
Chiroptical Sensing of Amino Acid Derivatives by Host–Guest Complexation with Cyclo[6]aramide

 ,
,  and
and
Abstract
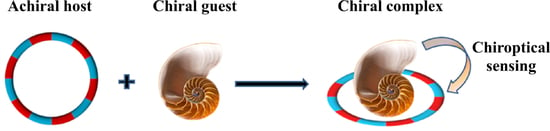
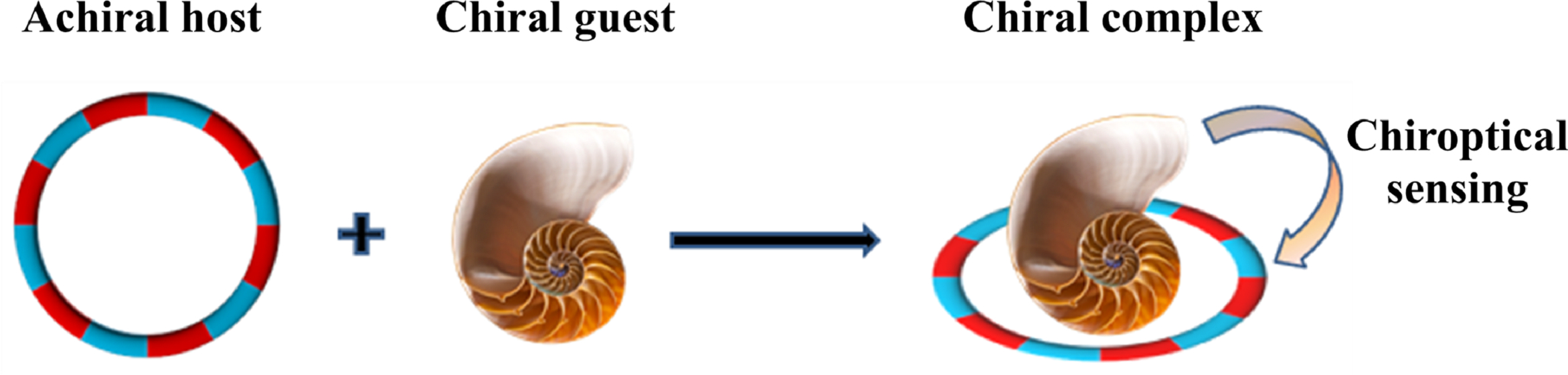
:
1. Introduction
2. Results and Discussion
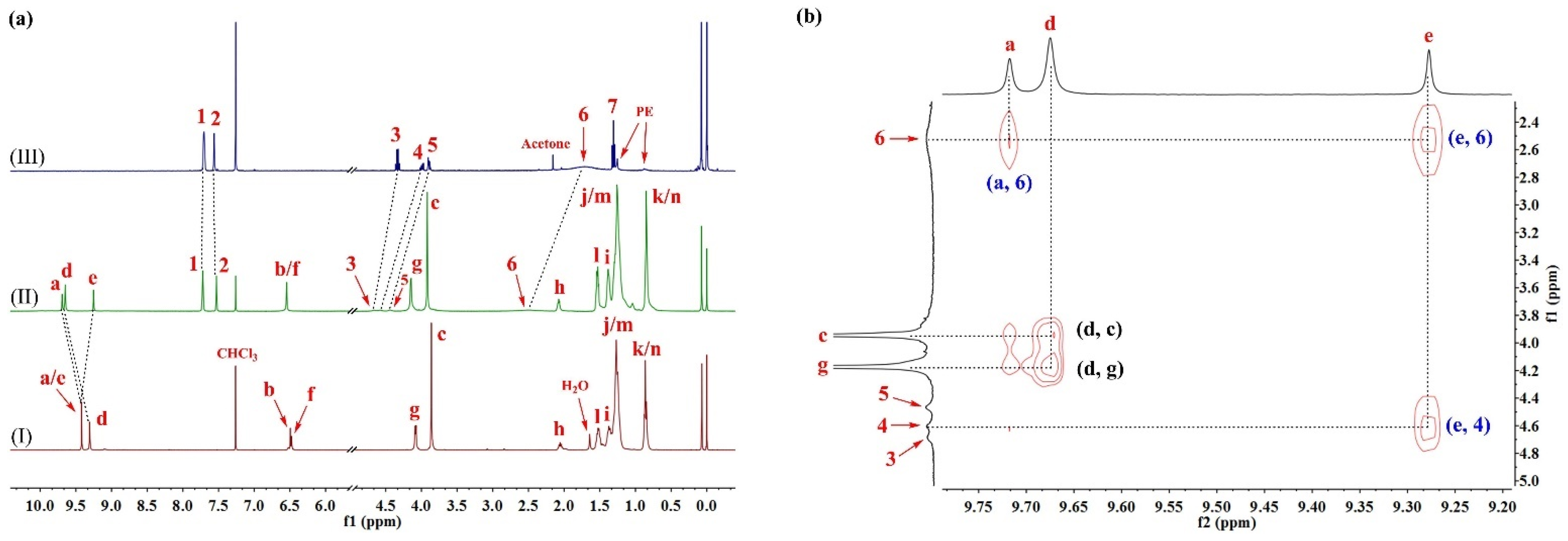
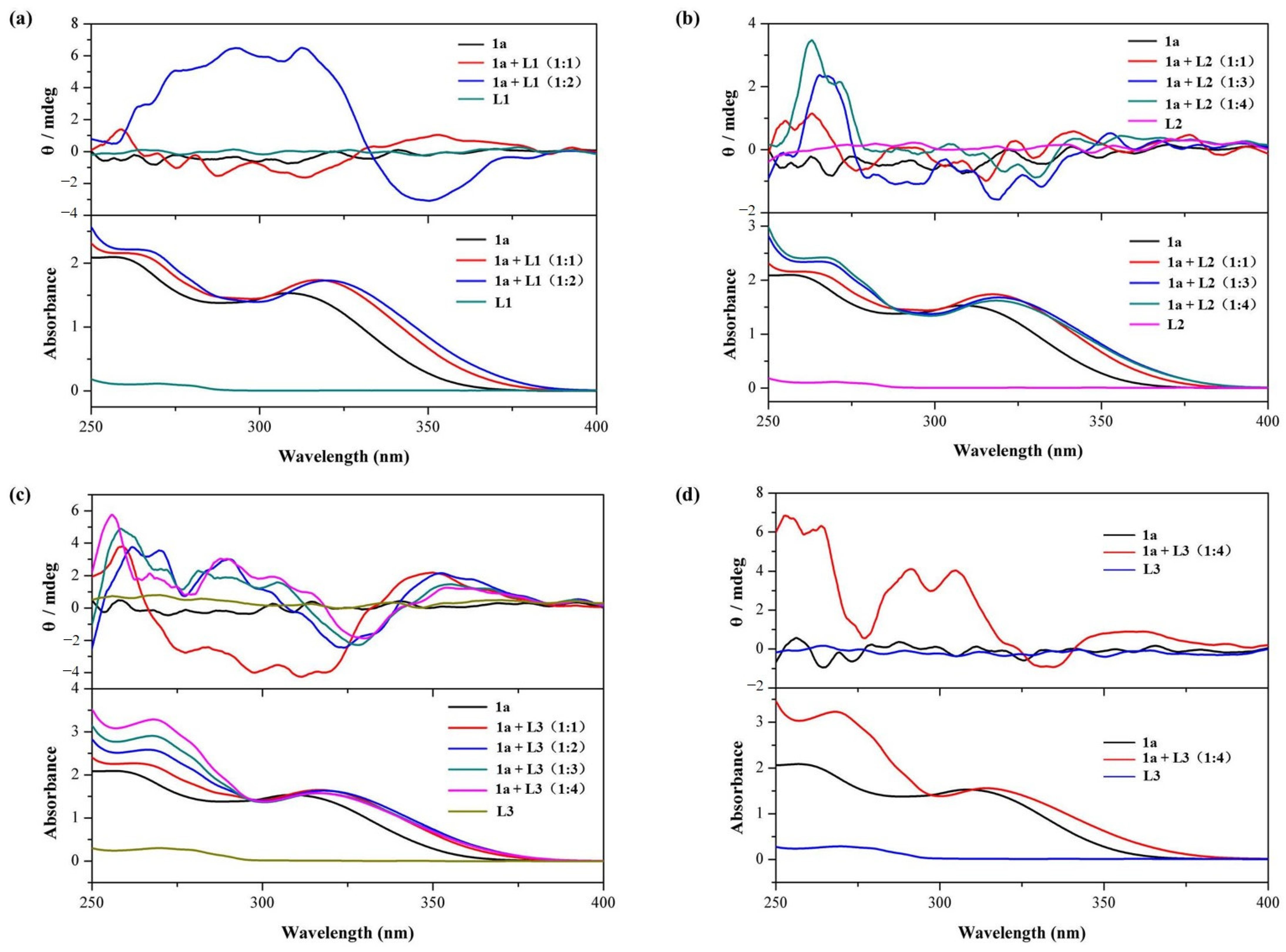
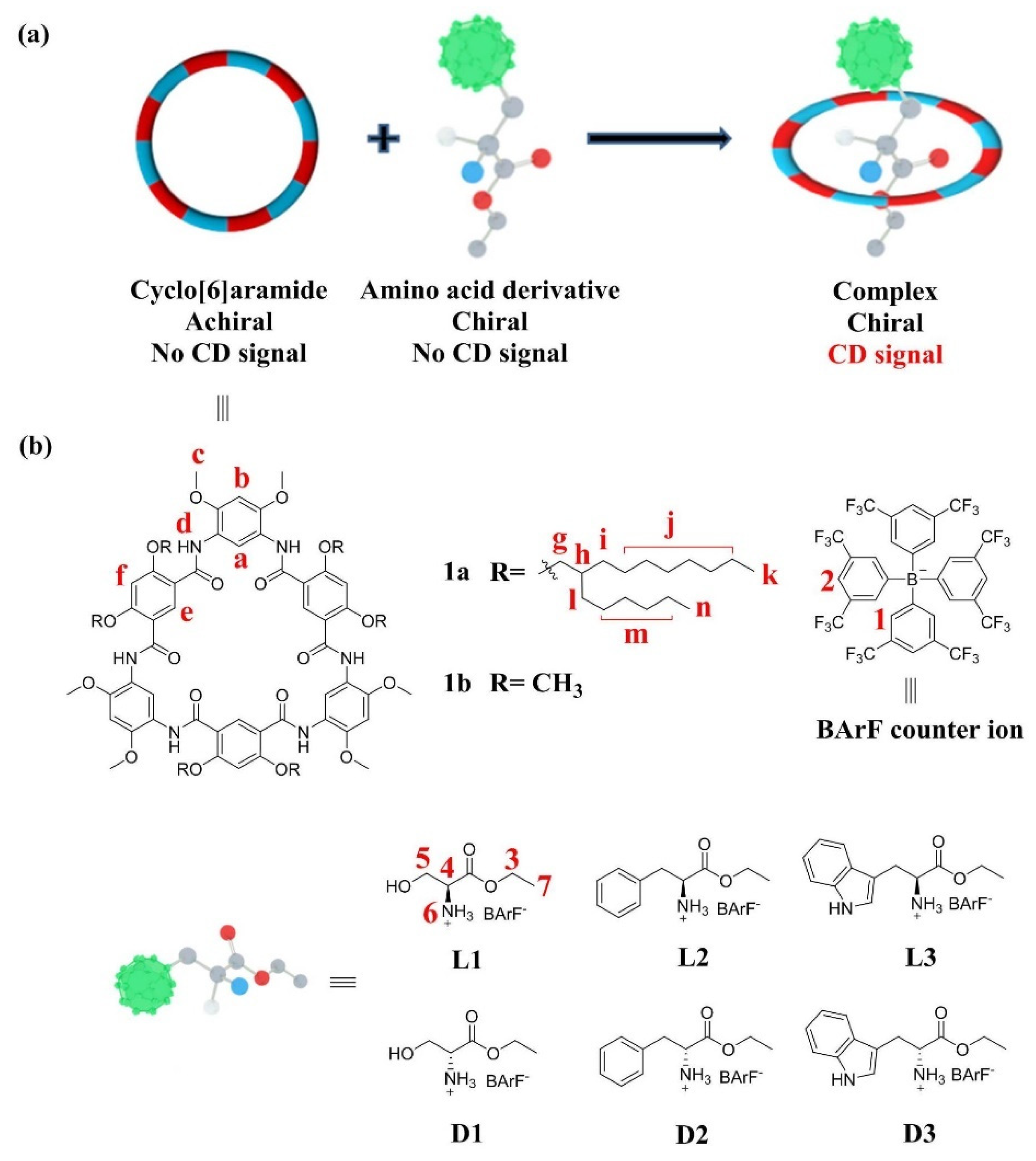
2.1. Host–Guest Interaction
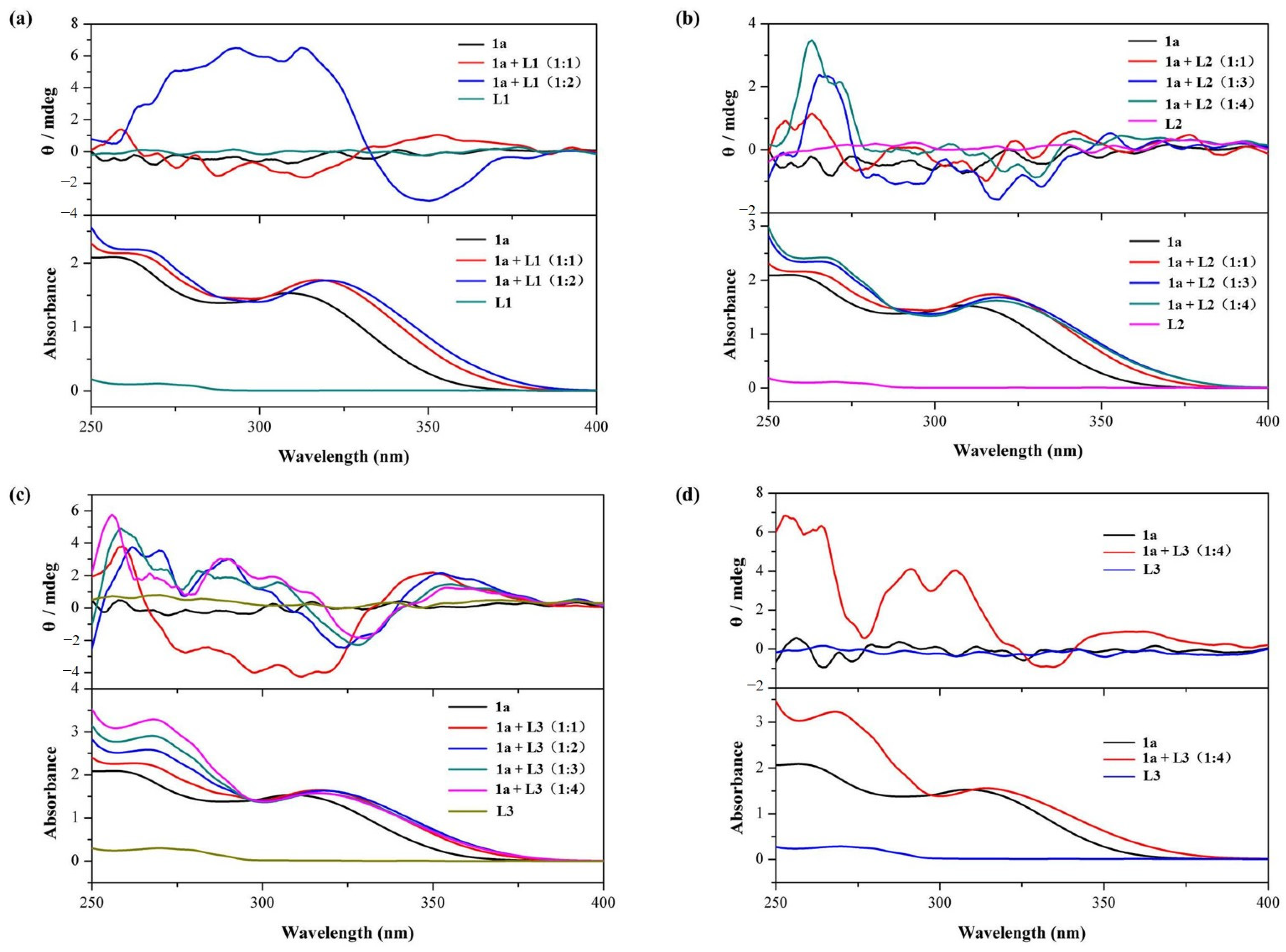
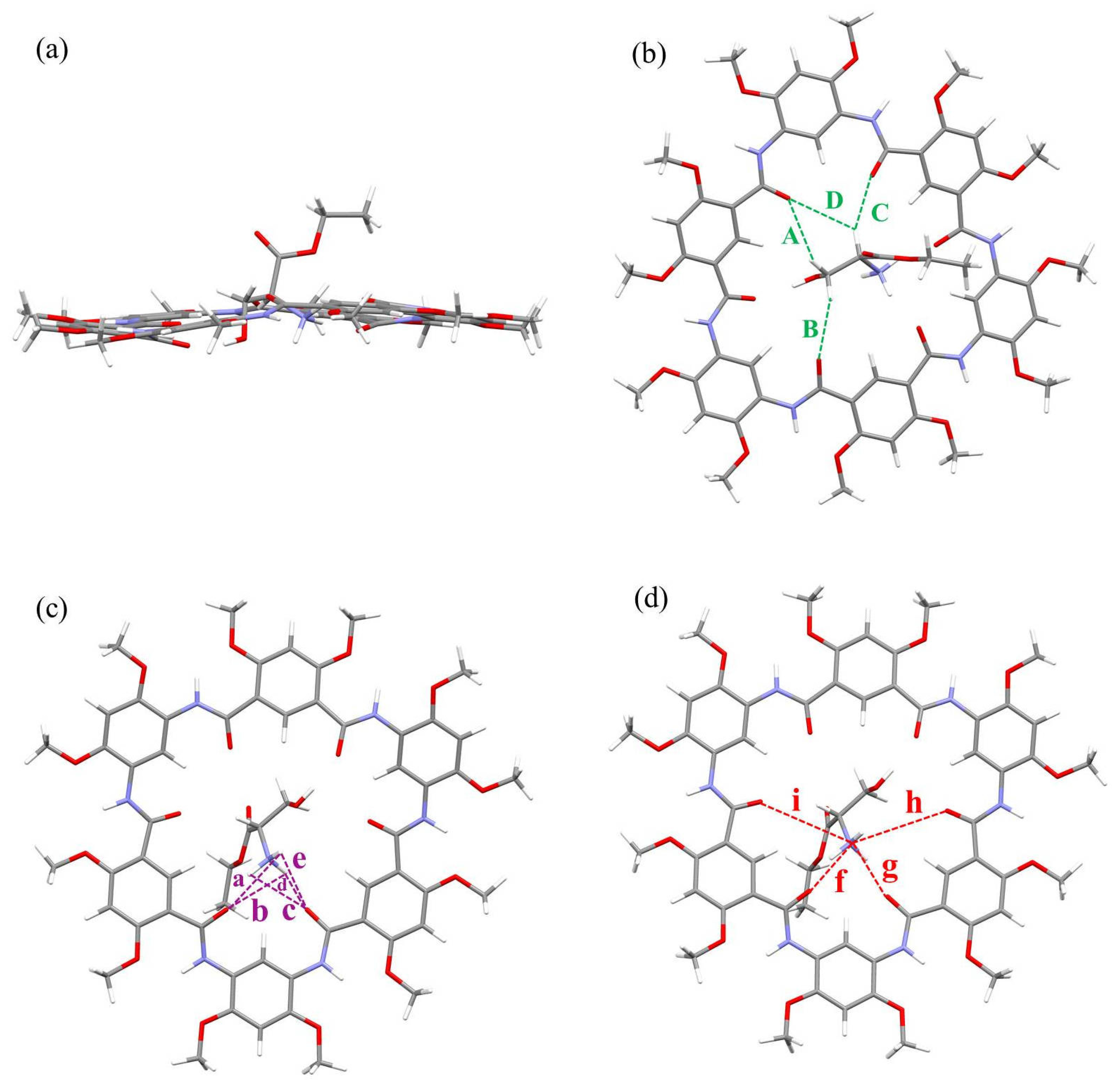
2.2. Pseduo-Achiral Host–Chiral Guest Complexation by CD Spectroscopy
3. Materials and Methods
3.1. Materials and Reagents
3.2. Experimental Methods
3.3. Synthesis of Cyclo[6]aramide and Amino Acid Derivatives
3.4. DFT Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Hegstrom, R.A.; Kondepudi, D.K. The Handedness of the Universe. Sci. Am. 1990, 262, 108–115. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, L.; Wang, T. Supramolecular Chirality in Self-Assembled Systems. Chem. Rev. 2015, 115, 7304–7397. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.-B.; Chen, J.-F.; Cheng, X.-B.; Li, H.; Han, B.-B.; Zhang, Y.-M.; Yao, H.; Lin, Q. A novel functionalized pillar[5]arene-based selective amino acid sensor for L-tryptophan. Org. Chem. Front. 2017, 4, 210–213. [Google Scholar] [CrossRef]
- Amako, Y.; Woo, C.M. A chiral trick to map protein ligandability. Nat. Chem. 2019, 11, 1080–1082. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wang, Q.; Wu, X.; Li, Z.; Jiang, Y.-B. Optical chirality sensing using macrocycles, synthetic and supramolecular oligomers/polymers, and nanoparticle based sensors. Chem. Soc. Rev. 2015, 44, 4249–4263. [Google Scholar] [CrossRef]
- Borovkov, V.V.; Fujii, I.; Muranaka, A.; Hembury, G.A.; Tanaka, T.; Ceulemans, A.; Kobayashi, N.; Inoue, Y. Rationalization of Supramolecular Chirality in a Bisporphyrin System. Angew. Chem. Int. Ed. 2004, 43, 5481–5485. [Google Scholar] [CrossRef]
- Borovkov, V.V.; Hembury, G.A.; Inoue, Y. Origin, Control, and Application of Supramolecular Chirogenesis in Bisporphyrin-Based Systems. Acc. Chem. Res. 2004, 37, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Dragna, J.M.; Pescitelli, G.; Tran, L.; Lynch, V.M.; Anslyn, E.V.; Bari, L.D. In Situ Assembly of Octahedral Fe(II) Complexes for the Enantiomeric Excess Determination of Chiral Amines Using Circular Dichroism Spectroscopy. J. Am. Chem. Soc. 2012, 134, 4398–4407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, L.; Berman, J.S.; Anslyn, E.V. Dynamic multi-component covalent assembly for the reversible binding of secondary alcohols and chirality sensing. Nat. Chem. 2011, 3, 943–948. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Liang, W.; Niu, T.; Wu, W.; Zhou, D.; Fan, C.; Ji, J.; Gao, G.; Men, J.; Yang, Y.; et al. Induced chirality sensing through formation and aggregation of the chiral imines double winged with pyrenes or perylenes. Chem. Commun. 2018, 54, 9206–9209. [Google Scholar] [CrossRef]
- Wang, H.-J.; Zhang, H.-Y.; Zhang, H.-Y.; Liu, G.; Dai, X.; Wu, H.; Liu, Y. Guest-induced supramolecular chirality transfer in [2]pseudorotaxanes: Experimental and computational study. Org. Biomol. Chem. 2020, 18, 7649–7655. [Google Scholar] [CrossRef] [PubMed]
- James, T.D.; Sandanayake, K.R.A.S.; Shinkai, S. Chiral discrimination of monosaccharides using a fluorescent molecular sensor. Nature 1995, 374, 345–347. [Google Scholar] [CrossRef]
- Pu, L. Simultaneous Determination of Concentration and Enantiomeric Composition in Fluorescent Sensing. Acc. Chem. Res. 2017, 50, 1032–1040. [Google Scholar] [CrossRef]
- Yashima, E.; Matsushima, T.; Okamoto, Y. Poly((4-carboxyphenyl)acetylene) as a Probe for Chirality Assignment of Amines by Circular Dichroism. J. Am. Chem. Soc. 1995, 117, 11596–11597. [Google Scholar] [CrossRef]
- Lim, J.Y.C.; Marques, I.; Félix, V.; Beer, P.D. A Chiral Halogen-Bonding [3]Rotaxane for the Recognition and Sensing of Biologically Relevant Dicarboxylate Anions. Angew. Chem. Int. Ed. 2018, 57, 584–588. [Google Scholar] [CrossRef]
- Jamieson, E.M.G.; Modicom, F.; Goldup, S.M. Chirality in rotaxanes and catenanes. Chem. Soc. Rev. 2018, 118, 5266–5311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gawroński, J.; Grajewski, J. The Significance of Induced Circular Dichroism. Org. Lett. 2003, 5, 3301–3303. [Google Scholar] [CrossRef]
- Vergura, S.; Scafato, P.; Belviso, S.; Superchi, S. Absolute Configuration Assignment from Optical Rotation Data by Means of Biphenyl Chiroptical Probes. Chem. Eur. J. 2019, 25, 5682–5690. [Google Scholar] [CrossRef]
- Santoro, E.; Vergura, S.; Scafato, P.; Belviso, S.; Masi, M.; Evidente, A.; Superchi, S. Absolute Configuration Assignment to Chiral Natural Products by Biphenyl Chiroptical Probes: The Case of the Phytotoxins Colletochlorin A and Agropyrenol. J. Nat. Prod. 2020, 83, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Mądry, T.; Czapik, A.; Kwit, M. “Double-Twist”-Based Dynamic Induction of Optical Activity in Multichromophoric System. Symmetry 2021, 13, 325. [Google Scholar] [CrossRef]
- Maeda, K.; Hirose, D.; Okoshi, N.; Shimomura, K.; Wada, Y.; Ikai, T.; Kanoh, S.; Yashima, E. Direct Detection of Hardly Detectable Hidden Chirality of Hydrocarbons and Deuterated Isotopomers by a Helical Polyacetylene through Chiral Amplification and Memory. J. Am. Chem. Soc. 2018, 140, 3270–3276. [Google Scholar] [CrossRef]
- Shi, Z.-M.; Chen, S.-G.; Zhao, X.; Jiang, X.-K.; Li, Z.-T. meta-Substituted benzamide oligomers that complex mono-, di- and tricarboxylates: Folding-induced selectivity and chirality. Org. Biomol. Chem. 2011, 9, 8122–8129. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-L.; Chen, Z.; Liu, W.-E.; Ke, H.; Wang, S.-H.; Jiang, W. Molecular Recognition and Chirality Sensing of Epoxides in Water Using Endo-Functionalized Molecular Tubes. J. Am. Chem. Soc. 2017, 139, 8436–8439. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, K.; Yamaguchi, M.; Yasui, M.; Fujiwara, S.; Hashimoto, T.; Hayashita, T. Guest-induced supramolecular chirality in a ditopic azoprobe–cyclodextrin complex in water. Chem. Commun. 2014, 50, 10059–10061. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Lin, L.-R.; Huang, Y.-J.; Li, Z.; Jiang, Y.-B. A 2: 2 stilbeneboronic acid–γ-cyclodextrin fluorescent ensemble highly selective for glucose in aqueous solutions. Chem. Commun. 2012, 48, 4362–4364. [Google Scholar] [CrossRef]
- Ji, J.; Li, Y.; Xiao, C.; Cheng, G.; Luo, K.; Gong, Q.; Zhou, D.; Chruma, J.J.; Wu, W.; Yang, C. Supramolecular enantiomeric and structural differentiation of amino acid derivatives with achiral pillar[5]arene homologs. Chem. Commun. 2020, 56, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Fu, L.; Sun, B.; Qian, C.; Wang, R.; Jiang, J.; Lin, C.; Ma, J.; Wang, L. Competitive Selection of Conformation Chirality of Water-Soluble Pillar[5]arene Induced by Amino Acid Derivatives. Org. Lett. 2020, 22, 2266–2270. [Google Scholar] [CrossRef]
- Zhu, H.; Li, Q.; Gao, Z.; Wang, H.; Shi, B.; Wu, Y.; Shangguan, L.; Hong, X.; Wang, F.; Huang, F. Pillararene Host–Guest Complexation Induced Chirality Amplification: A New Way to Detect Cryptochiral Compounds. Angew. Chem. Int. Ed. 2020, 59, 10868–10872. [Google Scholar] [CrossRef]
- Wolf, C.; Bentley, K.W. Chirality sensing using stereodynamic probes with distinct electronic circular dichroism output. Chem. Soc. Rev. 2013, 42, 5408–5424. [Google Scholar] [CrossRef]
- Gong, B.; Shao, Z. Self-Assembling Organic Nanotubes with Precisely Defined, Sub-nanometer Pores: Formation and Mass Transport Characteristics. Acc. Chem. Res. 2013, 46, 2856–2866. [Google Scholar] [CrossRef]
- Ong, W.Q.; Zeng, H. Rapid construction of shape-persistent H-bonded macrocycles via one-pot H-bonding-assisted macrocyclization. J. Incl. Phenom. Mol. Recognit. Chem. 2013, 76, 1–11. [Google Scholar] [CrossRef]
- Zhang, D.-W.; Zhao, X.; Hou, J.-L.; Li, Z.-T. Aromatic Amide Foldamers: Structures, Properties, and Functions. Chem. Rev. 2012, 112, 5271–5316. [Google Scholar] [CrossRef]
- Hu, J.C.; Chen, L.; Shen, J.; Luo, J.; Deng, P.C.; Ren, Y.; Zeng, H.Q.; Feng, W.; Yuan, L.H. Convergent heteroditopic cyclo[6]aramides as macrocyclic ion-pair receptors for constructing [2]pseudorotaxanes. Chem. Commun. 2014, 50, 8024–8027. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.W.; Yang, Y.Z.; Peng, Z.Y.; Cai, Y.M.; Feng, W.; Yuan, L.H. Highly efficient synthesis of hydrogen-bonded aromatic tetramers as macrocyclic receptors for selective recognition of lithium ions. Org. Chem. Front. 2019, 6, 2654–2661. [Google Scholar] [CrossRef]
- Peng, Z.Y.; Guo, X.W.; Xu, W.T.; Li, J.; Deng, P.C.; Xiao, X.; Feng, W.; Yuan, L.H. Strong positive allosteric cooperativity in ternary complexes based on hydrogen-bonded aromatic amide macrocycles. Chem. Commun. 2019, 55, 4869–4872. [Google Scholar] [CrossRef]
- Zhong, L.J.; Chen, L.; Feng, W.; Zou, S.L.; Yang, Y.Y.; Liu, N.; Yuan, L.H. Shape-persistent macrocycles: Efficient extraction towards lanthanide and actinide elements. J. Incl. Phenom. Macrocycl. Chem. 2012, 72, 367–373. [Google Scholar] [CrossRef]
- Ren, C.; Shen, J.; Zeng, H. One-Pot Synthesis of Strained Macrocyclic Pyridone Hexamers and Their High Selectivity toward Cu2+ Recognition. Org. Lett. 2015, 17, 5946–5949. [Google Scholar] [CrossRef]
- Helsel, A.J.; Brown, A.L.; Yamato, K.; Feng, W.; Yuan, L.H.; Clements, A.J.; Harding, S.V.; Szabo, G.; Shao, Z.F.; Gong, B. Highly Conducting Transmembrane Pores Formed by Aromatic Oligoamide Macrocycles. J. Am. Chem. Soc. 2008, 130, 15784–15785. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.; Lohrman, J.A.; Nagarajan, S.; Chen, L.X.; Deng, P.C.; Shen, X.; Fu, K.R.; Feng, W.; Johnson, D.W.; Yuan, L.H. Convergent Ditopic Receptors Enhance Anion Binding upon Alkali Metal Complexation for Catalyzing the Ritter Reaction. Org. Lett. 2019, 21, 652–655. [Google Scholar] [CrossRef]
- Pan, W.; Mao, L.J.; Shi, M.S.; Fu, Y.H.; Jiang, X.M.; Feng, W.; He, Y.Z.; Xu, D.G.; Yuan, L.H. The cytochrome c–cyclo[6]aramide complex as a supramolecular catalyst in methanol. New J. Chem. 2018, 42, 3857–3866. [Google Scholar] [CrossRef]
- Li, X.W.; Li, B.; Chen, L.; Hu, J.C.; Wen, C.D.Y.; Zheng, Q.D.; Wu, L.X.; Zeng, H.Q.; Gong, B.; Yuan, L.H. Liquid-Crystalline Mesogens Based on Cyclo[6]aramides: Distinctive Phase Transitions in Response to Macrocyclic Host-Guest Interactions. Angew. Chem. Int. Ed. 2015, 54, 11147–11152. [Google Scholar] [CrossRef]
- He, Y.Z.; Xu, M.; Gao, R.Z.; Li, X.W.; Li, F.X.; Wu, X.D.; Xu, D.G.; Zeng, H.Q.; Yuan, L.H. Two-Component Supramolecular Gels Derived from Amphiphilic Shape-Persistent Cyclo[6]aramides for Specific Recognition of Native Arginine. Angew. Chem. Int. Ed. 2014, 53, 11834–11839. [Google Scholar] [CrossRef]
- Xu, M.; Chen, L.; Jia, Y.M.; Mao, L.J.; Feng, W.; Ren, Y.; Yuan, L.H. A rare case for binding a diquat salt by two cyclo[6]aramides. Supramol. Chem. 2015, 27, 436–443. [Google Scholar] [CrossRef]
- Chen, L.; Peng, Z.Y.; Liu, S.; Li, X.W.; Chen, R.Z.; Ren, Y.; Feng, W.; Yuan, L.H. Cyclo[6]aramide-Tropylium Charge Transfer Complex as a Colorimetric Chemosensor for Differentiation of Intimate and Loose Ion Pairs. Org. Lett. 2015, 17, 5950–5953. [Google Scholar] [CrossRef]
- Li, X.W.; Yuan, X.Y.; Deng, P.C.; Chen, L.X.; Ren, Y.; Wang, C.Y.; Wu, L.X.; Feng, W.; Gong, B.; Yuan, L.H. Macrocyclic shape-persistency of cyclo[6]aramide results in enhanced multipoint recognition for the highly efficient template-directed synthesis of rotaxanes. Chem. Sci. 2017, 8, 2091–2100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Z.C.; Yang, Z.Y.; Wang, L.; Chen, L.X.; Cai, Y.M.; Deng, P.C.; Feng, W.; Li, X.P.; Yuan, L.H. A Dynamic Hydrogen-Bonded Azo-Macrocycle for Precisely Photo-Controlled Molecular Encapsulation and Release. Angew. Chem. Int. Ed. 2019, 58, 12519–12523. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yuan, X.Y.; Wang, Z.X.; Luo, Y.R.; Huang, W.; Zhang, S.; Yuan, W.L.; Qin, S.; Tao, G.H.; Yuan, L.H. A Redox-Responsive Complex System Based on 2 D Shape-Persistent Cyclo[6]aramide and Ferrocenium. Asian J. Org. Chem. 2016, 5, 966–970. [Google Scholar] [CrossRef]
- Kothapalli, S.S.K.; Kannekanti, V.K.; Ye, Z.C.; Yang, Z.Y.; Chen, L.X.; Cai, Y.M.; Zhu, B.C.; Feng, W.; Yuan, L.H. Light-controlled switchable complexation by a non-photoresponsive hydrogen-bonded amide macrocycle. Org. Chem. Front. 2020, 7, 846–855. [Google Scholar] [CrossRef]
- Hu, J.C.; Chen, L.; Ren, Y.; Deng, P.C.; Li, X.W.; Wang, Y.J.; Jia, Y.M.; Luo, J.; Yang, X.S.; Feng, W.; et al. Nonaggregational Shape-Persistent Cyclo[6]aramide and Its Macrocyclic Effect toward Binding Secondary Ammonium Salts in Moderately Polar Media. Org. Lett. 2013, 15, 4670–4673. [Google Scholar] [CrossRef] [PubMed]
- Labuta, J.; Hill, J.P.; Ishihara, S.; Hanyková, L.; Ariga, K. Chiral Sensing by Nonchiral Tetrapyrroles. Acc. Chem. Res. 2015, 48, 521–529. [Google Scholar] [CrossRef]
- Kuwahara, S.; Chamura, R.; Tsuchiya, S.; Ikeda, M.; Habata, Y. Chirality transcription and amplification by [2]pseudorotaxanes. Chem. Commun. 2013, 49, 2186–2188. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Li, Y.; Qin, Z.; Xu, L.; Zhu, D.; Li, Y. A chiral macrocyclic receptor for sulfate anions with CD signals. RSC Adv. 2014, 4, 2023–2028. [Google Scholar] [CrossRef]
- Yuan, L.H.; Sanford, A.R.; Feng, W.; Zhang, A.M.; Zhu, J.; Zeng, H.Q.; Yamato, K.; Li, M.F.; Ferguson, J.S.; Gong, B. Synthesis of Crescent Aromatic Oligoamides. J. Org. Chem. 2005, 70, 10660–10669. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.H.; Feng, W.; Yamato, K.; Sanford, A.R.; Xu, D.G.; Guo, H.; Gong, B. Highly Efficient, One-Step Macrocyclizations Assisted by the Folding and Preorganization of Precursor Oligomers. J. Am. Chem. Soc. 2004, 126, 11120–11121. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | Stoichiometry a | Ka (M−1) b | R2 | CD λmax/λmin (nm) c |
|---|---|---|---|---|
| 1a⊃L1 | 1:1 | (5.60 ± 0.01) × 102 | 0.9968 | 312/350 |
| 1a⊃L2 | 1:1 | (3.81 ± 0.03) × 103 | 0.9902 | 263/330 |
| 1a⊃L3 | 1:1 | (2.82 ± 0.01) × 103 | 0.9987 | 256/330 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Ji, J.; Liu, Z.; Cai, Y.; Tang, J.; Shi, Y.; Yang, C.; Yuan, L. Chiroptical Sensing of Amino Acid Derivatives by Host–Guest Complexation with Cyclo[6]aramide. Molecules 2021, 26, 4064. https://doi.org/10.3390/molecules26134064
Wang X, Ji J, Liu Z, Cai Y, Tang J, Shi Y, Yang C, Yuan L. Chiroptical Sensing of Amino Acid Derivatives by Host–Guest Complexation with Cyclo[6]aramide. Molecules. 2021; 26(13):4064. https://doi.org/10.3390/molecules26134064
Chicago/Turabian StyleWang, Xuebin, Jiecheng Ji, Zejiang Liu, Yimin Cai, Jialiang Tang, Yunzhi Shi, Cheng Yang, and Lihua Yuan. 2021. "Chiroptical Sensing of Amino Acid Derivatives by Host–Guest Complexation with Cyclo[6]aramide" Molecules 26, no. 13: 4064. https://doi.org/10.3390/molecules26134064
APA StyleWang, X., Ji, J., Liu, Z., Cai, Y., Tang, J., Shi, Y., Yang, C., & Yuan, L. (2021). Chiroptical Sensing of Amino Acid Derivatives by Host–Guest Complexation with Cyclo[6]aramide. Molecules, 26(13), 4064. https://doi.org/10.3390/molecules26134064