
Molecular Dynamics Simulations of a Cytochrome P450 from Tepidiphilus thermophilus (P450-TT) Reveal How Its Substrate-Binding Channel Opens



Abstract
:1. Introduction
2. Results
3. Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Sono, M.; Roach, M.P.; Coulter, E.D.; Dawson, J.H. Heme-containing oxygenases. Chem. Rev. 1996, 96, 2841–2888. [Google Scholar] [CrossRef] [PubMed]
- Groves, J.T. The bioinorganic chemistry of iron in oxygenases and supramolecular assemblies. Proc. Natl. Acad. Sci. USA 2003, 100, 3569–3574. [Google Scholar] [CrossRef] [Green Version]
- Denisov, I.G.; Makris, T.M.; Sligar, S.G.; Schlichting, I. Structure and chemistry of cytochrome P450. Chem. Rev. 2005, 105, 2253–2278. [Google Scholar] [CrossRef] [PubMed]
- Ruppel, J.; Fields, K.; Snyder, N.; Zhang, X.; Kadish, K.; Smith, K.; Guilard, R. Handbook of Porphyrin Science; World Scientific Publishing Co.: Singapore, 2010. [Google Scholar]
- Ortiz de Montellano, P.R. Hydrocarbon hydroxylation by cytochrome P450 enzymes. Chem. Rev. 2010, 110, 932–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meunier, B.; De Visser, S.P.; Shaik, S. Mechanism of oxidation reactions catalyzed by cytochrome P450 enzymes. Chem. Rev. 2004, 104, 3947–3980. [Google Scholar] [CrossRef] [PubMed]
- Nebert, D.W.; Russell, D.W. Clinical importance of the cytochromes P450. Lancet 2002, 360, 1155–1162. [Google Scholar] [CrossRef]
- Fleming, I.; Busse, R. Endothelium-derived epoxyeicosatrienoic acids and vascular function. Hypertension 2006, 47, 629–633. [Google Scholar] [CrossRef]
- Johnson, E.F.; Stout, C.D. Structural diversity of human xenobiotic-metabolizing cytochrome P450 monooxygenases. Biochem. Biophys. Res. Commun. 2005, 338, 331–336. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Yadav, J.S. Microbial P450 enzymes in bioremediation and drug discovery: Emerging potentials and challenges. Curr. Protein Pept. Sci. 2018, 19, 75–86. [Google Scholar] [CrossRef]
- Andersen, J.F.; Tatsuta, K.; Gunji, H.; Ishiyama, T.; Hutchinson, C.R. Substrate specificity of 6-deoxyerythronolide B hydroxylase, a bacterial cytochrome P450 of erythromycin A biosynthesis. Biochemistry 1993, 32, 1905–1913. [Google Scholar] [CrossRef]
- Wang, B.; Yang, L.-P.; Zhang, X.-Z.; Huang, S.-Q.; Bartlam, M.; Zhou, S.-F. New insights into the structural characteristics and functional relevance of the human cytochrome P450 2D6 enzyme. Drug Metab. Rev. 2009, 41, 573–643. [Google Scholar] [CrossRef] [PubMed]
- Guengerich, F.P. Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chem. Res. Toxicol. 2001, 14, 611–650. [Google Scholar] [CrossRef]
- Munro, A.W.; Lindsay, J.G. Bacterial cytochromes P-450. Molec. Microbiol. 1996, 20, 1115–1125. [Google Scholar] [CrossRef]
- Hanukoglu, I. Electron transfer proteins of cytochrome P450 systems. In Advances in Molecular and Cell Biology; Bittar, E.E., Ed.; Elsevier: Amsterdam, The Netherlands, 1996; Volume 14, pp. 29–56. [Google Scholar] [CrossRef]
- De Mot, R.; Parret, A.H. A novel class of self-sufficient cytochrome P450 monooxygenases in prokaryotes. Trends Microbiol. 2002, 10, 502–508. [Google Scholar] [CrossRef]
- Warman, A.J.; Robinson, J.W.; Luciakova, D.; Lawrence, A.D.; Marshall, K.R.; Warren, M.J.; Cheesman, M.R.; Rigby, S.E.; Munro, A.W.; McLean, K.J. Characterization of Cupriavidus metallidurans CYP116B1–A thiocarbamate herbicide oxygenating P450–phthalate dioxygenase reductase fusion protein. FEBS J. 2012, 279, 1675–1693. [Google Scholar] [CrossRef]
- Minerdi, D.; Sadeghi, S.J.; Di Nardo, G.; Rua, F.; Castrignanò, S.; Allegra, P.; Gilardi, G. CYP116B5: A new class VII catalytically self-sufficient cytochrome P 450 from A cinetobacter radioresistens that enables growth on alkanes. Molec. Microbiol. 2015, 95, 539–554. [Google Scholar] [CrossRef]
- Hrycay, E.G.; Bandiera, S.M. Monooxygenase, peroxidase and peroxygenase properties and reaction mechanisms of cytochrome P450 enzymes. In Monooxygenase, Peroxidase and Peroxygenase Properties and Mechanisms of Cytochrome P450; Hrycay, E.G., Bandiera, S.M., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 1–61. [Google Scholar] [CrossRef]
- Qiu, Y.; Tittiger, C.; Wicker-Thomas, C.; Le Goff, G.; Young, S.; Wajnberg, E.; Fricaux, T.; Taquet, N.; Blomquist, G.J.; Feyereisen, R. An insect-specific P450 oxidative decarbonylase for cuticular hydrocarbon biosynthesis. Proc. Natl. Acad. Sci. USA 2012, 109, 14858–14863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Ullrich, R.; Hofrichter, M.; Groves, J.T. Heme-thiolate ferryl of aromatic peroxygenase is basic and reactive. Proc. Natl. Acad. Sci. USA 2015, 112, 3686–3691. [Google Scholar] [CrossRef] [Green Version]
- Rittle, J.; Green, M.T. Cytochrome P450 compound I: Capture, characterization, and CH bond activation kinetics. Science 2010, 330, 933–937. [Google Scholar] [CrossRef] [Green Version]
- Nishida, C.R.; de Montellano, P.R.O. Thermophilic cytochrome P450 enzymes. Biochem. Biophys. Res. Commun. 2005, 338, 437–445. [Google Scholar] [CrossRef]
- Roberts, G.A.; Celik, A.; Hunter, D.J.; Ost, T.W.; White, J.H.; Chapman, S.K.; Turner, N.J.; Flitsch, S.L. A self-sufficient cytochrome P450 with a primary structural organization that includes a flavin domain and a [2Fe-2S] redox center. J. Biol. Chem. 2003, 278, 48914–48920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciaramella, A.; Catucci, G.; Gilardi, G.; Di Nardo, G. Crystal structure of bacterial CYP116B5 heme domain: New insights on class VII P450s structural flexibility and peroxygenase activity. Int. J. Biol. Macromol. 2019, 140, 577–587. [Google Scholar] [CrossRef] [PubMed]
- Manning, J.; Tavanti, M.; Porter, J.L.; Kress, N.; De Visser, S.P.; Turner, N.J.; Flitsch, S.L. Regio- and Enantio-selective Chemo-enzymatic C-H-Lactonization of Decanoic Acid to (S)-delta-Decalactone. Angew. Chem. Int. Ed. Engl. 2019, 58, 5668–5671. [Google Scholar] [CrossRef] [Green Version]
- Tavanti, M.; Porter, J.L.; Levy, C.W.; Castellanos, J.R.G.; Flitsch, S.L.; Turner, N.J. The crystal structure of P450-TT heme-domain provides the first structural insights into the versatile class VII P450s. Biochem. Biophys. Res. Commun. 2018, 501, 846–850. [Google Scholar] [CrossRef] [PubMed]
- Pravda, L.; Berka, K.; Vařeková, R.S.; Sehnal, D.; Banáš, P.; Laskowski, R.A.; Koča, J.; Otyepka, M. Anatomy of enzyme channels. BMC Bioinform. 2014, 15, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urban, P.; Lautier, T.; Pompon, D.; Truan, G. Ligand access channels in cytochrome P450 enzymes: A review. Int. J. Mol. Sci. 2018, 19, 1617. [Google Scholar] [CrossRef] [Green Version]
- Gora, A.; Brezovsky, J.; Damborsky, J. Gates of enzymes. Chem. Rev. 2013, 113, 5871–5923. [Google Scholar] [CrossRef]
- Kingsley, L.J.; Lill, M.A. Substrate tunnels in enzymes: Structure–function relationships and computational methodology. Proteins 2015, 83, 599–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, Z.; Treado, J.D.; Grigas, A.T.; Levine, Z.A.; Regan, L.; O’Hern, C.S. Analyses of protein cores reveal fundamental differences between solution and crystal structures. Proteins 2020, 88, 1154–1161. [Google Scholar] [CrossRef] [Green Version]
- Tien, M.Z.; Meyer, A.G.; Sydykova, D.K.; Spielman, S.J.; Wilke, C.O. Maximum allowed solvent accessibilites of residues in proteins. PLoS ONE 2013, 8, e80635. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09 Gaussian; Gaussian 09 Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Case, D.A.; Betz, R.M.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Homeyer, N.; et al. AMBER 2016; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012, 40, W537–W541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myers, J.; Grothaus, G.; Narayanan, S.; Onufriev, A. A simple clustering algorithm can be accurate enough for use in calculations of pKs in macromolecules. Proteins 2006, 63, 928–938. [Google Scholar] [CrossRef] [Green Version]
- Gordon, J.C.; Myers, J.B.; Folta, T.; Shoja, V.; Heath, L.S.; Onufriev, A. H++: A server for estimating p K as and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005, 33 (Suppl. S2), W368–W371. [Google Scholar] [CrossRef]
- Becke, A. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self—Consistent molecular orbital methods. XII. Further extensions of Gaussian—Type basis sets for use in molecular orbital studies of organic molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Li, P.; Merz, K.M., Jr. MCPB. py: A python based metal center parameter builder. J. Chem. Inf. Model. 2016, 56, 599–604. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
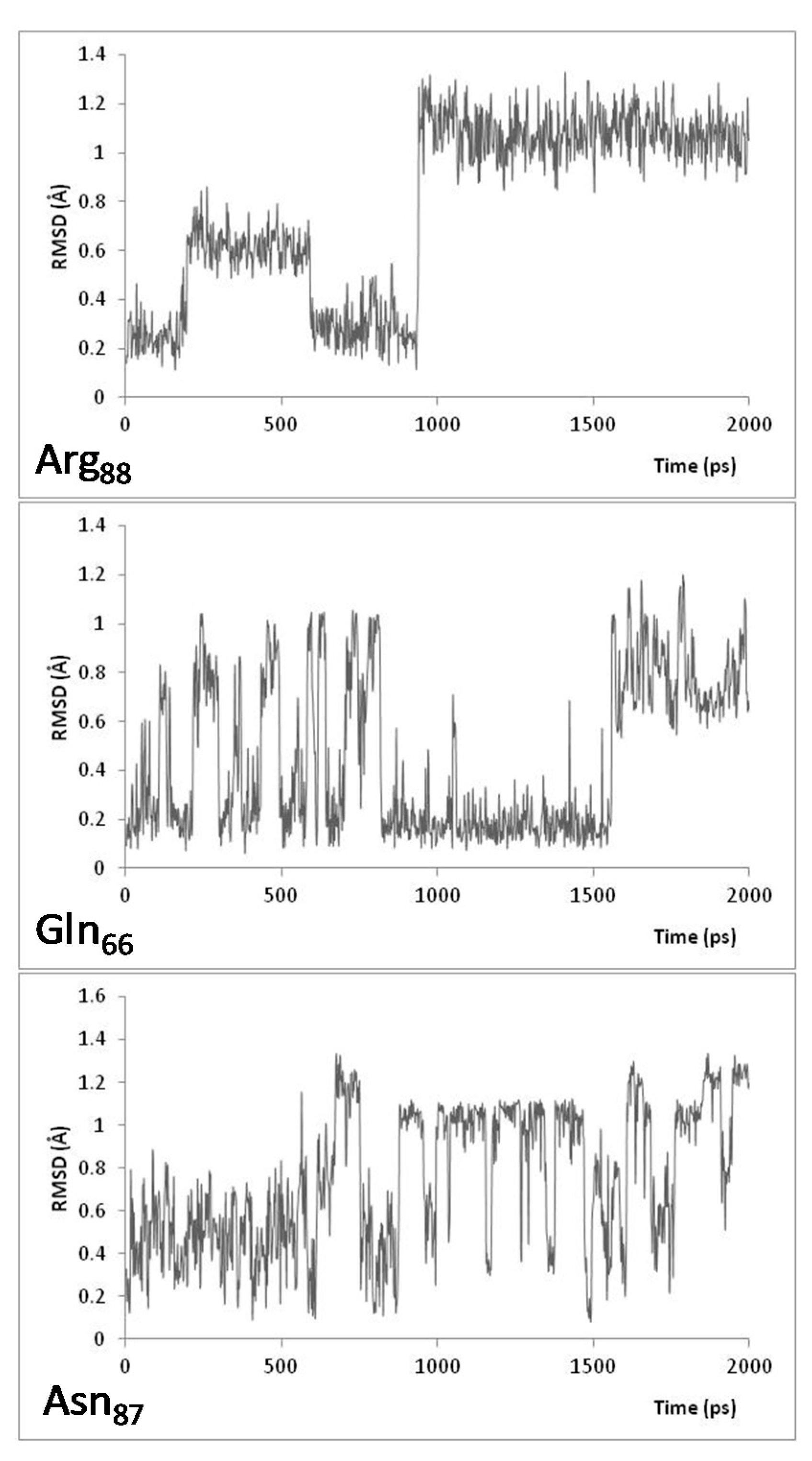


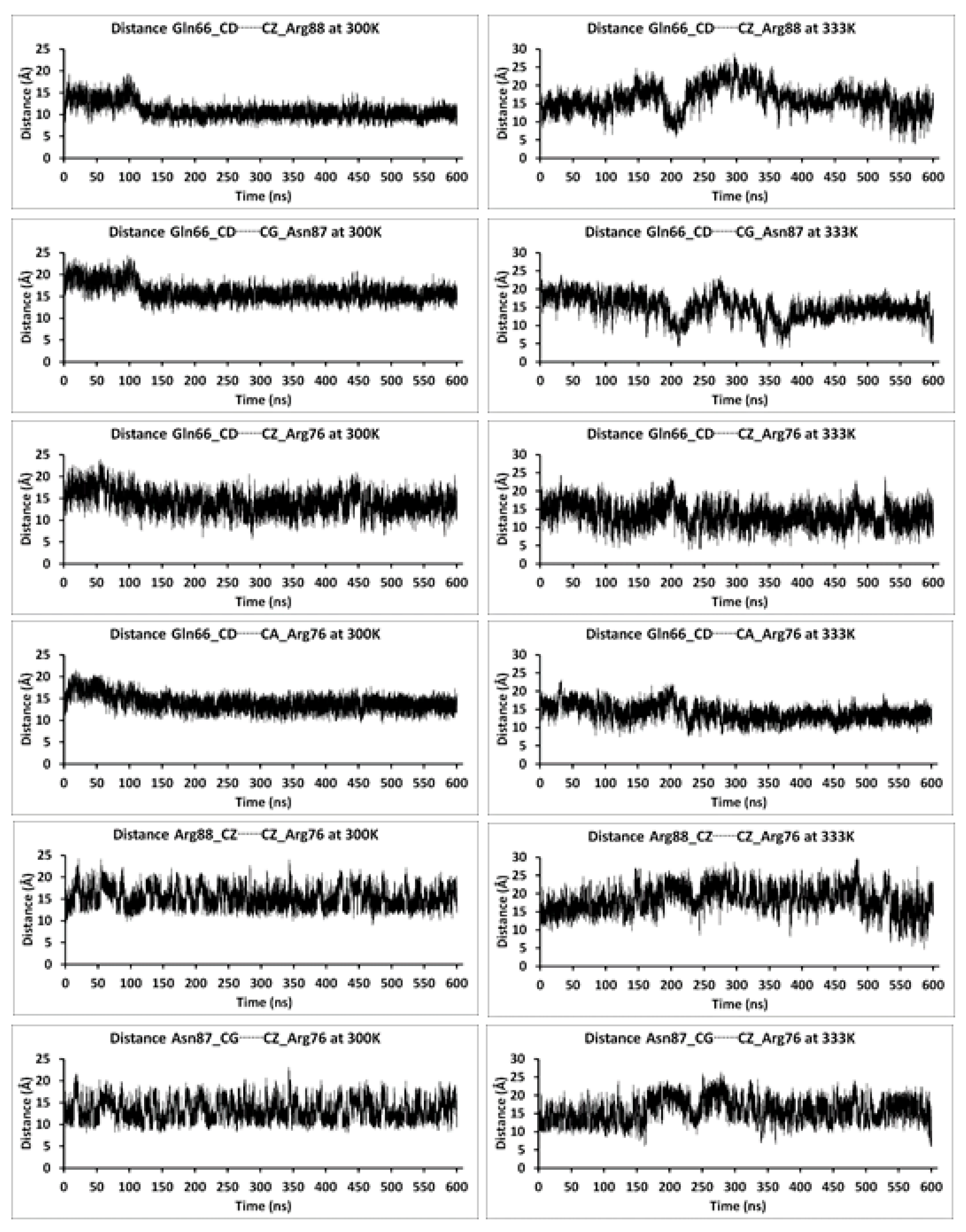




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Relative Solvent Accessibility (RAS) at 300 K | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Residues | Crystal Structure | Minimized Structure | Equilibrated Structure | 100 ns | 200 ns | 300 ns | 400 ns | 500 ns | 600 ns |
| Leu65 | 0.31 | 0.32 | 0.36 | 0.38 | 0.23 | 0.25 | 0.26 | 0.23 | 0.24 |
| Gln66 | 0.27 | 0.40 | 0.26 | 0.50 | 0.46 | 0.48 | 0.35 | 0.41 | 0.38 |
| Ser67 | 0.26 | 0.20 | 0.21 | 0.20 | 0.14 | 0.15 | 0.16 | 0.22 | 0.27 |
| Phe68 | 0.15 | 0.16 | 0.20 | 0.20 | 0.09 | 0.08 | 0.11 | 0.13 | 0.14 |
| Arg76 | 0.58 | 0.55 | 0.40 | 0.62 | 0.51 | 0.56 | 0.40 | 0.49 | 0.49 |
| Asn87 | 0.22 | 0.27 | 0.26 | 0.47 | 0.35 | 0.42 | 0.57 | 0.34 | 0.35 |
| Arg88 | 0.47 | 0.39 | 0.55 | 0.38 | 0.36 | 0.36 | 0.36 | 0.37 | 0.36 |
| Val91 | 0.27 | 0.12 | 0.14 | 0.35 | 0.11 | 0.16 | 0.17 | 0.17 | 0.14 |
| Val246 | 0.25 | 0.26 | 0.21 | 0.11 | 0.03 | 0.02 | 0.03 | 0.03 | 0.01 |
| Phe397 | 0.05 | 0.03 | 0.04 | 0.06 | 0.07 | 0.04 | 0.09 | 0.06 | 0.08 |
| Relative Solvent Accessibility (RAS) at 333 K | |||||||||
| Residues | Crystal Structure | Minimized Structure | Equilibrated Structure | 100 ns | 200 ns | 300 ns | 400 ns | 500 ns | 600 ns |
| Leu65 | 0.31 | 0.32 | 0.32 | 0.39 | 0.11 | 0.27 | 0.20 | 0.24 | 0.22 |
| Gln66 | 0.27 | 0.40 | 0.42 | 0.60 | 0.13 | 0.42 | 0.26 | 0.38 | 0.54 |
| Ser67 | 0.26 | 0.20 | 0.23 | 0.30 | 0.30 | 0.16 | 0.20 | 0.13 | 0.16 |
| Phe68 | 0.15 | 0.16 | 0.23 | 0.42 | 0.56 | 0.17 | 0.14 | 0.11 | 0.22 |
| Arg76 | 0.58 | 0.55 | 0.48 | 0.38 | 0.38 | 0.59 | 0.57 | 0.31 | 0.46 |
| Phe86 | 0.32 | 0.45 | 0.32 | 0.34 | 0.32 | 0.59 | 0.38 | 0.31 | 0.17 |
| Asn87 | 0.22 | 0.27 | 0.23 | 0.56 | 0.05 | 0.15 | 0.18 | 0.20 | 0.65 |
| Arg88 | 0.47 | 0.39 | 0.35 | 0.40 | 0.41 | 0.32 | 0.52 | 0.80 | 0.46 |
| Val91 | 0.27 | 0.12 | 0.18 | 0.34 | 0.36 | 0.78 | 0.14 | 0.28 | 0.26 |
| Val246 | 0.25 | 0.26 | 0.14 | 0.32 | 0.11 | 0.27 | 0.04 | 0.02 | 0.04 |
| Phe397 | 0.05 | 0.03 | 0.03 | 0.14 | 0.02 | 0.04 | 0.05 | 0.05 | 0.06 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faponle, A.S.; Roy, A.; Adelegan, A.A.; Gauld, J.W. Molecular Dynamics Simulations of a Cytochrome P450 from Tepidiphilus thermophilus (P450-TT) Reveal How Its Substrate-Binding Channel Opens. Molecules 2021, 26, 3614. https://doi.org/10.3390/molecules26123614
Faponle AS, Roy A, Adelegan AA, Gauld JW. Molecular Dynamics Simulations of a Cytochrome P450 from Tepidiphilus thermophilus (P450-TT) Reveal How Its Substrate-Binding Channel Opens. Molecules. 2021; 26(12):3614. https://doi.org/10.3390/molecules26123614
Chicago/Turabian StyleFaponle, Abayomi S., Anupom Roy, Ayodeji A. Adelegan, and James W. Gauld. 2021. "Molecular Dynamics Simulations of a Cytochrome P450 from Tepidiphilus thermophilus (P450-TT) Reveal How Its Substrate-Binding Channel Opens" Molecules 26, no. 12: 3614. https://doi.org/10.3390/molecules26123614
APA StyleFaponle, A. S., Roy, A., Adelegan, A. A., & Gauld, J. W. (2021). Molecular Dynamics Simulations of a Cytochrome P450 from Tepidiphilus thermophilus (P450-TT) Reveal How Its Substrate-Binding Channel Opens. Molecules, 26(12), 3614. https://doi.org/10.3390/molecules26123614