
4-Phenethyl-1-Propargylpiperidine-Derived Dual Inhibitors of Butyrylcholinesterase and Monoamine Oxidase B




Abstract
:1. Introduction
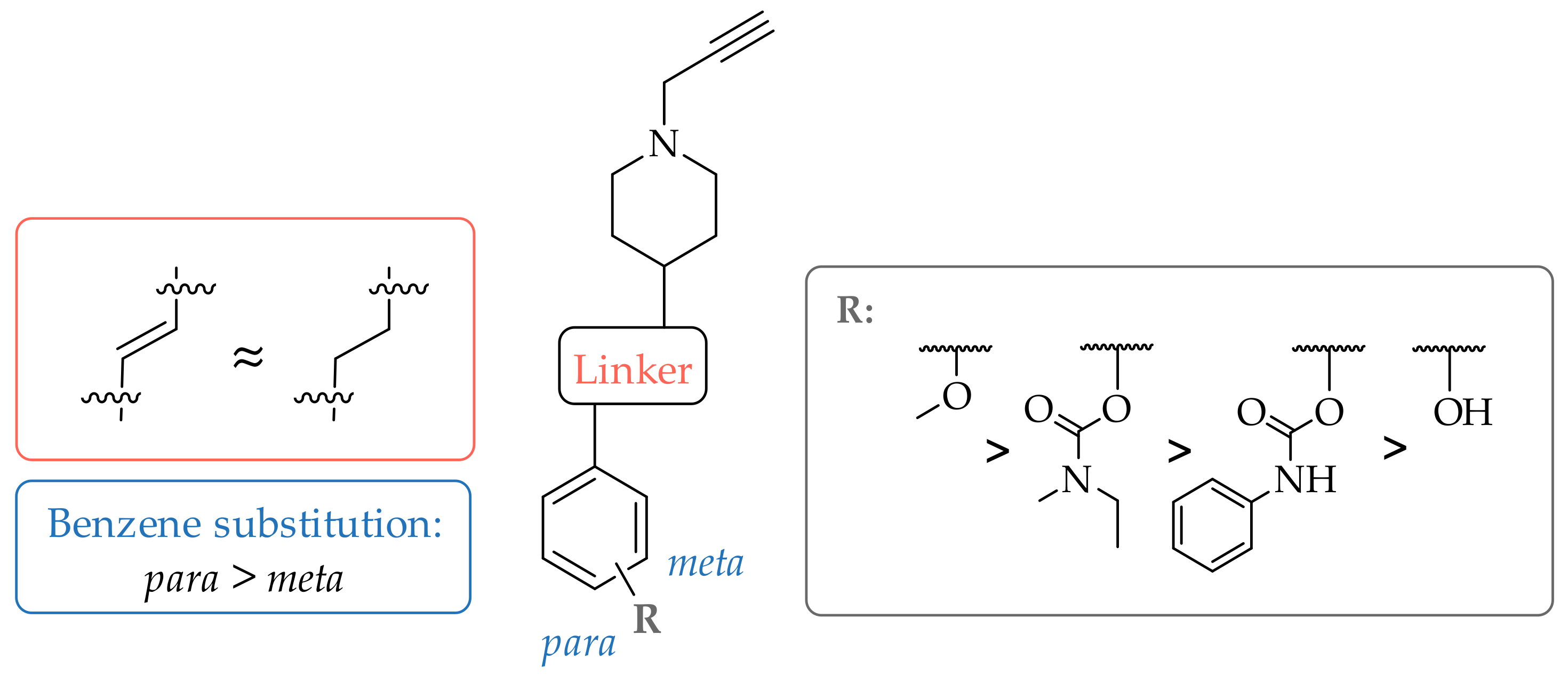
2. Results and Discussion
2.1. Chemistry
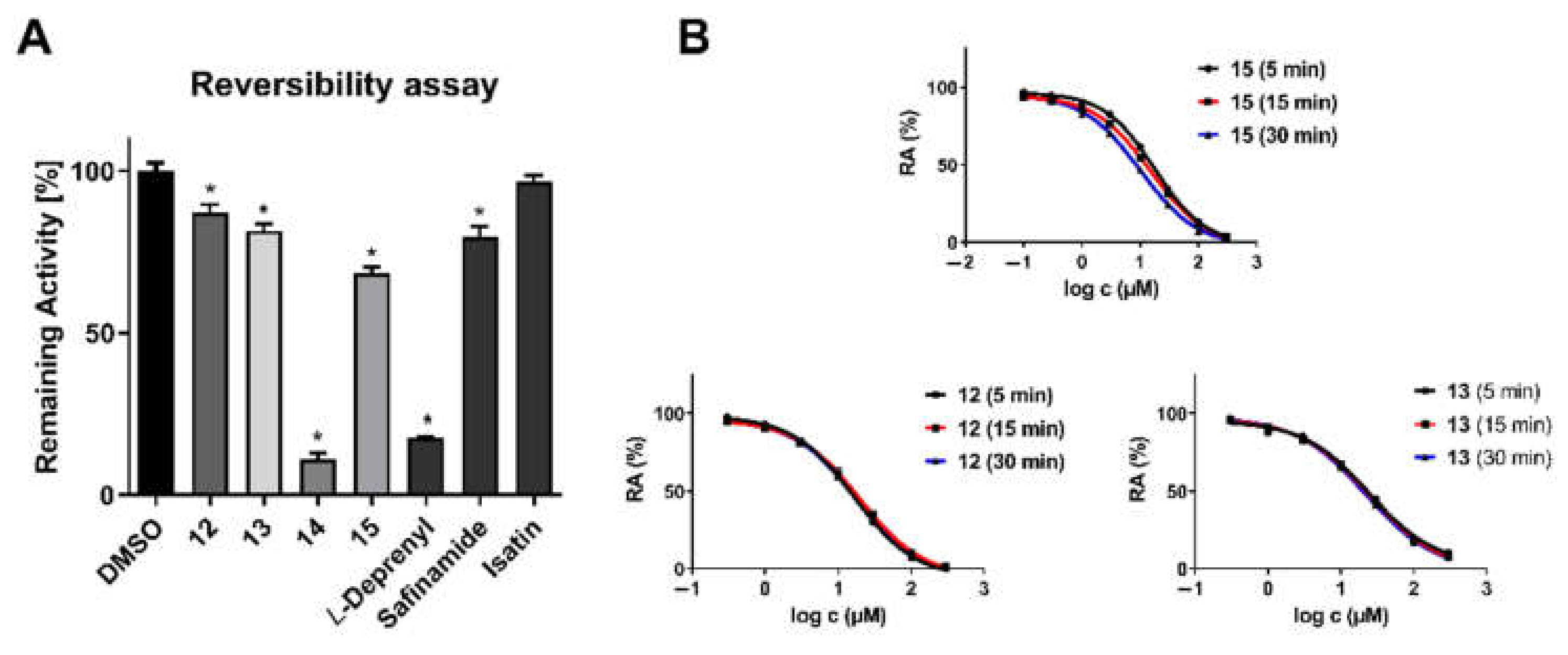
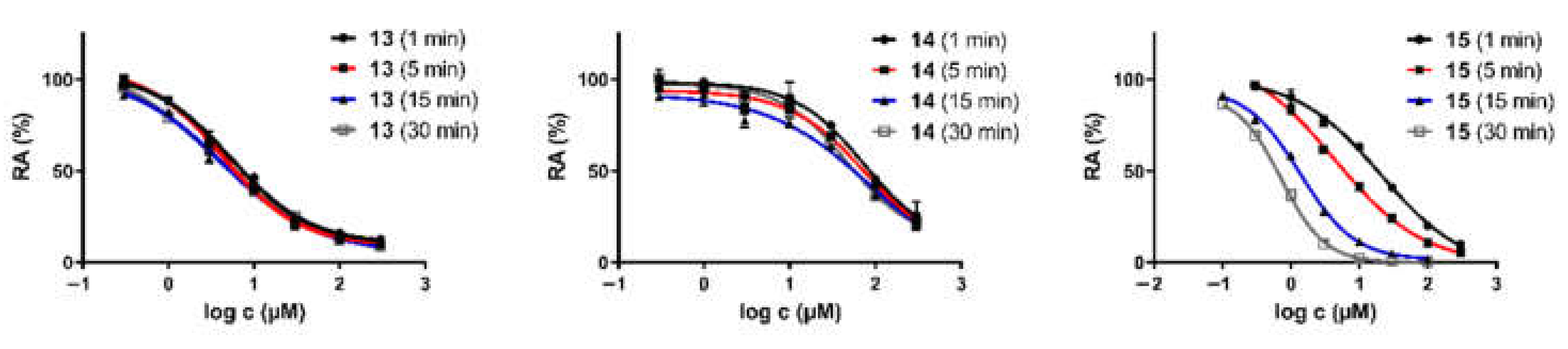
2.2. Monoamine Oxidase and Cholinesterase Inhibitory Potencies
3. Materials and Methods
3.1. Chemistry
3.1.1. General Information
3.1.2. General Experimental Procedures
General Procedure A: Synthesis of Phosphonium Salts
General Procedure B: Wittig Reaction
General Procedure C: Reduction of Double Bond
General Procedure D: Boc-Protection Removal
General Procedure E: Alkylation of Secondary Amine
General Procedure F: Demethylation
General Procedure G: Carbamate Synthesis with Phenyl Isocyanate
General Procedure H: Carbamate Synthesis with N-ethyl-N-methylcarbamoyl chloride
3.1.3. Synthetic and Analytical Data for Intermediates and Inhibitors
tert-Butyl (E/Z)-4-(4-methoxystyryl)piperidine-1-carboxylate (isomer (E)) (2)
tert-Butyl (E/Z)-4-(3-methoxystyryl)piperidine-1-carboxylate (isomer (E)) (3)
tert-Butyl 4-(4-methoxyphenethyl)piperidine-1-carboxylate (4)
tert-Butyl 4-(3-methoxyphenethyl)piperidine-1-carboxylate (5)
(E)-4-(4-methoxystyryl)-1-(prop-2-yn-1-yl)piperidine (6)
4-(4-methoxyphenethyl)-1-(prop-2-yn-1-yl)piperidine (7)
4-(3-methoxyphenethyl)-1-(prop-2-yn-1-yl)piperidine (8)
(E)-4-(2-(1-(prop-2-yn-1-yl)piperidin-4-yl)vinyl)phenol (9)
4-(2-(1-(prop-2-yn-1-yl)piperidin-4-yl)ethyl)phenol (10)
3-(2-(1-(prop-2-yn-1-yl)piperidin-4-yl)ethyl)phenol (11)
4-(2-(1-(prop-2-yn-1-yl)piperidin-4-yl)ethyl)phenyl phenylcarbamate (12)
3-(2-(1-(prop-2-yn-1-yl)piperidin-4-yl)ethyl)phenyl phenylcarbamate (13)
4-(2-(1-(prop-2-yn-1-yl)piperidin-4-yl)ethyl)phenyl ethyl(methyl)carbamate (14)
3-(2-(1-(prop-2-yn-1-yl)piperidin-4-yl)ethyl)phenyl ethyl(methyl)carbamate (15)
3.2. Biological Evaluation
3.2.1. In Vitro Cholinesterase Assay
3.2.2. In Vitro MAO-A/B Assay
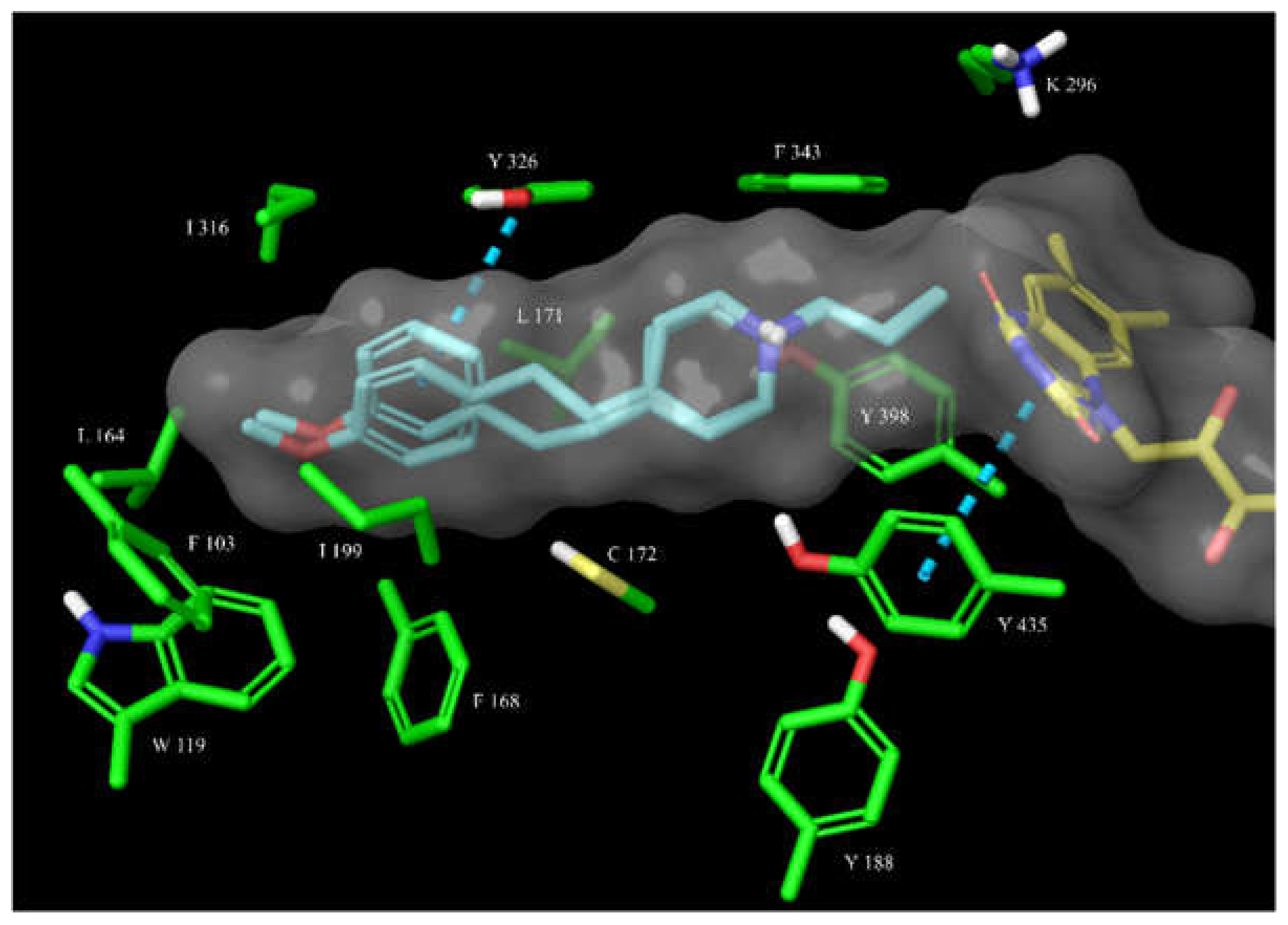
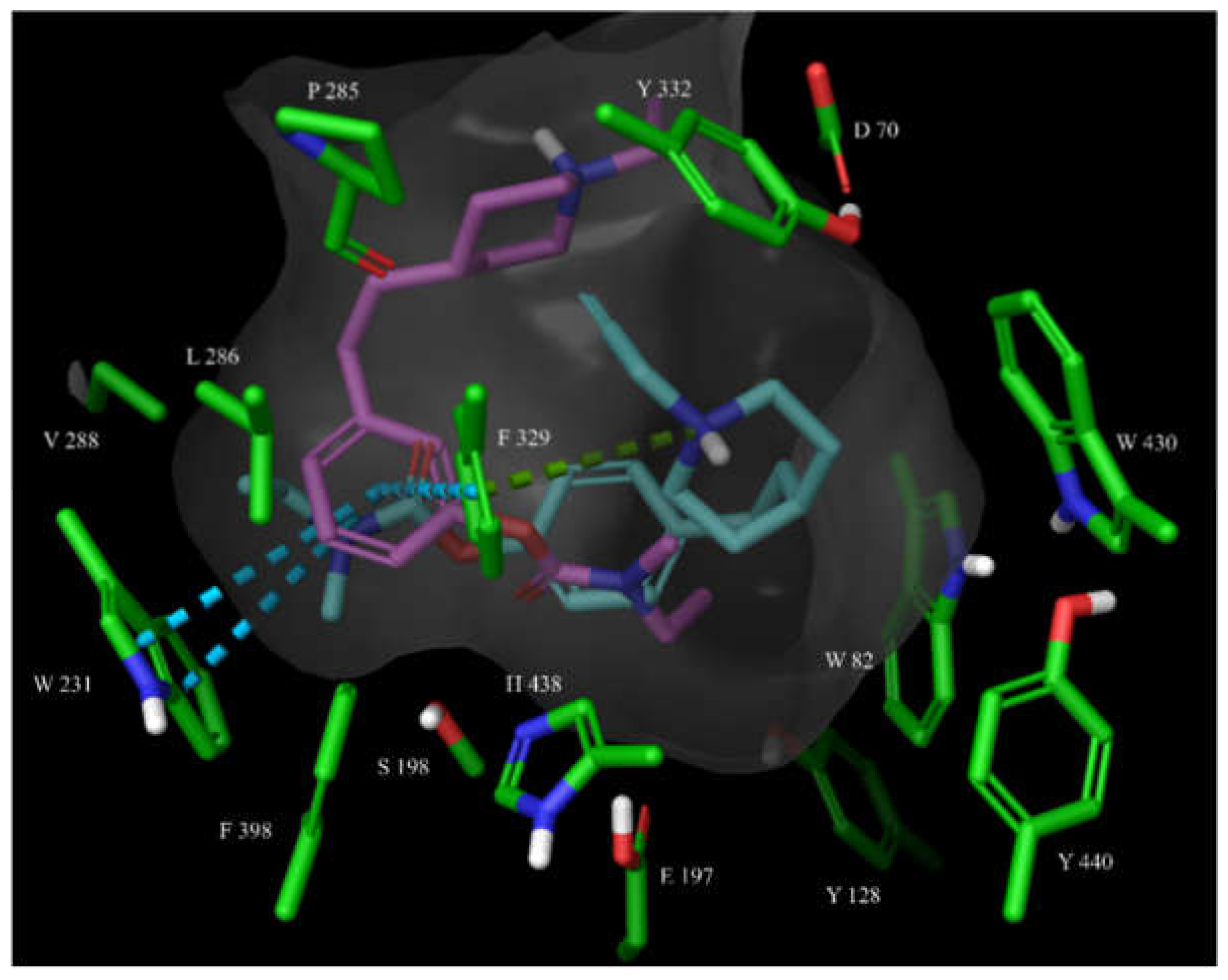
3.2.3. Molecular Docking
3.2.4. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Abeysinghe, A.; Deshapriya, R.; Udawatte, C. Alzheimer’s disease; a review of the pathophysiological basis and therapeutic interventions. Life Sci. 2020, 256, 117996. [Google Scholar] [CrossRef]
- Scheltens, P.; De Strooper, B.; Kivipelto, M.; Holstege, H.; Chételat, G.; Teunissen, C.E.; Cummings, J.; van der Flier, W.M. Alzheimer’s disease. Lancet 2021, 397, 1577–1590. [Google Scholar] [CrossRef]
- Mullane, K.; Williams, M. Alzheimer’s disease (AD) therapeutics—2: Beyond amyloid—Re-defining AD and its causality to discover effective therapeutics. Biochem. Pharmacol. 2018, 158, 376–401. [Google Scholar] [CrossRef]
- Zhou, Y.; Sun, Y.; Ma, Q.-H.; Liu, Y. Alzheimer’s disease: Amyloid-based pathogenesis and potential therapies. Cell Stress 2018, 2, 150–161. [Google Scholar] [CrossRef]
- Busche, M.A.; Hyman, B.T. Synergy between amyloid-β and tau in Alzheimer’s disease. Nat. Neurosci. 2020, 23, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Misrani, A.; Tabassum, S.; Yang, L. Mitochondrial Dysfunction and Oxidative Stress in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 57. [Google Scholar] [CrossRef]
- Wang, L.; Yin, Y.-L.; Liu, X.-Z.; Shen, P.; Zheng, Y.-G.; Lan, X.-R.; Lu, C.-B.; Wang, J.-Z. Current understanding of metal ions in the pathogenesis of Alzheimer’s disease. Transl. Neurodegener. 2020, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Weidling, I.W.; Swerdlow, R.H. Mitochondria in Alzheimer’s disease and their potential role in Alzheimer’s proteostasis. Exp. Neurol. 2020, 330, 113321. [Google Scholar] [CrossRef] [PubMed]
- John, A.; Reddy, P.H. Synaptic basis of Alzheimer’s disease: Focus on synaptic amyloid beta, P-tau and mitochondria. Ageing Res. Rev. 2021, 65, 101208. [Google Scholar] [CrossRef] [PubMed]
- Vainchtein, I.D.; Molofsky, A.V. Astrocytes and Microglia: In Sickness and in Health. Trends Neurosci. 2020, 43, 144–154. [Google Scholar] [CrossRef]
- Arranz, A.M.; De Strooper, B. The role of astroglia in Alzheimer’s disease: Pathophysiology and clinical implications. Lancet Neurol. 2019, 18, 406–414. [Google Scholar] [CrossRef]
- Ferreira-Vieira, T.H.; Guimaraes, I.M.; Silva, F.R.; Ribeiro, F.M. Alzheimer’s disease: Targeting the Cholinergic System. Curr. Neuropharmacol. 2016, 14, 101–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hampel, H.; Mesulam, M.-M.; Cuello, A.C.; Farlow, M.R.; Giacobini, E.; Grossberg, G.T.; Khachaturian, A.S.; Vergallo, A.; Cavedo, E.; Snyder, P.J.; et al. The cholinergic system in the pathophysiology and treatment of Alzheimer’s disease. Brain 2018, 141, 1917–1933. [Google Scholar] [CrossRef]
- Agatonovic-Kustrin, S.; Kettle, C.; Morton, D. A molecular approach in drug development for Alzheimer’s disease. Biomed. Pharmacother. 2018, 106, 553–565. [Google Scholar] [CrossRef]
- Greig, N.H.; Utsuki, T.; Yu, Q.-S.; Zhu, X.; Holloway, H.W.; Perry, T.; Lee, B.; Ingram, D.K.; Lahiri, D.K. A New Therapeutic Target in Alzheimer’s Disease Treatment: Attention to Butyrylcholinesterase. Curr. Med. Res. Opin. 2001, 17, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Giacobini, E. Cholinesterase inhibitors: New roles and therapeutic alternatives. Pharmacol. Res. 2004, 50, 433–440. [Google Scholar] [CrossRef]
- Ghimire, S.; Flury, M.; Scheenstra, E.J.; Miles, C.A. Sampling and degradation of biodegradable plastic and paper mulches in field after tillage incorporation. Sci. Total Environ. 2020, 703, 135577. [Google Scholar] [CrossRef]
- Kim, D.; Baik, S.H.; Kang, S.; Cho, S.W.; Bae, J.; Cha, M.-Y.; Sailor, M.J.; Mook-Jung, I.; Ahn, K.H. Close Correlation of Monoamine Oxidase Activity with Progress of Alzheimer’s Disease in Mice, Observed by in Vivo Two-Photon Imaging. ACS Cent. Sci. 2016, 2, 967–975. [Google Scholar] [CrossRef]
- Mathew, B.; Parambi, D.G.T.; Mathew, G.E.; Uddin, S.; Inasu, S.T.; Kim, H.; Marathakam, A.; Unnikrishnan, M.K.; Carradori, S. Emerging therapeutic potentials of dual-acting MAO and AChE inhibitors in Alzheimer’s and Parkinson’s diseases. Arch. Der Pharm. 2019, 352, e1900177. [Google Scholar] [CrossRef]
- Zagórska, A.; Jaromin, A. Perspectives for New and More Efficient Multifunctional Ligands for Alzheimer′s Disease Therapy. Molecules 2020, 25, 3337. [Google Scholar] [CrossRef] [PubMed]
- Devenish, S.R. The current landscape in Alzheimer’s disease research and drug discovery. Drug Discov. Today 2020, 25, 943–945. [Google Scholar] [CrossRef]
- Long, J.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Bolognesi, M.L. Harnessing Polypharmacology with Medicinal Chemistry. ACS Med. Chem. Lett. 2019, 10, 273–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uddin, S.; Kabir, T.; Rahman, M.; Mathew, B.; Shah, M.A.; Ashraf, G.M. TV 3326 for Alzheimer’s dementia: A novel multimodal ChE and MAO inhibitors to mitigate Alzheimer’s-like neuropathology. J. Pharm. Pharmacol. 2020, 72, 1001–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinreb, O.; Amit, T.; Bar-Am, O.; Youdim, M.B. Ladostigil: A Novel Multimodal Neuroprotective Drug with Cholinesterase and Brain-Selective Monoamine Oxidase Inhibitory Activities for Alzheimers Disease Treatment. Curr. Drug Targets 2012, 13, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Carreiras, M.D.C.; Ismaili, L.; Marco-Contelles, J. Propargylamine-derived multi-target directed ligands for Alzheimer’s disease therapy. Bioorganic Med. Chem. Lett. 2020, 30, 126880. [Google Scholar] [CrossRef]
- Schneider, L.S.; Geffen, Y.; Rabinowitz, J.; Thomas, R.G.; Schmidt, R.; Ropele, S.; Weinstock, M.; Ladostigil Study Group. Low-dose ladostigil for mild cognitive impairment: A phase 2 placebo-controlled clinical trial. Neurology 2019, 93, e1474–e1484. [Google Scholar] [CrossRef] [PubMed]
- Guieu, B.; Lecoutey, C.; Legay, R.; Davis, A.; Santos, J.S.D.O.; Altomare, C.D.; Catto, M.; Rochais, C.; Dallemagne, P. First Synthesis of Racemic Trans Propargylamino-Donepezil, a Pleiotrope Agent Able to Both Inhibit AChE and MAO-B, with Potential Interest against Alzheimer’s Disease. Molecules 2020, 26, 80. [Google Scholar] [CrossRef]
- Knez, D.; Colettis, N.; Iacovino, L.G.; Sova, M.; Pišlar, A.; Konc, J.; Lešnik, S.; Higgs, J.; Kamecki, F.; Mangialavori, I.C.; et al. Stereoselective Activity of 1-Propargyl-4-styrylpiperidine-like Analogues That Can Discriminate between Monoamine Oxidase Isoforms A and B. J. Med. Chem. 2020, 63, 1361–1387. [Google Scholar] [CrossRef]
- Košak, U.; Strašek, N.; Knez, D.; Jukič, M.; Žakelj, S.; Zahirović, A.; Pišlar, A.; Brazzolotto, X.; Nachon, F.; Kos, J.; et al. N-alkylpiperidine carbamates as potential anti-Alzheimer’s agents. Eur. J. Med. Chem. 2020, 197, 112282. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.; Andres, V.; Featherstone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Košak, U.; Knez, D.; Coquelle, N.; Brus, B.; Pišlar, A.; Nachon, F.; Brazzolotto, X.; Kos, J.; Colletier, J.-P.; Gobec, S. N-Propargylpiperidines with naphthalene-2-carboxamide or naphthalene-2-sulfonamide moieties: Potential multifunctional anti-Alzheimer’s agents. Bioorg. Med. Chem. 2017, 25, 633–645. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Voloshina, N.P.-A. One-Step Fluorometric Method for the Continuous Measurement of Monoamine Oxidase Activity. Anal. Biochem. 1997, 253, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Iacovino, L.G.; Magnani, F.; Binda, C. The structure of monoamine oxidases: Past, present, and future. J. Neural Transm. 2018, 125, 1567–1579. [Google Scholar] [CrossRef] [PubMed]
- Meden, A.; Knez, D.; Malikowska-Racia, N.; Brazzolotto, X.; Nachon, F.; Svete, J.; Sałat, K.; Grošelj, U.; Gobec, S. Structure-activity relationship study of tryptophan-based butyrylcholinesterase inhibitors. Eur. J. Med. Chem. 2020, 208, 112766. [Google Scholar] [CrossRef]
- Von der Eltz, H.; Guder, H.-J.; Muhlegger, K. New Hydrolase Substrates. U.S. Patent 4,900,822, 13 February 1990. [Google Scholar]
- Greenwood, J.R.; Calkins, D.; Sullivan, A.; Shelley, J.C. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput. Mol. Des. 2010, 24, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Brazzolotto, X.; Nachon, F.; Harst, M.; Knez, D.; Gobec, S. Human Butyrylcholinesterase in Complex with (S)-2-(butylamino)-N-(2-cycloheptylethyl)-3-(1H-indol-3-yl)propanamide. 2019. Available online: https://www.rcsb.org/structure/6QAA (accessed on 25 April 2021).
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein−Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Zhu, K.; Borrelli, K.W.; Greenwood, J.R.; Day, T.; Abel, R.; Farid, R.S.; Harder, E. Docking Covalent Inhibitors: A Parameter Free Approach To Pose Prediction and Scoring. J. Chem. Inf. Model. 2014, 54, 1932–1940. [Google Scholar] [CrossRef]
- Iacovino, L.; Knez, D.; Colettis, N.; Sova, M.; Pislar, A.; Higgs, J.; Kamecki, F.; Mangialavori, I.; Dolsak, A.; Zakelj, S.; et al. Crystal Structure of Human Monoamine Oxidase B in Complex with Styrylpiperidine Analogue 84. 2020. Available online: https://www.wwpdb.org/pdb?id=pdb_00006rkp (accessed on 25 April 2021).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | RA ± SD (%) at 100 µM or IC50 ± SEM | ||||
|---|---|---|---|---|---|
| Compound | Substituent | hAChE | hBChE | hMAO-A | hMAO-B |
| 6 | 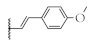 | 61.9 ± 1.2% | 68.8 ± 0.6% | 52.6 ± 0.7% | 72.3 ± 7.5 nM |
| 7 | 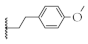 | 73.1 ± 4.3% | 51.2 ± 1.6% | 62.2 ± 1.8% | 93.8 ± 4.1 nM |
| 8 | 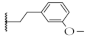 | 73.2 ± 8.9% | 55.5 ± 0.2% | 59.8 ± 3.7% | 238.4 ± 26.9 nM |
| 9 | 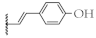 | 65.9 ± 3.1% | 64.9 ± 2.0% | 15.5 ± 0.9 µM | 12.9 ± 1.1 µM |
| 10 | 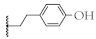 | 61.4 ± 2.7% | 53.8 ± 0.6% | 34.0 ± 5.8 µM | 12.6 ± 2.8 µM |
| 11 | 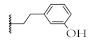 | 54.7 ± 2.9% | 72.9 ± 2.8% | 63.3 ± 0.4% | 52.5 ± 14.8 µM |
| 12 | 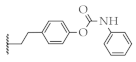 | 52.2 ± 1.7% | 60.2 ± 1.8% | 56.7 ± 0.9% | 9.7 ± 1.2 µM |
| 13 | 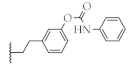 | 51.9 ± 1.4% | 4.4 ± 0.8 µM | 54.0 ± 0.4% | 23.6 ± 3.0 µM |
| 14 | 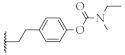 | 66.1 ± 4.8% | 75.5 ± 8.4 µM | 51.2 ± 1.5% | 181.4 ± 28.7 nM |
| 15 | 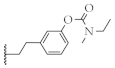 | 60.2 ± 1.8% | 4.3 ± 0.8 µM | 69.4 ± 9.4% | 8.5 ± 0.9 µM |
| PDB Crystal Structure | RMSD of Highest CovDock-Scored vs. Cognate Pose |
|---|---|
| 1S2Q | 3.3049 |
| 1S2Y | 2.4053 |
| 1S3B | 1.7822 |
| 1S3E | 1.9623 |
| 2BYB | 0.9706 |
| 2C65 | 1.2612 |
| 2C66 | 1.5559 |
| 4CRT | 1.0416 |
| 5MRL | 1.6187 |
| 6RKB | 0.5906 |
| 6RKP | 1.0418 |
| 6RLE | 0.7104 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazej, T.; Knez, D.; Meden, A.; Gobec, S.; Sova, M. 4-Phenethyl-1-Propargylpiperidine-Derived Dual Inhibitors of Butyrylcholinesterase and Monoamine Oxidase B. Molecules 2021, 26, 4118. https://doi.org/10.3390/molecules26144118
Mazej T, Knez D, Meden A, Gobec S, Sova M. 4-Phenethyl-1-Propargylpiperidine-Derived Dual Inhibitors of Butyrylcholinesterase and Monoamine Oxidase B. Molecules. 2021; 26(14):4118. https://doi.org/10.3390/molecules26144118
Chicago/Turabian StyleMazej, Tjaša, Damijan Knez, Anže Meden, Stanislav Gobec, and Matej Sova. 2021. "4-Phenethyl-1-Propargylpiperidine-Derived Dual Inhibitors of Butyrylcholinesterase and Monoamine Oxidase B" Molecules 26, no. 14: 4118. https://doi.org/10.3390/molecules26144118
APA StyleMazej, T., Knez, D., Meden, A., Gobec, S., & Sova, M. (2021). 4-Phenethyl-1-Propargylpiperidine-Derived Dual Inhibitors of Butyrylcholinesterase and Monoamine Oxidase B. Molecules, 26(14), 4118. https://doi.org/10.3390/molecules26144118