
Synthesis of 4-Substituted-1,2-Dihydroquinolines by Means of Gold-Catalyzed Intramolecular Hydroarylation Reaction of N-Ethoxycarbonyl-N-Propargylanilines

 , , , , and
, , , , and
Abstract
:
1. Introduction
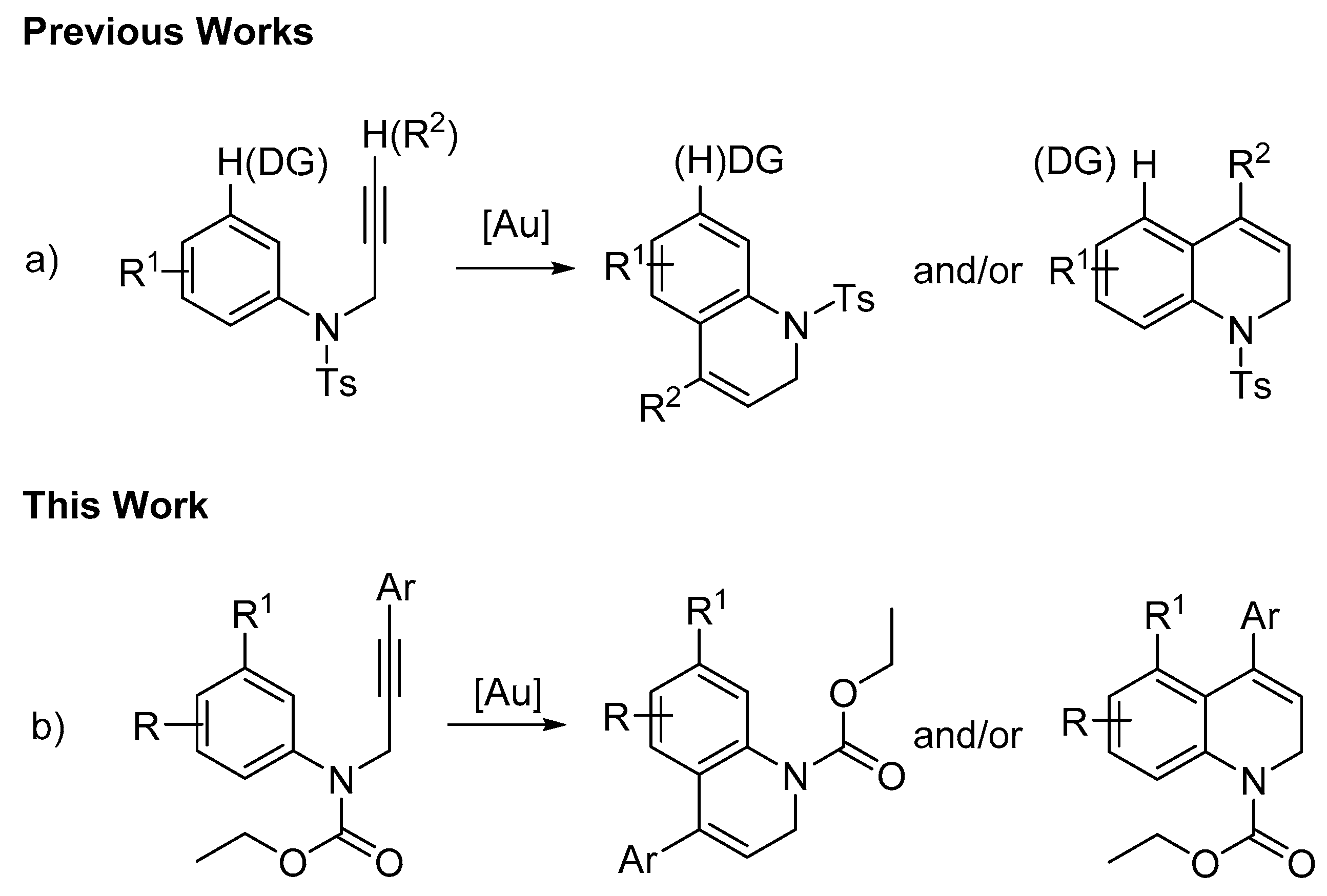
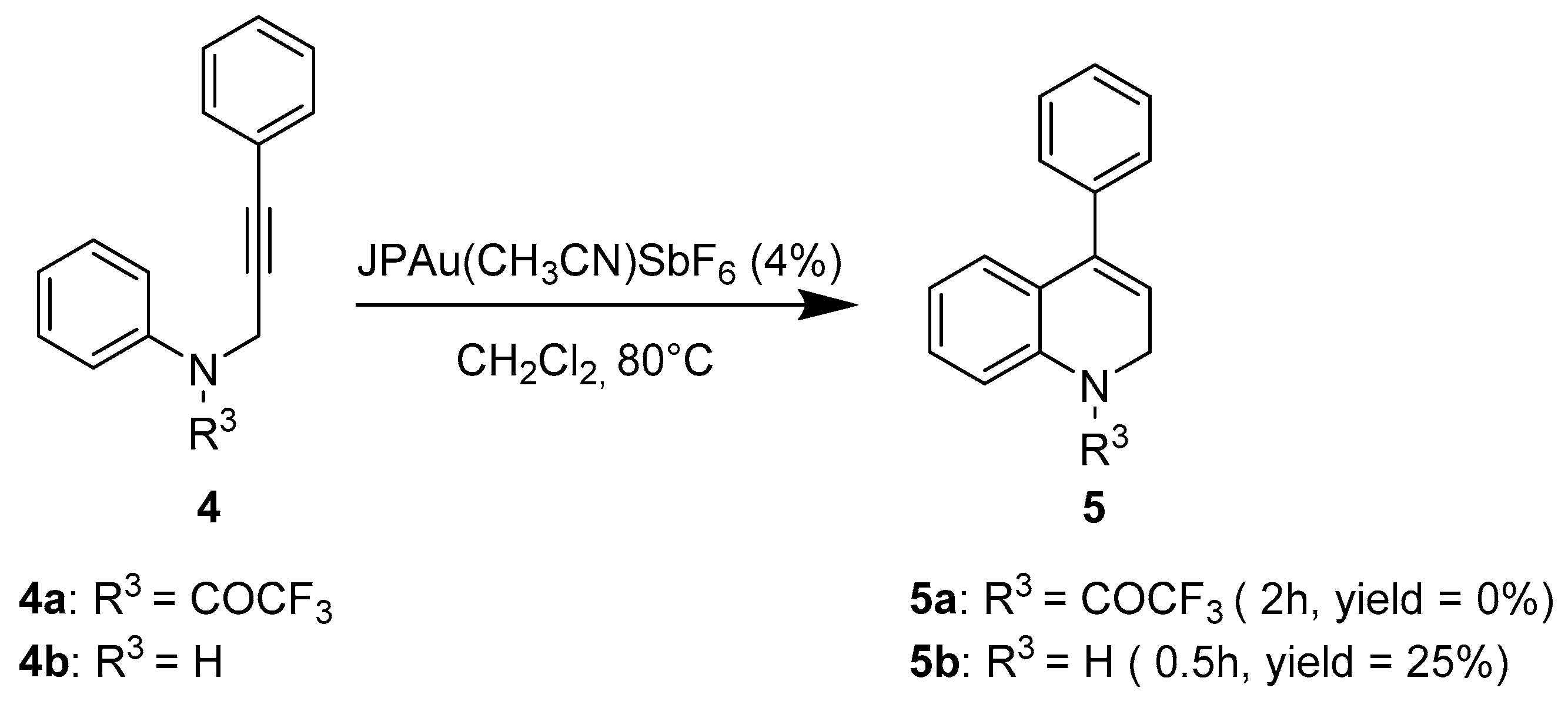
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. Synthetic Procedures
3.2.1. Preparation of Substrates 1
3.2.2. Preparation of Derivatives 2: Typical Procedure for the Preparation of the Ethyl 4-Phenylquinoline-1(2H)-Carboxylate 2a
3.3. Characterization Data
3.3.1. Characterization Data of Compound 1a–o
3.3.2. Characterization Data of Compound 2b–j, 2l–o, 2′l–2′o
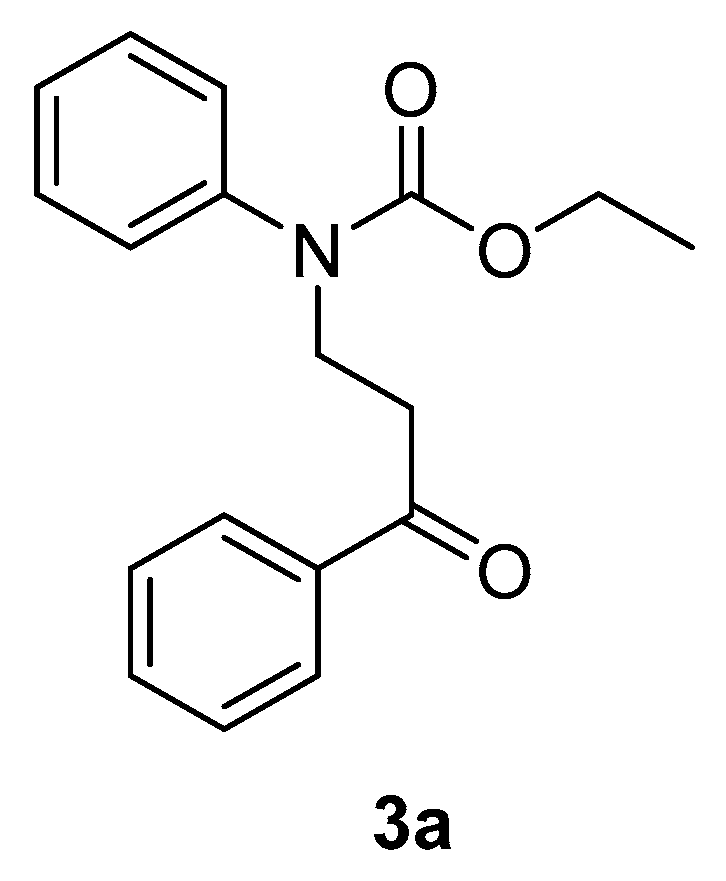
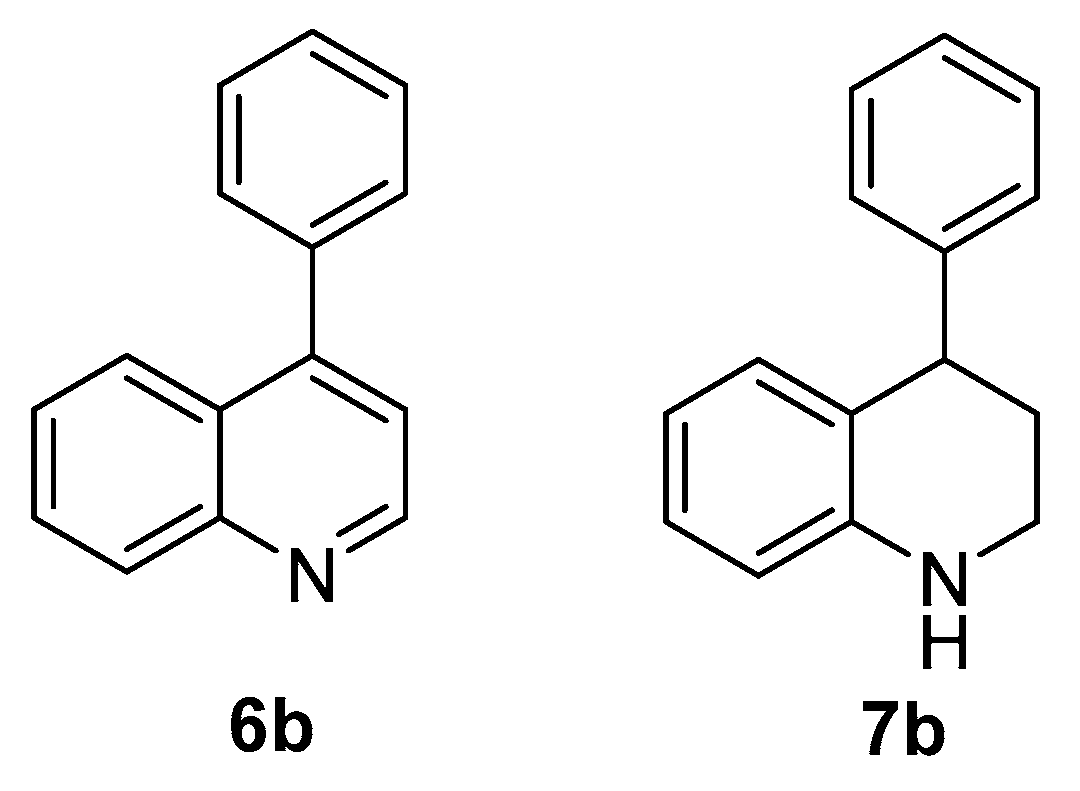
3.3.3. Characterization Data of Compounds 3a, 5b, 6b, 7b
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Vessally, E.; Edjlali, L.; Hosseinian, A.; Bekhradnia, A.; Esrafili, M.D. Novel routes to quinoline derivatives from N-propargylamines. Rsc. Adv. 2016, 6, 49730–49746. [Google Scholar] [CrossRef]
- Hussain, H.; Al-Harrasi, A.; Al-Rawahi, A.; Green, I.R.; Gibbons, S. Fruitful Decade for Antileishmanial Compounds from 2002 to Late 2011. Chem. Rev. 2014, 114, 10369–10428. [Google Scholar] [CrossRef]
- Pratap, R.; Ram, V.J. Natural and Synthetic Chromenes, Fused Chromenes, and Versatility of Dihydrobenzo[h]chromenes in Organic Synthesis. Chem. Rev. 2014, 114, 10476–10526. [Google Scholar] [CrossRef]
- Bhanja, C.; Jena, S.; Nayak, S.; Mohapatra, S. Organocatalytic tandem Michael addition reactions: A powerful access to the enantioselective synthesis of functionalized chromenes, thiochromenes and 1,2-dihydroquinolines. Beilstein J. Org. Chem 2012, 8, 1668–1694. [Google Scholar] [CrossRef]
- Barluenga, J.; Rodriguez, F.; Fananas, F.J. Recent Advances in the Synthesis of Indole and Quinoline Derivatives through Cascade Reactions. Chem-Asian J. 2009, 4, 1036–1048. [Google Scholar] [CrossRef]
- Amrutham, V.; Radikovich, A.M.; Mameda, N.; Gajula, K.S.; Grigor’eva, N.G.; Ivanovich, K.B.; Akula, V.; Nama, N. A heterogeneous catalytic and solvent-free approach to 1,2-dihydroquinoline derivatives from aromatic amines and alkynes by tandem hydroarylation-hydroamination. Catal. Commun. 2020, 135, 105888. [Google Scholar] [CrossRef]
- Pucheta, A.; Mendieta, A.; Madrigal, D.A.; Hernández-Benitez, R.I.; Romero, L.; Garduño-Siciliano, L.; Rugerio-Escalona, C.; Cruz-López, M.C.; Jiménez, F.; Ramírez-Villalva, A.; et al. Synthesis and biological activity of fibrate-based acyl- and alkyl-phenoxyacetic methyl esters and 1,2-dihydroquinolines. Med. Chem. Res. 2020, 29, 459–478. [Google Scholar] [CrossRef]
- Das, R.; Khot, N.P.; Deshpande, A.S.; Kapur, M. Catalyst Control in Switching the Site Selectivity of C−H Olefinations of 1,2-Dihydroquinolines: An Approach to Positional-Selective Functionalization of Quinolines. Chem. Eur. J. 2020, 26, 927–938. [Google Scholar] [CrossRef]
- El-Harairy, A.; Yiliqi; Lai, B.; Vaccaro, L.; Li, M.; Gu, Y. A Sulfone-Containing Imidazolium-Based Brønsted Acid Ionic Liquid Catalyst Enables Replacing Dipolar Aprotic Solvents with Butyl Acetate. Adv. Synth. Catal. 2019, 361, 3342–3350. [Google Scholar] [CrossRef]
- Wu, S.; Liu, C.; Luo, G.; Jin, Z.; Zheng, P.; Chi, Y.R. NHC-Catalyzed Chemoselective Reactions of Enals and Aminobenzaldehydes for Access to Chiral Dihydroquinolines. Angew. Chem. Int. Edit. 2019, 58, 18410–18413. [Google Scholar] [CrossRef]
- El-Remaily, M.A.E.A.A.A.; Abu-Dief, A.M.; Elhady, O. Green synthesis of TiO2 nanoparticles as an efficient heterogeneous catalyst with high reusability for synthesis of 1,2-dihydroquinoline derivatives. Appl. Organomet. Chem. 2019, 33, e5005. [Google Scholar] [CrossRef]
- Goulart, T.A.C.; Kazmirski, J.A.G.; Back, D.F.; Zeni, G. Iron(III)-Promoted Synthesis of 3-(Organoselanyl)-1,2-Dihydroquinolines from Diorganyl Diselenides and N-Arylpropargylamines by Sequential Carbon-Carbon and Carbon-Selenium Bond Formation. Adv. Synth. Catal. 2019, 361, 96–104. [Google Scholar] [CrossRef]
- Nevado, C.; Echavarren, A.M. Intramolecular Hydroarylation of Alkynes Catalyzed by Platinum or Gold: Mechanism and endo Selectivity. Chem. Eur. J. 2005, 11, 3155–3164. [Google Scholar] [CrossRef] [PubMed]
- Johannsen, T.; Golz, C.; Alcarazo, M. α-Cationic Phospholes: Synthesis and Applications as Ancillary Ligands. Angew. Chem. Int. Ed. 2020, 59, 22779–22784. [Google Scholar] [CrossRef] [PubMed]
- Ding, D.; Mou, T.; Feng, M.; Jiang, X. Utility of Ligand Effect in Homogenous Gold Catalysis: Enabling Regiodivergent π-Bond-Activated Cyclization. J. Am. Chem. Soc. 2016, 138, 5218–5221. [Google Scholar] [CrossRef]
- Menon, R.S.; Findlay, A.D.; Bissember, A.C.; Banwell, M.G. The Au(I)-Catalyzed Intramolecular Hydroarylation of Terminal Alkynes Under Mild Conditions: Application to the Synthesis of 2H-Chromenes, Coumarins, Benzofurans, and Dihydroquinolines. J. Org. Chem. 2009, 74, 8901–8903. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Marañón, L.; Sarandeses, L.A.; Martínez, M.M.; Pérez Sestelo, J. Synthesis of fused chromenes by the indium(iii)-catalyzed cascade hydroarylation/cycloisomerization reactions of polyyne-type aryl propargyl ethers. Org. Chem. Front. 2018, 5, 2308–2312. [Google Scholar] [CrossRef]
- Alonso-Marañón, L.; Sarandeses, L.A.; Martínez, M.M.; Pérez Sestelo, J. Sequential In-catalyzed intramolecular hydroarylation and Pd-catalyzed cross-coupling reactions using bromopropargyl aryl ethers and amines. Org. Chem. Front. 2017, 4, 500–505. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Larock, R.C. Synthesis of Spiro[4.5]trienones by Intramolecular ipso-Halocyclization of 4-(p-Methoxyaryl)-1-alkynes. J. Am. Chem. Soc. 2005, 127, 12230–12231. [Google Scholar] [CrossRef]
- Arcadi, A.; Fabrizi, G.; Fochetti, A.; Franzini, R.; Ghirga, F.; Goggiamani, A.; Iazzetti, A.; Marrone, F.; Serraiocco, A. Synthesis of Polycyclic Chromene Cores through Gold (I)-Catalyzed Intramolecular Hydroarylation Reaction (IMHA). Eur. J. Org. Chem. 2021, 1676–1687. [Google Scholar] [CrossRef]
- Arcadi, A.; Ciogli, A.; Fabrizi, G.; Fochetti, A.; Franzini, R.; Ghirga, F.; Goggiamani, A.; Iazzetti, A. Synthesis of pyrano[2,3-f]chromen-2-ones vs. pyrano[3,2-g]chromen-2-ones through site controlled gold-catalyzed annulations. Org. Biomol Chem 2019, 17, 10065–10072. [Google Scholar] [CrossRef]
- Arcadi, A.; Blesi, F.; Cacchi, S.; Fabrizi, G.; Goggiamani, A.; Marinelli, F. Gold versus silver catalyzed intramolecular hydroarylation reactions of [(3-arylprop-2-ynyl)oxy]benzene derivatives. Org. Biomol Chem 2012, 10, 9700–9708. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Brindisi, M. Organic Carbamates in Drug Design and Medicinal Chemistry. J. Med. Chem. 2015, 58, 2895–2940. [Google Scholar] [CrossRef] [Green Version]
- Alfonsi, M.; Arcadi, A.; Chiarini, M.; Marinelli, F. Sequential alkylation/gold-catalyzed annulation reactions of anilines with propargylic bromide derivatives. Tetrahedron Lett. 2011, 52, 5145–5148. [Google Scholar] [CrossRef]
- Arcadi, A.; Alfonsi, M.; Chiarini, M.; Marinelli, F. Sequential gold-catalyzed reactions of 1-phenylprop-2-yn-1-ol with 1,3-dicarbonyl compounds. J. Organomet. Chem. 2009, 694, 576–582. [Google Scholar] [CrossRef]
- Martín-Matute, B.; Nevado, C.; Cárdenas, D.J.; Echavarren, A.M. Intramolecular Reactions of Alkynes with Furans and Electron Rich Arenes Catalyzed by PtCl2: The Role of Platinum Carbenes as Intermediates. J. Am. Chem. Soc. 2003, 125, 5757–5766. [Google Scholar] [CrossRef]
- Abbiati, G.; Arcadi, A.; Bianchi, G.; Di Giuseppe, S.; Marinelli, F.; Rossi, E. Sequential Amination/Annulation/Aromatization Reaction of Carbonyl Compounds and Propargylamine: A New One-Pot Approach to Functionalized Pyridines. J. Org. Chem. 2003, 68, 6959–6966. [Google Scholar] [CrossRef]
- Chintawar, C.C.; Yadav, A.K.; Kumar, A.; Sancheti, S.P.; Patil, N.T. Divergent Gold Catalysis: Unlocking Molecular Diversity through Catalyst Control. Chem. Rev. 2021. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Liu, Y.; Lv, P.; Zhu, R.; Liu, C.; Zhang, D. Theoretical Insight into the Mechansim and Origin of Ligand-Controlled Regioselectivity in Homogenous Gold-Catalyzed Intramolecular Hydroarylation of Alkynes. J. Org. Chem. 2018, 83, 2763–2772. [Google Scholar] [CrossRef]
- Vacala, T.; Bejcek, L.P.; Williams, G.; Williamson, A.C.; Vadola, P.A. Gold-Catalyzed Hydroarylation of N-Aryl Alkynamides for the Synthesis of 2-Quinolinones. J. Org. Chem. 2017, 82, 2558–2569. [Google Scholar] [CrossRef]

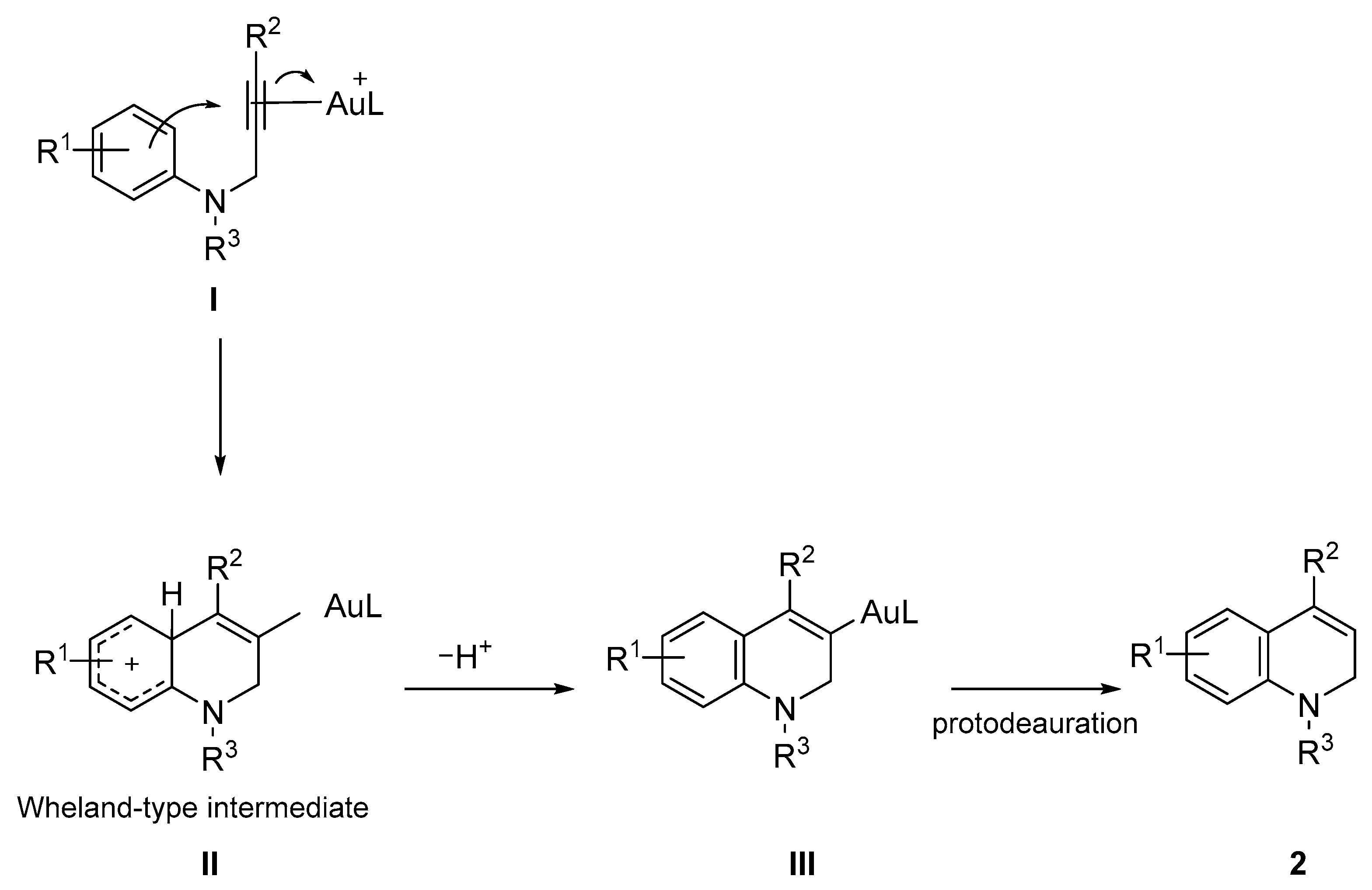

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
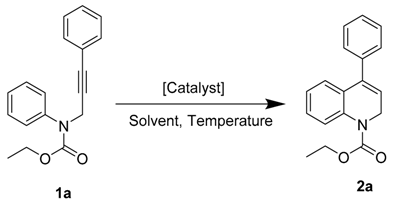
| Entry | Catalyst | T (°C) | Solvent | Time (h) | 2a (Yield %) |
|---|---|---|---|---|---|
| 1 | PtCl2 | 80 | EtOH | 24 | - b |
| 2 | NaAuCl4 | 80 | EtOH | 48 | - c |
| 3 | JPAu(CH3CN)SbF6 | 80 | DCM d | 1 | 98 |
| 4 | JPAu(CH3CN)SbF6 | 100 | CHCl3 | 4.5 | 70 e |
| 5 | JPAu(CH3CN)SbF6 | 100 | CHCl3 d | 0.75 | 86 f |
| 6 | JPAuCl/AgNTf2 | 80 | CH2Cl2 | 3.5 | 94 |
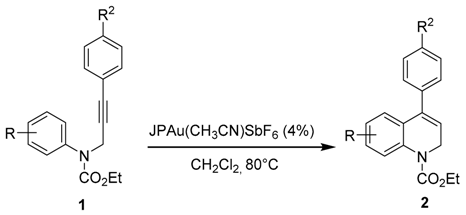
| Entry | R | R2 | 1 | Time (h) | 2 (Yield %) |
|---|---|---|---|---|---|
| 1 | H | H | a | 1 | 2a (98) |
| 2 | H | OMe | b | 18 | 2b (82) |
| 3 | H | COMe | c | 2 | 2c (99) |
| 4 | 4-OMe | H | d | 2 | 2d (82) |
| 5 | 4-Me | H | e | 1 | 2e (99) |
| 6 | 4-Me | OMe | f | 5 | 2f (68) b |
| 7 | 4-Me | COMe | g | 1 | 2g (99) |
| 8 | 3,5-(Me)2 | H | h | 1 | 2h (99) |
| 9 | 4-Cl | H | i | 24 | 2i (56) c |
| 10 | 4-CF3 | OMe | j | 24 | 2j (10) d |
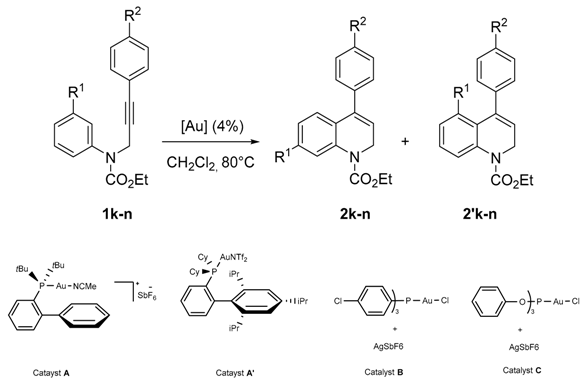
| Entry | R1 | R2 | Catalyst | Overall Yield (%) b | 2/2′ | (Ratio) c |
|---|---|---|---|---|---|---|
| 1 | OMe | H | A | 99 | 2k/2′k | (67/33) |
| 2 | A′ | 99 | (94/6) | |||
| 3 | B | 90 | (44/56) | |||
| 4 | C | 67 | (46/54) | |||
| 5 | OMe | COMe | A | 99 | 2l/2′l | (61/39) |
| 6 | A′ | 99 | (75/25) | |||
| 7 | B | 99 | (54/46) | |||
| 8 | C | 86 | (54/46) | |||
| 9 | OMe | OMe | A | 70 | 2m/2′m | (91/9) |
| 10 | A′ | 73 | (91/9) | |||
| 11 | B | 83 | (63/37) | |||
| 12 | C | 77 | (51/49) | |||
| 13 | COMe | COOMe | A | 85 | 2n/2′n | (88/12) |
| 14 | A′ | 75 | (64/36) | |||
| 15 | B | 99 | (40/60) | |||
| 16 | C | 72 | (19/81) | |||
| 17 | COMe | COMe | A | 88 | 2o/2′o | (88/12) |
| 18 | A′ | 90 | (65/35) | |||
| 19 | B | 70 | (33/67) | |||
| 20 | C | 82 | (20/80) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arcadi, A.; Calcaterra, A.; Fabrizi, G.; Fochetti, A.; Goggiamani, A.; Iazzetti, A.; Marrone, F.; Marsicano, V.; Mazzoccanti, G.; Serraiocco, A. Synthesis of 4-Substituted-1,2-Dihydroquinolines by Means of Gold-Catalyzed Intramolecular Hydroarylation Reaction of N-Ethoxycarbonyl-N-Propargylanilines. Molecules 2021, 26, 3366. https://doi.org/10.3390/molecules26113366
Arcadi A, Calcaterra A, Fabrizi G, Fochetti A, Goggiamani A, Iazzetti A, Marrone F, Marsicano V, Mazzoccanti G, Serraiocco A. Synthesis of 4-Substituted-1,2-Dihydroquinolines by Means of Gold-Catalyzed Intramolecular Hydroarylation Reaction of N-Ethoxycarbonyl-N-Propargylanilines. Molecules. 2021; 26(11):3366. https://doi.org/10.3390/molecules26113366
Chicago/Turabian StyleArcadi, Antonio, Andrea Calcaterra, Giancarlo Fabrizi, Andrea Fochetti, Antonella Goggiamani, Antonia Iazzetti, Federico Marrone, Vincenzo Marsicano, Giulia Mazzoccanti, and Andrea Serraiocco. 2021. "Synthesis of 4-Substituted-1,2-Dihydroquinolines by Means of Gold-Catalyzed Intramolecular Hydroarylation Reaction of N-Ethoxycarbonyl-N-Propargylanilines" Molecules 26, no. 11: 3366. https://doi.org/10.3390/molecules26113366