
Using Design of Experiments to Optimize a Screening Analytical Methodology Based on Solid-Phase Microextraction/Gas Chromatography for the Determination of Volatile Methylsiloxanes in Water

 ,
,  ,
,
Abstract
:1. Introduction
2. Results and Discussion
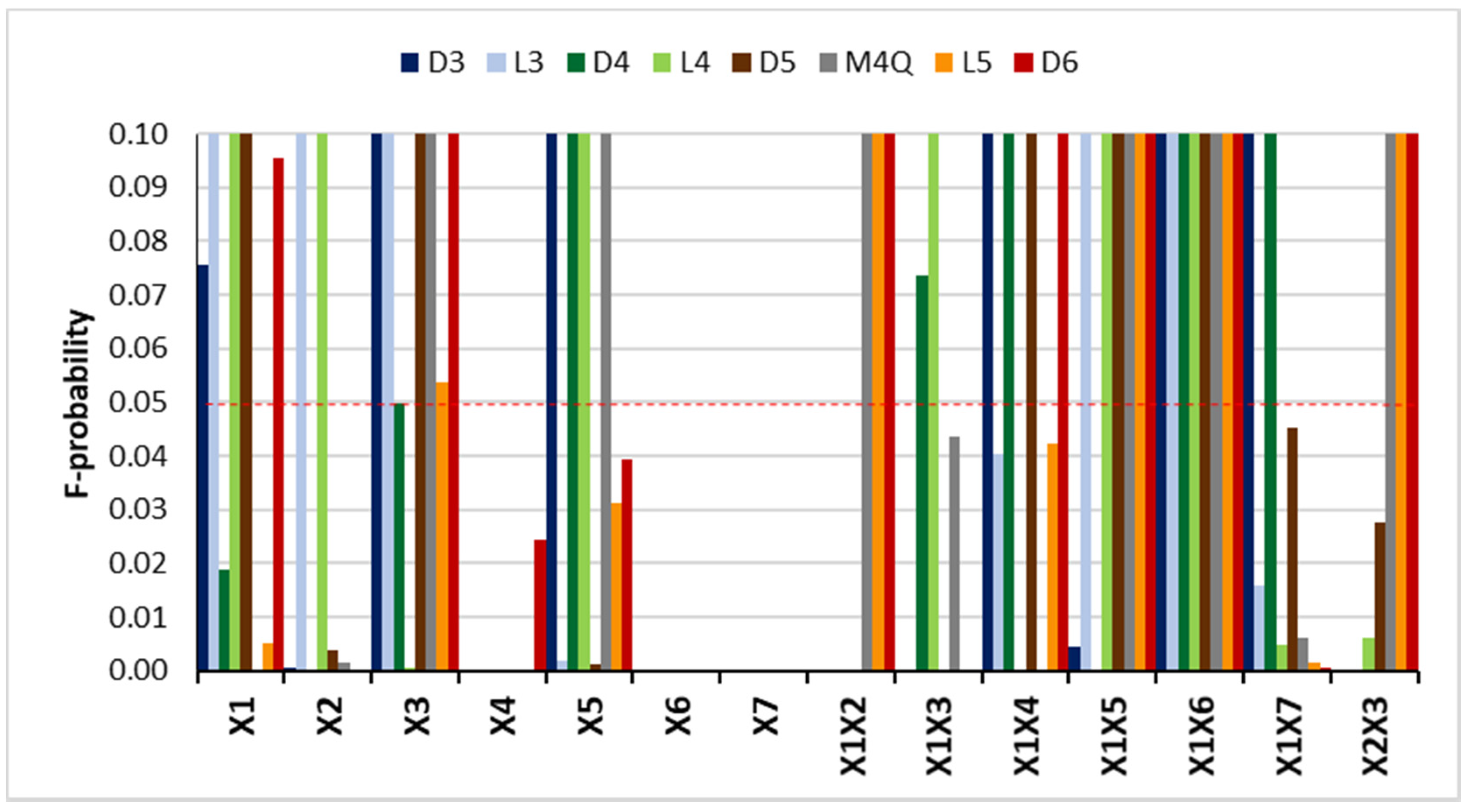
2.1. Optimization of the Extraction Procedure
2.1.1. Screening Design
2.1.2. Central Composite Design
2.2. Method Validation
2.3. Application of the Developed Method to Environmental Samples
3. Materials and Methods
3.1. Materials
3.2. Solid-Phase Microextraction Procedure
3.3. GC-FID Analysis
3.4. Quality Assurance and Control (QA/QC)
3.5. Design of Experiments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Rücker, C.; Kümmerer, K. Environmental Chemistry of Organosiloxanes. Chem. Rev. 2014, 115, 466–524. [Google Scholar] [CrossRef] [PubMed]
- Lassen, C.; Hansen, C.; Mikkelsen, S.; Maag, J. Siloxanes-Consumption, Toxicity and Alternatives, 1st ed.; COWI A/S: Kongens Lyngby, Denmark, 2005; Volume 1031. [Google Scholar]
- Alleni, R.B.; Kochs, P.; Chandra, G. Industrial Organosilicon Materials, Their Environmental Entry and Predicted Fate. In Organosilicon Materials: The Handbook of Environmental Chemistry (Anthropogenic Compounds. Part H); Chandra, G., Ed.; Springer: Berlin/Heidelberg, Germany, 1997; Volume 3/3H. [Google Scholar]
- Montemayor, B.; Price, B.; Egmond, R. Accounting for intended use application in characterizing the contributions of cyclopentasiloxane (D5) to aquatic loadings following personal care product use: Antiperspirants, skin care products and hair care products. Chemosphere 2013, 93, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.G.; Steer, H.; Tait, T.; Williams, Z.; Pacepavicius, G.; Young, T.; Ng, T.; Smyth, S.A.; Kinsman, L.; Alaee, M. Concentrations of cyclic volatile methylsiloxanes in biosolid amended soil, influent, effluent, receiving water, and sediment of wastewater treatment plants in Canada. Chemosphere 2013, 93, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Bletsou, A.A.; Asimakopoulos, A.G.; Stasinakis, A.S.; Thomaidis, N.S.; Kannan, K. Mass Loading and Fate of Linear and Cyclic Siloxanes in a Wastewater Treatment Plant in Greece. Environ. Sci. Technol. 2013, 47, 1824–1832. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Shen, M.; Tian, Y.; Zeng, G. Cyclic volatile methylsiloxanes in sediment, soil, and surface water from Dongting Lake, China. J. Soils Sediments 2018, 18, 2063–2071. [Google Scholar] [CrossRef]
- European Commission. Commission Regulation (EU) 2018/35 of 10 January 2018 Amending Annex XVII to Regulation (EC) No 1907/2006 of the European Parliament and of the Council Concerning the Registration, Evaluation, Authorization and Restriction of Chemicals (REACH) as Regards Octamethylcyclotetrasiloxane (‘D4’) and Decamethylcyclopentasiloxane (‘D5’). 2018. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32018R0035&from=EN (accessed on 4 January 2020).
- European Chemicals Agency (ECHA). Proposal for a Restriction of Octamethylcyclotetrasiloxane (D4), Decamethylcyclopentasiloxane (D5) and Dodecamethylcyclohexasiloxane (D6)—Annex XV Restriction Report; European Chemicals Agency (ECHA): Helsinki, Finland, 2019; Available online: https://echa.europa.eu/documents/10162/4157b383-d51a-0219-7715-a63a3299fd2c (accessed on 4 January 2020).
- Capela, D.; Ratola, N.; Alves, A.; Homem, V. Volatile methylsiloxanes through wastewater treatment plants—A review of levels and implications. Environ. Int. 2017, 102, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Strange, R.S. Introduction to experiment design for chemists. J. Chem. Educ. 1990, 67, 113. [Google Scholar] [CrossRef]
- Xu, L.; Shi, Y.; Cai, Y. Occurrence and fate of volatile siloxanes in a municipal Wastewater Treatment Plant of Beijing, China. Water Res. 2013, 47, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Companioni-Damas, E.Y.; Santos, F.J.; Galceran, M.T. Analysis of linear and cyclic methylsiloxanes in water by headspace-solid phase microextraction and gas chromatography–mass spectrometry. Talanta 2012, 89, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.N.; Miller, J.C. Statistics and Chemometrics for Analytical Chemistry, 6th ed.; Pearson/Prentice Hall: Harlow, UK, 2010. [Google Scholar]
- Ellison, S.; Rosslein, M.; Williams, A. EURACHEM/CITAC Guide, Quantifying Uncertainty in Analytical Measurement, 3rd ed.; EURACHEM: Teddington, UK, 2012. [Google Scholar]
- Konieczka, P.; Namieśnik, J. Estimating uncertainty in analytical procedures based on chromatographic techniques. J. Chromatogr. A 2010, 1217, 882–891. [Google Scholar] [CrossRef] [PubMed]
- Capela, D.; Homem, V.; Alves, A.; Santos, L. Volatile methylsiloxanes in personal care products—Using QuEChERS as a “green” analytical approach. Talanta 2016, 155, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Ratola, N.; Santos, L.; Herbert, P.; Alves, A. Uncertainty associated to the analysis of organochlorine pesticides in water by solid-phase microextraction/gas chromatography-electron capture detection—Evaluation using two different approaches. Anal. Chim. Acta 2006, 573–574, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Mousavi, L.; Tamiji, Z.; Khoshayand, M.R. Applications and opportunities of experimental design for the dispersive liquid-liquid microextraction method—A review. Talanta 2018, 190, 335–356. [Google Scholar] [CrossRef] [PubMed]
- Homem, V.; Alves, A.; Santos, L. Ultrasound-assisted dispersive liquid–liquid microextraction for the determination of synthetic musk fragrances in aqueous matrices by gas chromatography–mass spectrometry. Talanta 2016, 148, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Ramos, S.; Homem, V.; Santos, L. Development and optimization of a QuEChERS-GC-MS/MS methodology to analyse ultraviolet-filters and synthetic musks in sewage sludge. Sci. Total Environ. 2019, 651, 2606–2614. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| i | Factor | Coded Values (xi) | |
|---|---|---|---|
| Low (−1) | High (+1) | ||
| 1 | Ionic strength (% NaCl) | 0 | 20 |
| 2 | Extraction time (min) | 5 | 45 |
| 3 | Desorption time (min) | 1 | 10 |
| 4 | Extraction temperature (°C) | 25 | 80 |
| 5 | Desorption temperature (°C) | 200 | 250 |
| 6 | Fiber type | PDMS | PDMS/DVB |
| 7 | Sample volume (mL) | 5 | 10 |
| Run | X1 Ionic Strength (% w/v) | X2 Extraction Time (min) | X3 Desorption Time (min) | X4 Extraction Temperature (°C) | X5 Desorption Temperature (°C) | X6 Fiber Type | X7 Sample Volume (mL) |
|---|---|---|---|---|---|---|---|
| 1 | 0 | 5 | 1 | 25 | 200 | PDMS | 5 |
| 2 | 20 | 45 | 10 | 25 | 200 | PDMS | 5 |
| 3 | 0 | 45 | 10 | 25 | 200 | PDMS/DVB | 10 |
| 4 | 20 | 5 | 1 | 25 | 200 | PDMS/DVB | 10 |
| 5 | 0 | 45 | 1 | 25 | 250 | PDMS | 10 |
| 6 | 20 | 5 | 10 | 25 | 250 | PDMS | 10 |
| 7 | 0 | 5 | 10 | 25 | 250 | PDMS/DVB | 5 |
| 8 | 20 | 45 | 1 | 25 | 250 | PDMS/DVB | 5 |
| 9 | 0 | 5 | 10 | 80 | 200 | PDMS | 10 |
| 10 | 20 | 45 | 1 | 80 | 200 | PDMS | 10 |
| 11 | 0 | 45 | 1 | 80 | 200 | PDMS/DVB | 5 |
| 12 | 20 | 5 | 10 | 80 | 200 | PDMS/DVB | 5 |
| 13 | 0 | 45 | 10 | 80 | 250 | PDMS | 5 |
| 14 | 20 | 5 | 1 | 80 | 250 | PDMS | 5 |
| 15 | 0 | 5 | 1 | 80 | 250 | PDMS/DVB | 10 |
| 16 | 20 | 45 | 10 | 80 | 250 | PDMS/DVB | 10 |
| Analyte | Linearity Range (µg/L) (n = 8) | Correlation Factor of the Calibration Curve (R) | Limit of Detection (µg/L) | Limit of Quantification (µg/L) |
|---|---|---|---|---|
| L3 | 0.125–5 | 0.998 | 0.024 | 0.080 |
| L4 | 0.125–5 | 0.997 | 0.014 | 0.047 |
| L5 | 0.125–5 | 0.998 | 0.018 | 0.061 |
| D3 | 0.125–5 | 0.992 | 0.015 | 0.050 |
| D4 | 0.125–5 | 0.997 | 0.015 | 0.049 |
| D5 | 0.125–5 | 0.996 | 0.018 | 0.059 |
| D6 | 0.125–5 | 0.993 | 0.014 | 0.046 |
| Analyte | Intra-Day Precision, n = 4 (%RSD) | Inter-Day Precision, n = 3 (%RSD) | Accuracy (% Mean Recovery ± SD) | ||||
|---|---|---|---|---|---|---|---|
| 1 µg/L | 5 µg/L | 1 µg/L | 5 µg/L | Wastewater | Tap Water | River Water | |
| L3 | 10 | 10 | 12 | 15 | 102 ± 3 | 100 ± 27 | 104 ± 12 |
| L4 | 10 | 12 | 12 | 18 | 79 ± 8 | 81 ± 4 | 100 ± 8 |
| L5 | 10 | 12 | 11 | 19 | 94 ± 5 | 88 ± 18 | 75 ± 16 |
| D3 | 10 | 14 | 13 | 17 | 84 ± 14 | 102 ± 26 | 87 ± 11 |
| D4 | 14 | 12 | 14 | 17 | 89 ± 5 | 82 ± 16 | 101 ± 12 |
| D5 | 13 | 11 | 12 | 18 | 76 ± 10 | 62 ± 8 | 85 ± 13 |
| D6 | 10 | 11 | 12 | 19 | 93 ± 15 | 70 ± 10 | 74 ± 18 |
| Average | 11 | 12 | 12 | 17 | 88 ± 8 | 84 ± 16 | 89 ± 13 |
| Analyte | Wastewater (µg/L) | Tap Water (µg/L) | River Water (µg/L) |
|---|---|---|---|
| L3 | 0.14 ± 0.23 | nd | nd |
| L4 | 0.44 ± 0.10 | nd | nd |
| L5 | 0.27 ± 0.12 | nd | nd |
| D3 | 0.67 ± 0.11 | nd | nd |
| D4 | 0.39 ± 0.16 | nd | nd |
| D5 | 0.34 ± 0.19 | nd | nd |
| D6 | 0.70 ± 0.13 | nd | nd |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bernardo, F.; González-Hernández, P.; Ratola, N.; Pino, V.; Alves, A.; Homem, V. Using Design of Experiments to Optimize a Screening Analytical Methodology Based on Solid-Phase Microextraction/Gas Chromatography for the Determination of Volatile Methylsiloxanes in Water. Molecules 2021, 26, 3429. https://doi.org/10.3390/molecules26113429
Bernardo F, González-Hernández P, Ratola N, Pino V, Alves A, Homem V. Using Design of Experiments to Optimize a Screening Analytical Methodology Based on Solid-Phase Microextraction/Gas Chromatography for the Determination of Volatile Methylsiloxanes in Water. Molecules. 2021; 26(11):3429. https://doi.org/10.3390/molecules26113429
Chicago/Turabian StyleBernardo, Fábio, Providencia González-Hernández, Nuno Ratola, Verónica Pino, Arminda Alves, and Vera Homem. 2021. "Using Design of Experiments to Optimize a Screening Analytical Methodology Based on Solid-Phase Microextraction/Gas Chromatography for the Determination of Volatile Methylsiloxanes in Water" Molecules 26, no. 11: 3429. https://doi.org/10.3390/molecules26113429