachariasen, W.H. Bond lengths in oxygen and halogen compounds of d and f elements. J. Less Common Met. 1978, 62, 1–7. [Google Scholar] [CrossRef]
- Brown, I.D. Influence of Chemical and Spatial Constraints on the Structures of Inorganic Compounds. Acta Crystallogr. Sect. B Struct. Sci. 1997, 53, 381–393. [Google Scholar] [CrossRef]
- Shields, P.G.; Raithby, P.R.; Allen, F.H.; Motherwell, W.D.S. The assignment and validation of metal oxidation states in the Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. 2000, 56, 455–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, I.D. Chemical and steric constraints in inorganic solids. Acta Crystallogr. Sect. B Struct. Sci. 1992, 48, 553–572. [Google Scholar] [CrossRef]
- Sieron, L.; Bukowska-Strzyzewska, M.; Korabik, M.; Mrozinski, J. X-ray structure of poly[aquabis(benzimidazole-N3)copper(II)-μ-trans-2-butene-1,4-dicarboxylato-O,O′:O″,O‴] (I) and magnetic and spectroscopic characterization of I and the isostructural copper(II)-μ-adipato complex II. Polyhedron 2002, 21, 2473–2479. [Google Scholar] [CrossRef]
- Nadeem, M.A.; Bhadhade, M.; Bircher, R.; Stride, J.A. Three isolated structural motifs in one crystal: Penetration of two 1D chains through large cavities within 2D polymeric sheets. CrystEngComm 2010, 12, 1391–1393. [Google Scholar] [CrossRef]
- Valach, F.; Grobelny, R.; Glowiak, T.; Mrozinski, J.; Lukes, V.; Blahova, Z. Structural study of semi-coordination in a seven-coordinate copper(II) complex: Distortion isomerism of [Cu(CH3COO)2(4-aminopyridine)2(H2O)]. J. Coord. Chem. 2010, 63, 1645–1651. [Google Scholar] [CrossRef]
- Favas, M.C.; Kepert, D.L. Aspects of the Stereochemistry of Four-Coordination and Five-Coordination. In Progress in Inorganic Chemistry; Lippard, S.J., Ed.; John Wiley: New York, NY, USA, 1980; Volume 27, pp. 325–463. [Google Scholar]
- Cao, J.; Huang, Z.; Cao, C.; Cheng, C.; Sun, C. catena-Poly[[tetra-μ-formato-κ8O:O’-dicopper(II)]-μ-hexa-methyl-ene-tetra mine-κ2N1:N5]. Acta Crystallogr. Sect. E Struct. Rep. Online 2013, 69, m690. [Google Scholar] [CrossRef] [Green Version]
- Pickardt, J. Struktur von catena-μ-(Hexamethylentetramin-N,N’)-[tetra-μ-acetato-dikupfer(II)]. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1981, 37, 1753–1756. [Google Scholar] [CrossRef]
- Hou-Qun, Y.; You-Fa, X.; Wei, X.; Chun-Yan, H.; Guang-Ming, B. Crystal structure of catena-poly[(μ2-hexamethylenetetramine-κ2N:N′)-tetrakis(μ2-2,6-difluorobenzoato-κ2O:O′)dicopper(II)], C34H24Cu2F8N4O8. Z. Kristallogr. New Cryst. Struct. 2017, 232, 823–825. [Google Scholar] [CrossRef] [Green Version]
- Valach, F.; Tokarcik, M.; Maris, T.; Watkin, D.J.; Prout, C.K. Bond-valence approach to the copper-copper and copper-oxygen bonding in binuclear copper(II) complexes: Structure of tetrakis(2-fluorobenzoato-O,O’)-bis(2-fluorobenzoate-O) dicopper(II). Z. Kristallogr. 2000, 215, 56–60. [Google Scholar] [CrossRef]
- Melnik, M. Study of the relation between the structural data and magnetic interactions in oxo-bridged binuclear copper(II) compounds. Coord. Chem. Rev. 1982, 42, 259–293. [Google Scholar] [CrossRef]
- Kawata, T.; Uekusa, H.; Ohba, S. Magneto-Structural Correlation in Dimeric Copper(II) Benzoates. Acta Crystallogr. Sect. B Struct. Sci. 1992, 48, 253–261. [Google Scholar] [CrossRef]
- Świątkowski, M.; Lanka, S.; Czylkowska, A.; Gas, K.; Sawicki, M. Structural, Spectroscopic, Thermal, and Magnetic Properties of a New Dinuclear Copper Coordination Compound with Tiglic Acid. Materials 2021, 14, 2148. [Google Scholar] [CrossRef]
- Ren, Z.; Ma, D.; Wang, Y.; Zhao, G. Molecular structure and hydrogen bonds in solid dimethylol propionic acid (DMPA). Spectrochim. Acta A 2003, 59, 2713–2722. [Google Scholar] [CrossRef]
- Jensen, J.O. Vibrational frequencies and structural determinations of hexamethylenetetramine. Spectrochim. Acta A 2002, 58, 1347–1364. [Google Scholar] [CrossRef]
- Van Leeuwen, R. Key Concepts in Time-Dependent Density-Functional Theory. Int. J. Mod. Phys. B 2001, 15, 1969–2023. [Google Scholar] [CrossRef]
- Welcher, F.J. Analityczne Zastosowanie Kwasu Wersenowego (Eng. The Analytical Uses of Etylenediamineteraacetic Acid); WNT: Warsaw, Poland, 1963; pp. 241–242. [Google Scholar]
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Cryst. 2020, 53, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Georgiev, G.T.; Butler, J.J. Long-term calibration monitoring of Spectralon di_users BRDF in the air-ultraviolet. Appl. Opt. 2007, 46, 7892–7899. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision, E.01; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Mardirossian, N.; Head-Gordon, M. Thirty years of density functional theory in computational chemistry: An overview and extensive assessment of 200 density functionals. Mol. Phys. 2017, 115, 2315–2372. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr.; Hay, P.J. Gaussian Basis Sets for Molecular Calculations. In Modern Theoretical Chemistry; Schaefer, H.F., III, Ed.; Plenum: New York, NY, USA, 1977; Volume 3, pp. 1–28. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| i—j | dij (Å) | νij (v.u.) | i—j—k | αijk (°) | i—j—k | αijk (°) |
|---|---|---|---|---|---|---|
| compound 1 | ||||||
| Cu1—O1 | 1.9654 (13) | 0.432 | O1—Cu1—O2 | 47.90 (5) | O11—Cu1—O12 | 50.38 (5) |
| Cu1—O2 | 2.9953 (15) | 0.027 | O1—Cu1—O11 | 169.04 (6) | O11—Cu1—O41 | 94.59 (5) |
| Cu1—O11 | 1.9684 (13) | 0.429 | O1—Cu1—O12 | 118.70 (5) | O11—Cu1—N21 | 90.41 (5) |
| Cu1—O12 | 2.8616 (16) | 0.038 | O1—Cu1—O41 | 96.25 (5) | O11—Cu1—N31 | 90.03 (5) |
| Cu1—O41 | 2.1931 (13) | 0.234 | O1—Cu1—N21 | 90.87 (5) | O12—Cu1—O41 | 144.86 (5) |
| Cu1—N21 | 2.1154 (14) | 0.337 | O1—Cu1—N31 | 89.44 (5) | O12—Cu1—N21 | 91.55 (5) |
| Cu1—N31 | 2.1272 (15) | 0.326 | O2—Cu1—O11 | 121.35 (5) | O12—Cu1—N31 | 91.72 (5) |
| O2—Cu1—O12 | 71.14 (4) | O41—Cu1—N21 | 91.36 (5) | |||
| O2—Cu1—O41 | 144.00 (4) | O41—Cu1—N31 | 84.73 (5) | |||
| O2—Cu1—N21 | 86.66 (5) | N21—Cu1—N31 | 176.09 (6) | |||
| O2—Cu1—N31 | 96.42 (5) | |||||
| compound 2 | ||||||
| Cu1—O1 | 1.9652 (12) | 0.431 | O1—Cu1—O2(i) | 169.67 (5) | O2(i)—Cu1—O12 (i) | 89.22 (6) |
| Cu1—O2(i) | 1.9752 (13) | 0.424 | O1—Cu1—O11 | 88.12 (6) | O2(i)—Cu1—N21 | 99.10 (5) |
| Cu1—O11 | 1.9562 (13) | 0.440 | O1—Cu1—O12(i) | 89.42 (6) | O11—Cu1—O12(i) | 169.53 (5) |
| Cu1—O12(i) | 1.9631 (13) | 0.435 | O1—Cu1—N21 | 91.21 (5) | O11—Cu1—N21 | 95.78 (5) |
| Cu1—N21 | 2.1839 (14) | 0.280 | O2(i)—Cu1—O11 | 91.37 (6) | O12(i)—Cu1—N21 | 94.45 (5) |
| Cu1•••Cu1(i) | 2.5927 (5) | |||||
| compound 3 | ||||||
| Cu1—O11 | 1.9624 (16) | 0.436 | O11—Cu1—O12(ii) | 168.86 (7) | O12(ii)—Cu1—O22(ii) | 88.06 (7) |
| Cu1—O12(ii) | 1.9761 (16) | 0.420 | O11—Cu1—O21 | 90.17 (7) | O12(ii)—Cu1—N1 | 88.76 (7) |
| Cu1—O21 | 1.9688 (16) | 0.428 | O11—Cu1—O22(ii) | 88.67 (7) | O21—Cu1—O22(ii) | 169.29 (7) |
| Cu1—O22(ii) | 1.9691 (16) | 0.428 | O11—Cu1—N1 | 102.21 (7) | O21—Cu1—N1 | 93.73 (7) |
| Cu1—N1 | 2.2135 (18) | 0.259 | O12(ii)—Cu1—O21 | 91.06 (7) | O22(ii)—Cu1—N1 | 96.92 (7) |
| Cu1•••Cu1(ii) | 2.6027 (6) | |||||
| Cu2—O31 | 1.9503 (17) | 0.450 | O31—Cu1—O32(i) | 169.32 (7) | O32(i)—Cu1—O42(i) | 89.87 (8) |
| Cu2—O32(i) | 1.9595 (17) | 0.439 | O31—Cu1—O41 | 89.25 (9) | O32(i)—Cu1—N2 | 96.71 (7) |
| Cu2—O41 | 1.9626 (17) | 0.435 | O31—Cu1—O42(i) | 90.22 (8) | O41—Cu1—O42(i) | 169.54 (7) |
| Cu2—O42(i) | 1.9717 (16) | 0.425 | O31—Cu1—N2 | 93.95 (7) | O41—Cu1—N2 | 96.72 (7) |
| Cu2—N2 | 2.1973 (18) | 0.270 | O32(i)—Cu1—O41 | 88.73 (9) | O42(i)—Cu1—N2 | 93.74 (7) |
| Cu2•••Cu2(i) | 2.5897 (6) | |||||
| D—H•••A | d(D—H) (Å) | d(H•••A) (Å) | d(D•••A) (Å) | <(DHA) (°) | Graph-Set |
|---|---|---|---|---|---|
| compound 1 | |||||
| O3—H3•••N22(i) | 0.84 | 2.17 | 2.942 (2) | 153 | C (10) |
| O4—H4•••N24(ii) | 0.84 | 2.01 | 2.840 (2) | 170 | R22 (20) |
| O13—H13•••N33(iii) | 0.84 | 2.01 | 2.850 (2) | 177 | R22 (20) |
| O14—H14•••N34(iv) | 0.84 | 2.16 | 2.936 (3) | 154 | C (10) |
| O41—H41O•••O12(v) | 0.86 | 1.83 | 2.636 (2) | 157 | C (6) |
| O41—H41P•••O2(v) | 0.87 | 1.82 | 2.6373 (19) | 155 | C (6) |
| compound 2 | |||||
| O3—H3•••O13(vi) | 0.84 | 2.05 | 2.867 (3) | 164 | C (12) |
| O4—H4•••O14(vii) | 0.84 | 1.90 | 2.737 (3) | 179 | C (12) |
| O13—H13•••N22(viii) | 0.84 | 1.92 | 2.759 (3) | 179 | C (10) |
| O14—H14•••O4 | 0.84 | 1.96 | 2.770 (3) | 162 | S (12) |
| compound 3 | |||||
| O13—H13•••N3(v) | 0.84 | 1.97 | 2.791 (3) | 166 | C (10) |
| O14—H14•••O24(ix) | 0.84 | 1.87 | 2.711 (4) | 176 | S (12) |
| O23—H23•••O34(x) | 0.84 | 2.09 | 2.786 (4) | 140 | C (16) |
| O24—H24•••O13(vi) | 0.84 | 1.89 | 2.727 (3) | 172 | C (12) |
| O33A—H33D•••O43A(xi) | 0.84 | 2.36 | 2.769 (5) | 111 | R22 (32) |
| O34—H34•••O14(xii) | 0.84 | 1.94 | 2.765 (4) | 166 | R22 (32) |
| O43A—H44D•••O23(xiii) | 0.84 | 2.03 | 2.855 (4) | 169 | S (16) |
| 1 | 2 | 3 | Dmpa [37] | Hmta [38] | Assignment |
|---|---|---|---|---|---|
| 3336 | 3407 | 3436 | 3368 | ν OH | |
| 3228 | 3230 | ν OH | |||
| 2973 | 2966 | ν CH | |||
| 2944 | 2941 | 2955 | ν CH | ||
| 2925 | 2919 | ν CH | |||
| 2873 | 2879 | 2885 | 2872 | ν CH | |
| 1703 | 1691 | ν C=O, δ OH(water) | |||
| 1701 | δ OH(water) | ||||
| 1627 | 1621 | νas COO, δ OH(water) | |||
| 1599 | 1570 | ν CN | |||
| 1460 | 1466 | 1467 | 1456 | 1456 | δ CH |
| 1423 | 1421 | νa COO | |||
| 1407 | 1403 | ν C-O | |||
| 1374 | 1367 | 1383 | 1370 | δ CH | |
| 1323 | 1298 | 1294 | 1309 | δ OH(hydroxyl) | |
| 1237 | 1236 | 1246 | 1236 | 1240 | ν CO(hydroxyl), δ CH, ν CN |
| 1232 | ν CN | ||||
| 1056 | 1052 | δ OH(hydroxyl), ν CC | |||
| 1026 | ν CN | ||||
| 1004 | 1001 | 1000 | 1007 | ν CN | |
| 925 | 939 | δ OH(hydroxyl) | |||
| 888 | 891 | 891 | 870 | ν CC | |
| 812 | 818 | 812 | ν CN | ||
| 776 | 782 | 787 | σ COO | ||
| 671 | 677 | 674 | 672 | δ NCN | |
| 512 | 514 | 506 | 512 | δ NCN | |
| 434 | 444 | 420 | ν Cu–N | ||
| 417 | 420 | 406 | ν Cu–O |
| Experimental λ (nm) | Theoretical λ (nm) | Orbitals Involved in Electronic Transitions | Character of Transition | ||||
|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 1 | 2 | 3 | ||
| 194 | 263 | 275 | 217 | αH − 2→αL + 1 αH − 1→αL + 1 | d(Cu)/n&σ(hmta)→σ*(-OH of dmp)/σ*(H2O) | ||
| 219 | αH→αL + 1 βH − 1→βL + 2 | ||||||
| 235 | αH − 5→αL + 4 βH − 4→βL + 5 | n(dmp)→σ*&π*(dmp) n&σ(dmp)→σ*&π *(dmp) | |||||
| 258 | αH − 18→αL βH − 18→βL | d(Cu)/n&σ(dmp)→d(Cu)/σ*(dmp-Cu) | |||||
| 262 | αH − 16→αL βH − 16→βL | ||||||
| 279 | αH − 13→αL βH − 13→βL | ||||||
| 381 | βH − 9→βL βH − 10→βL | d(Cu)/n&σ(dmp)/n&σ(hmta)→d(Cu)/σ*(dmp-Cu) d(Cu)/n&σ(dmp)/n(hmta)→d(Cu)/σ*(dmp-Cu) | |||||
| 383 | αH − 9→αL | d(Cu)/n&σ(dmp)→d(Cu)/σ*(dmp-Cu) | |||||
| 301 | 382 | 381 | 300 | βH − 8→βL | d(Cu)/n&σ(dmp)/n&σ(hmta)→d(Cu)/σ*(dmp-Cu)/σ *(hmta-Cu) | ||
| 308 | βH − 9→βL βH − 10→βL | ||||||
| 350 | βH − 6→βL βH − 8→βL | d(Cu)/n(-COO of dmp)n&σ(hmta)→d(Cu)/σ*(dmp-Cu)/σ*(hmta-Cu) d(Cu)/n&σ(dmp)/n&σ(hmta)→d(Cu)/σ*(dmp-Cu)/σ*(hmta-Cu) | |||||
| 361 | αH − 6→αL βH − 6→βL | d(Cu)/n&σ(dmp)→d(Cu)/σ*(dmp-Cu) | |||||
| 461 | αH − 4→αL | n&σ(hmta)→d(Cu)/σ*(dmp-Cu) | |||||
| 471 | αH − 3→αL | ||||||
| 483 | βH→βL | ||||||
| 621 | βH − 19→βL | d(Cu)/σ(dmp)→d(Cu)/σ*(dmp-Cu)/σ*(hmta-Cu) | |||||
| 733 | 688 | 686 | 604 | αH − 21→αL βH − 21→βL | d(Cu)/n(dmp-Cu)→d(Cu)/σ*(dmp-Cu) | ||
| 607 | αH − 33→αL βH − 33→βL | d(Cu)/n&σ(dmp)→d(Cu)/σ*(dmp-Cu) | |||||
| 614 | αH − 34→αL βH − 34→βL | ||||||
| 606 | βH − 63→βL + 1 | d(Cu)/n&σ(dmp)→d(Cu)/σ*(dmp-Cu) | |||||
| 632 | βH − 25→βL βH − 67→βL | d(Cu)/n&σ(dmp)→d(Cu)/σ*(dmp-Cu) d(Cu)/n&σ(dmp)/σ(hmta)→d(Cu)/σ*(dmp-Cu) | |||||
| Process/Final Product | 1 | 2 | 3 |
|---|---|---|---|
| Dehydration | 105–158 °C 1.9%/2.9% | - | - |
| Simultaneous disintegration of dmp and hmta | 158–315 °C 67.6%/84.5% | Substage 1 154–232 °C 27.6%/− | Substage 1 156–237 °C 26.3%/− |
| Substage 2 232–305 °C 39.8%/− | Substage 2 237–316 °C 54.7%/− | ||
| Totally 67.4%/83.1% | Totally 81.0%/80.1% | ||
| Combustion of pyrolitic soot | 315–565 °C 25.6%/− | 305–485 °C 18.4%/− | 316–495 °C 10.1%/− |
| CuO + Cu2O | 565 °C 4.9%/12.7% * | 485 °C 14.2%/16.9% * | 495 °C 8.9%/19.9% * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rauf, S.; Trzesowska-Kruszynska, A.; Sierański, T.; Świątkowski, M. Copper(II) 2,2-Bis(Hydroxymethyl)Propionate Coordination Compounds with Hexamethylenetetramine: From Mononuclear Complex to One-Dimensional Coordination Polymer. Molecules 2021, 26, 3358. https://doi.org/10.3390/molecules26113358
Rauf S, Trzesowska-Kruszynska A, Sierański T, Świątkowski M. Copper(II) 2,2-Bis(Hydroxymethyl)Propionate Coordination Compounds with Hexamethylenetetramine: From Mononuclear Complex to One-Dimensional Coordination Polymer. Molecules. 2021; 26(11):3358. https://doi.org/10.3390/molecules26113358
Chicago/Turabian StyleRauf, Sadaf, Agata Trzesowska-Kruszynska, Tomasz Sierański, and Marcin Świątkowski. 2021. "Copper(II) 2,2-Bis(Hydroxymethyl)Propionate Coordination Compounds with Hexamethylenetetramine: From Mononuclear Complex to One-Dimensional Coordination Polymer" Molecules 26, no. 11: 3358. https://doi.org/10.3390/molecules26113358