
Discovery of Novel 2,4-Dianilinopyrimidine Derivatives Containing 4-(Morpholinomethyl)phenyl and N-Substituted Benzamides as Potential FAK Inhibitors and Anticancer Agents

 , , ,
, , ,
Abstract
:
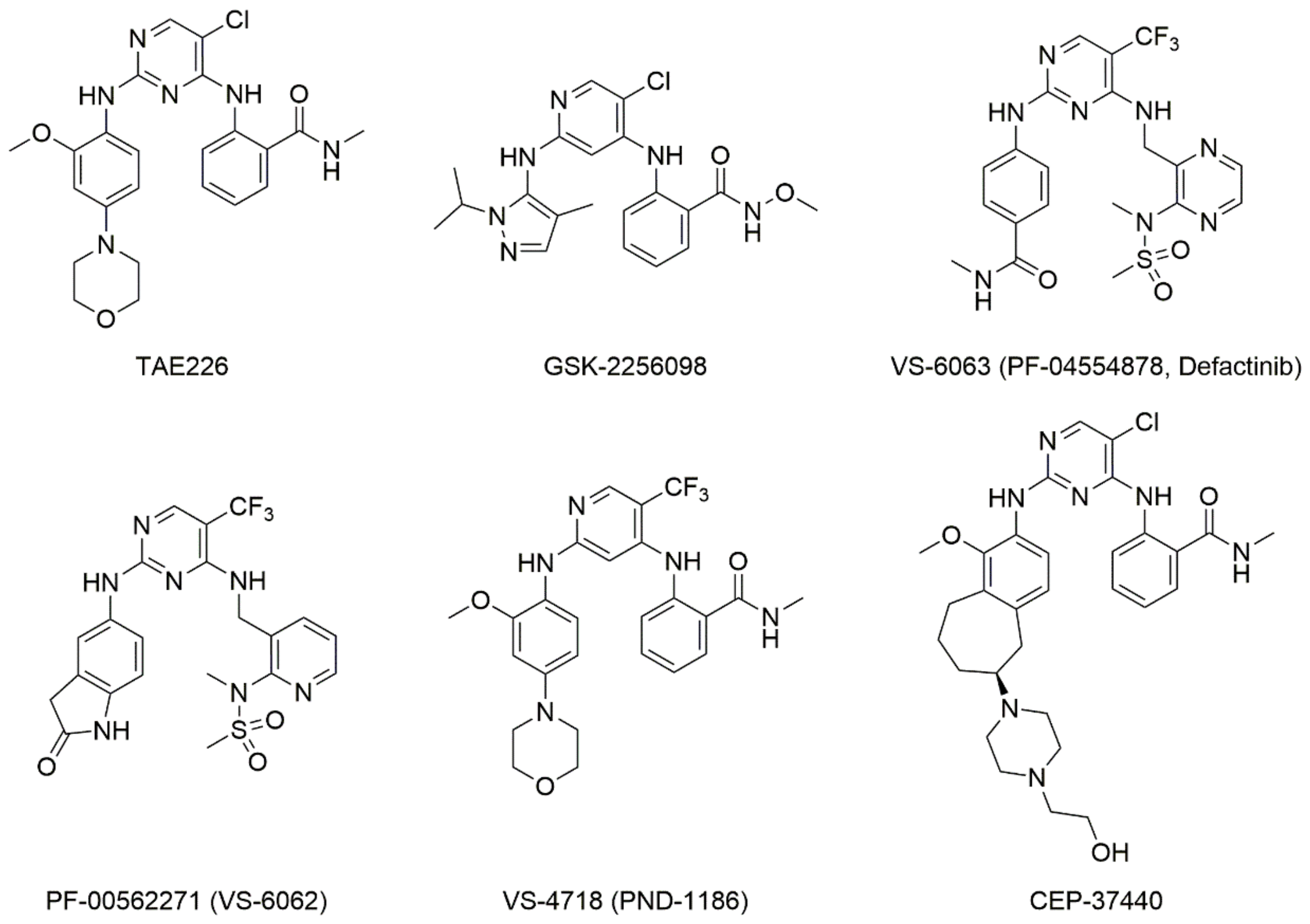
1. Introduction
2. Results and Discussion
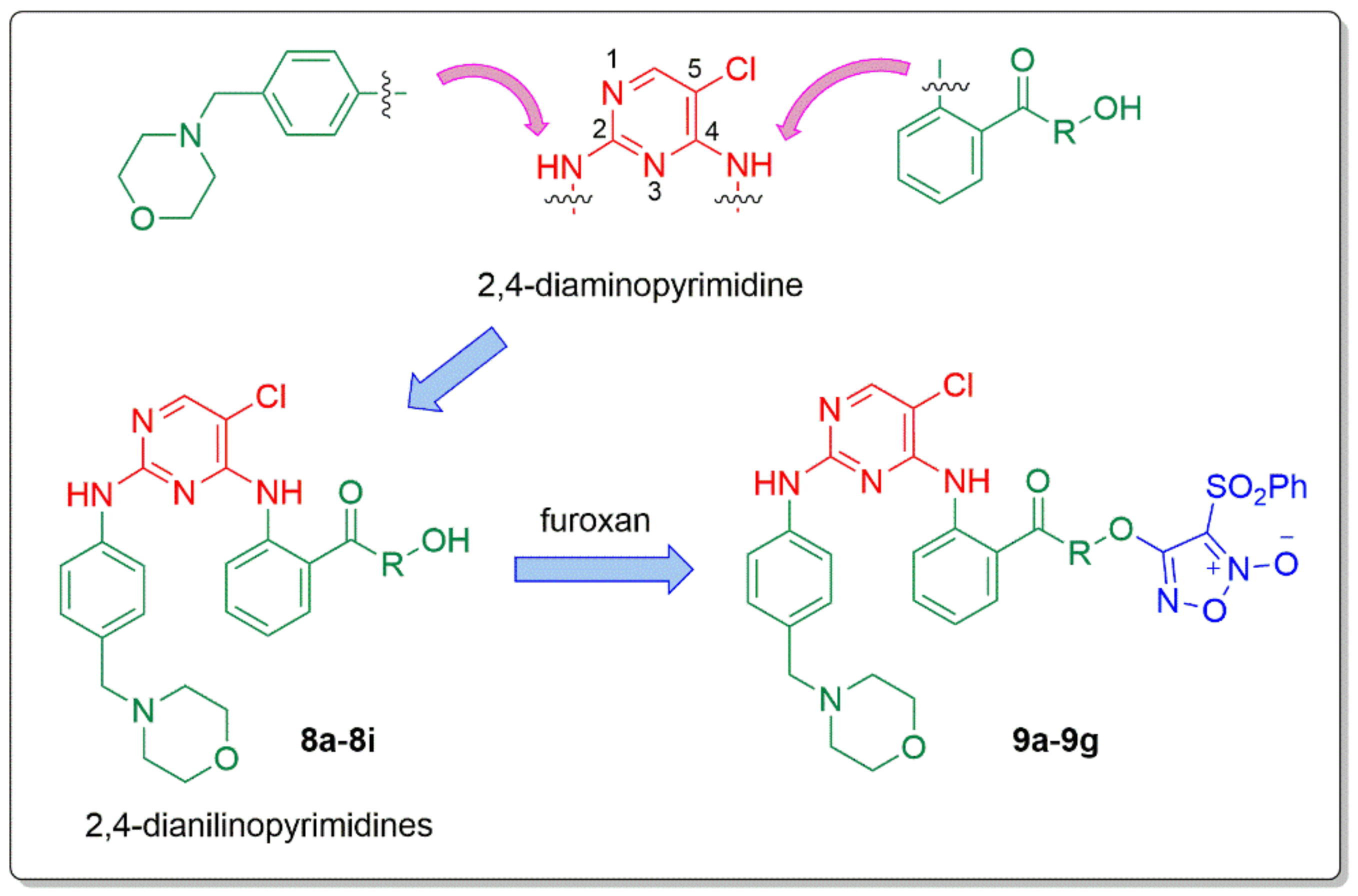
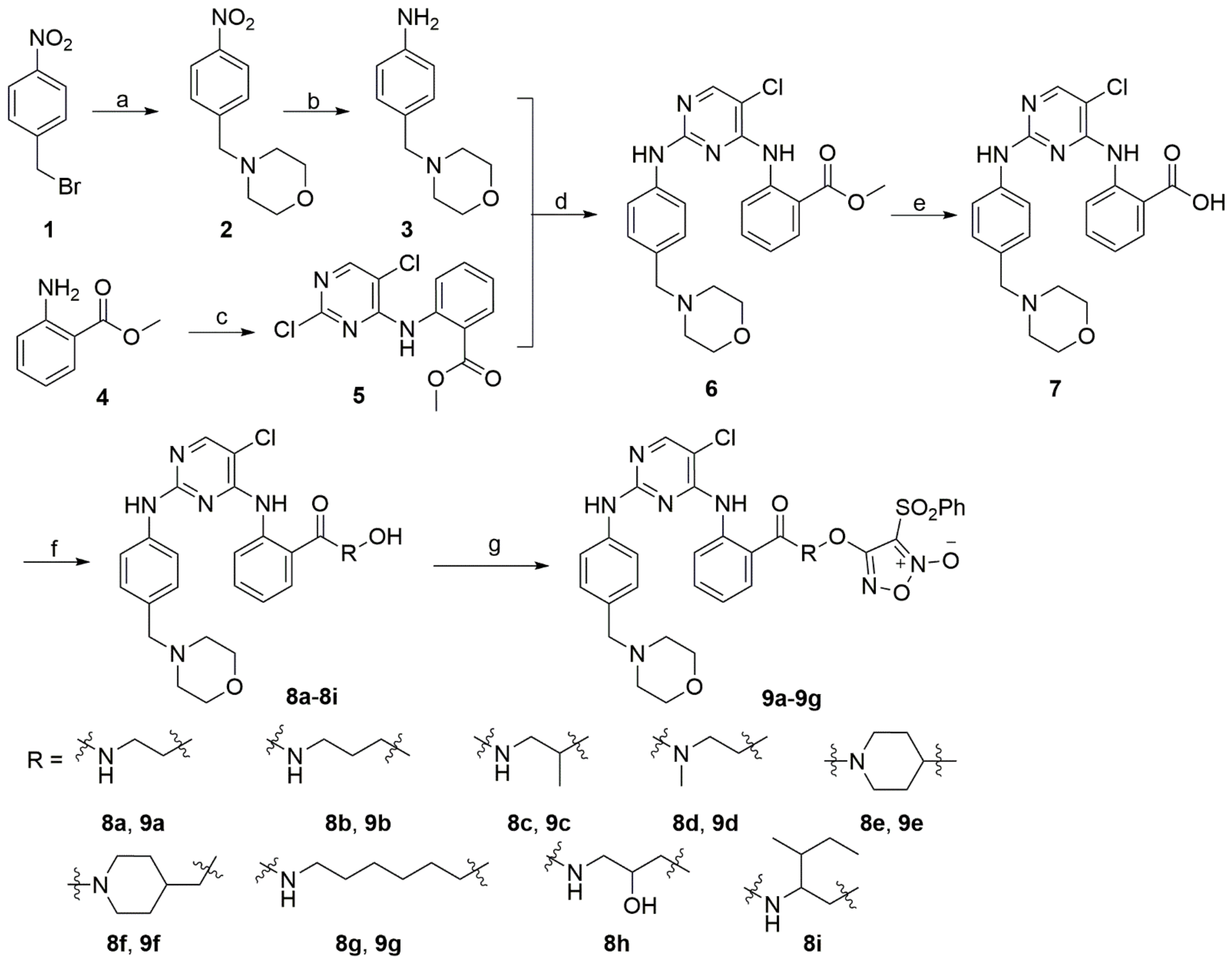
2.1. Synthetic Pathways
2.2. Kinase Inhibitory Activity and Structure–Activity Relationships
2.3. Antiproliferative Activity against Tumor Cells
2.4. Cellular Selectivity Assay
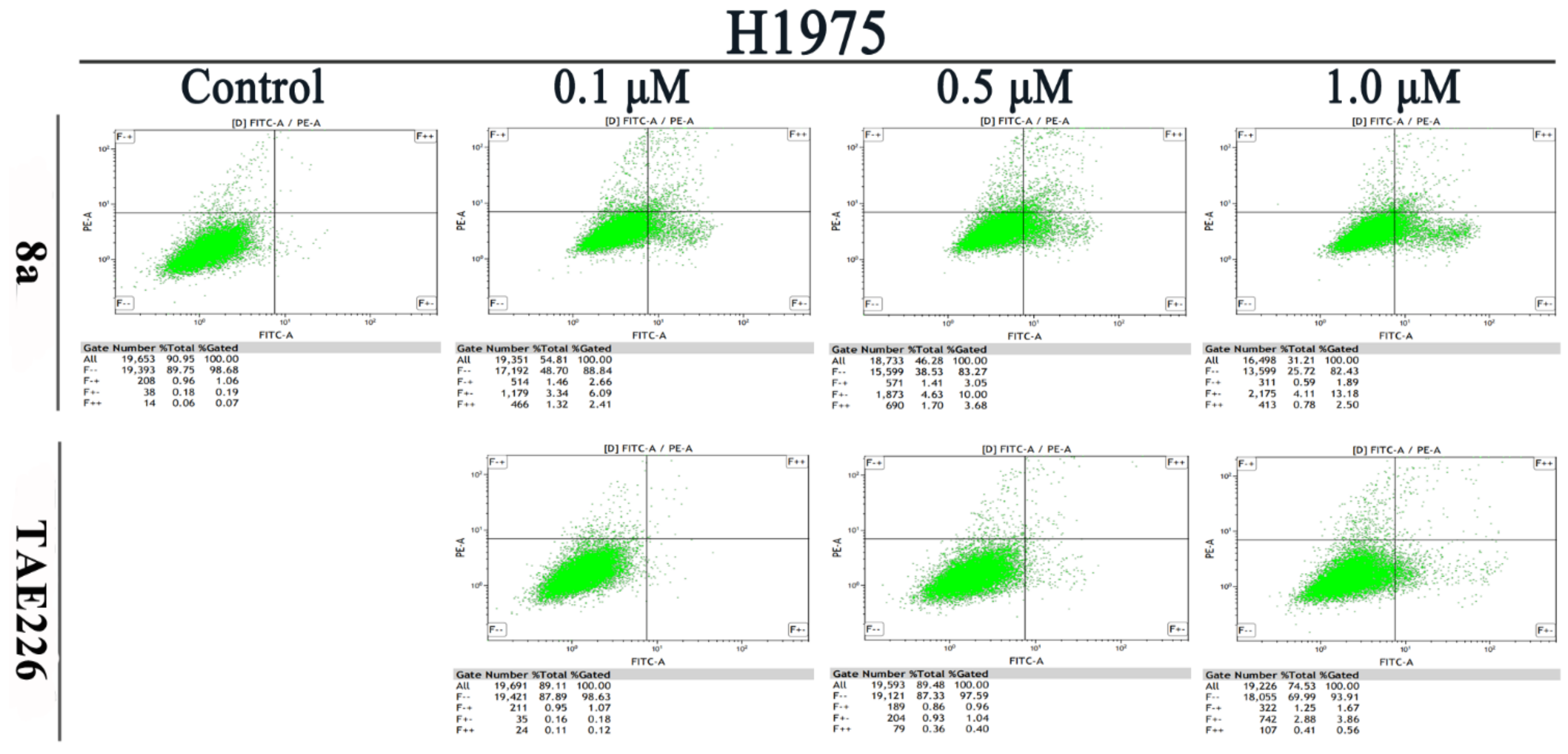
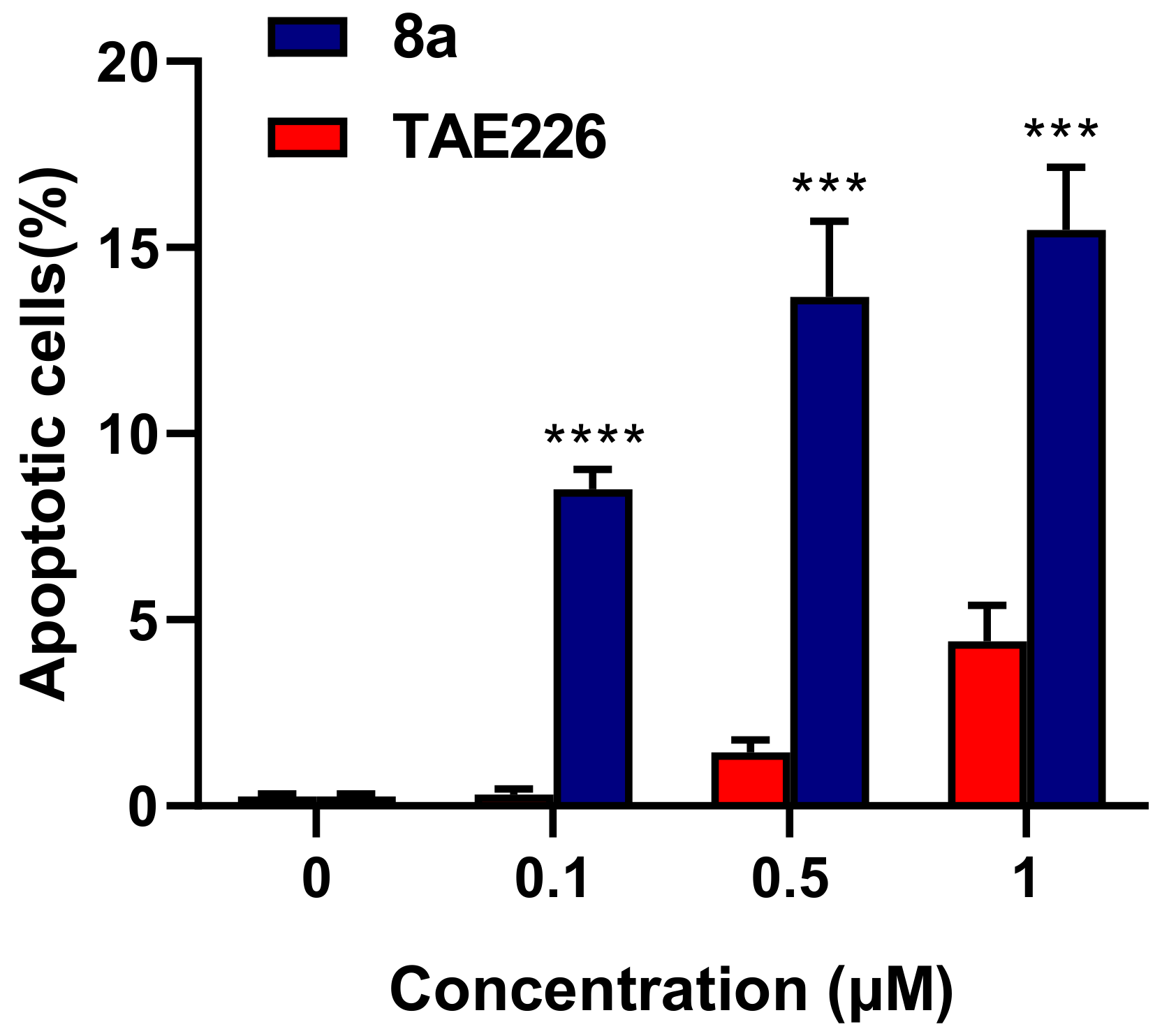
2.5. Cell Apoptosis Assay
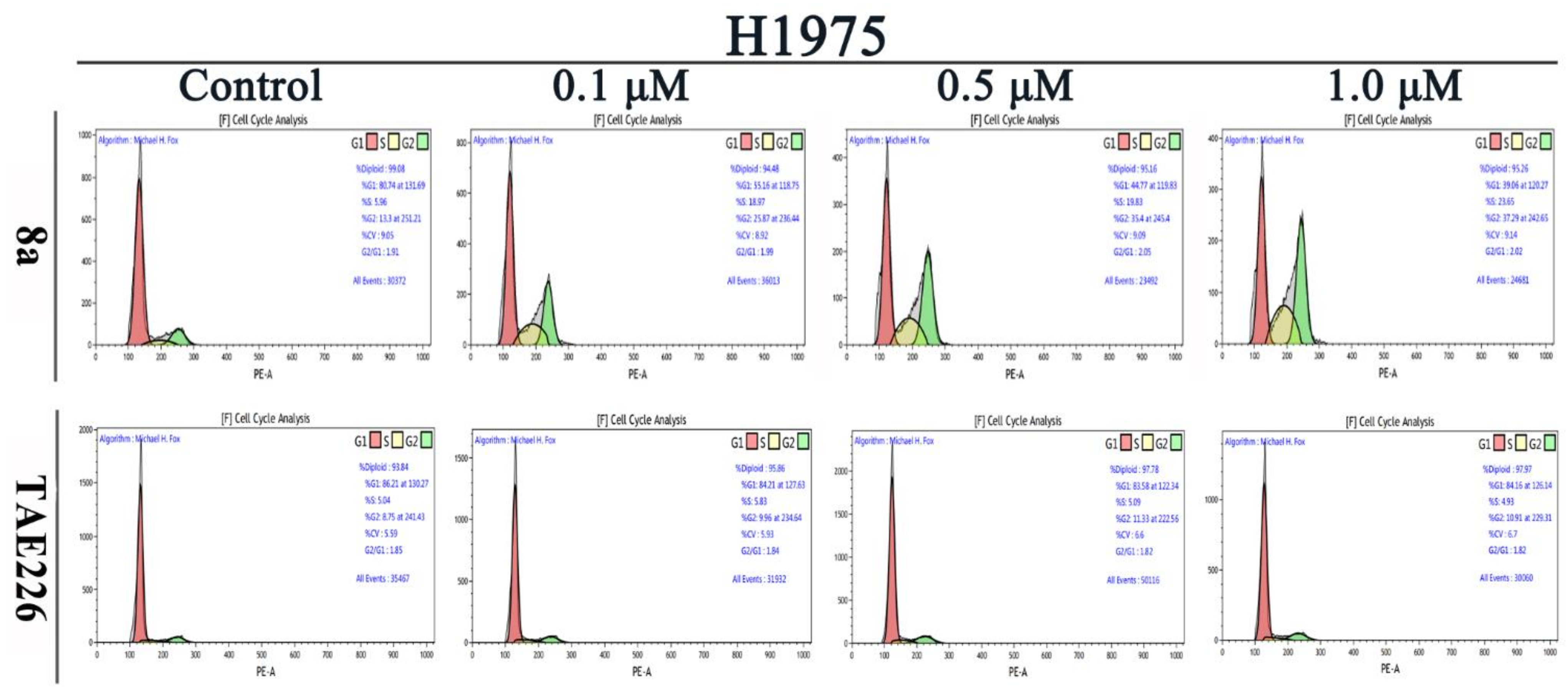
2.6. Cell Cycle Analysis
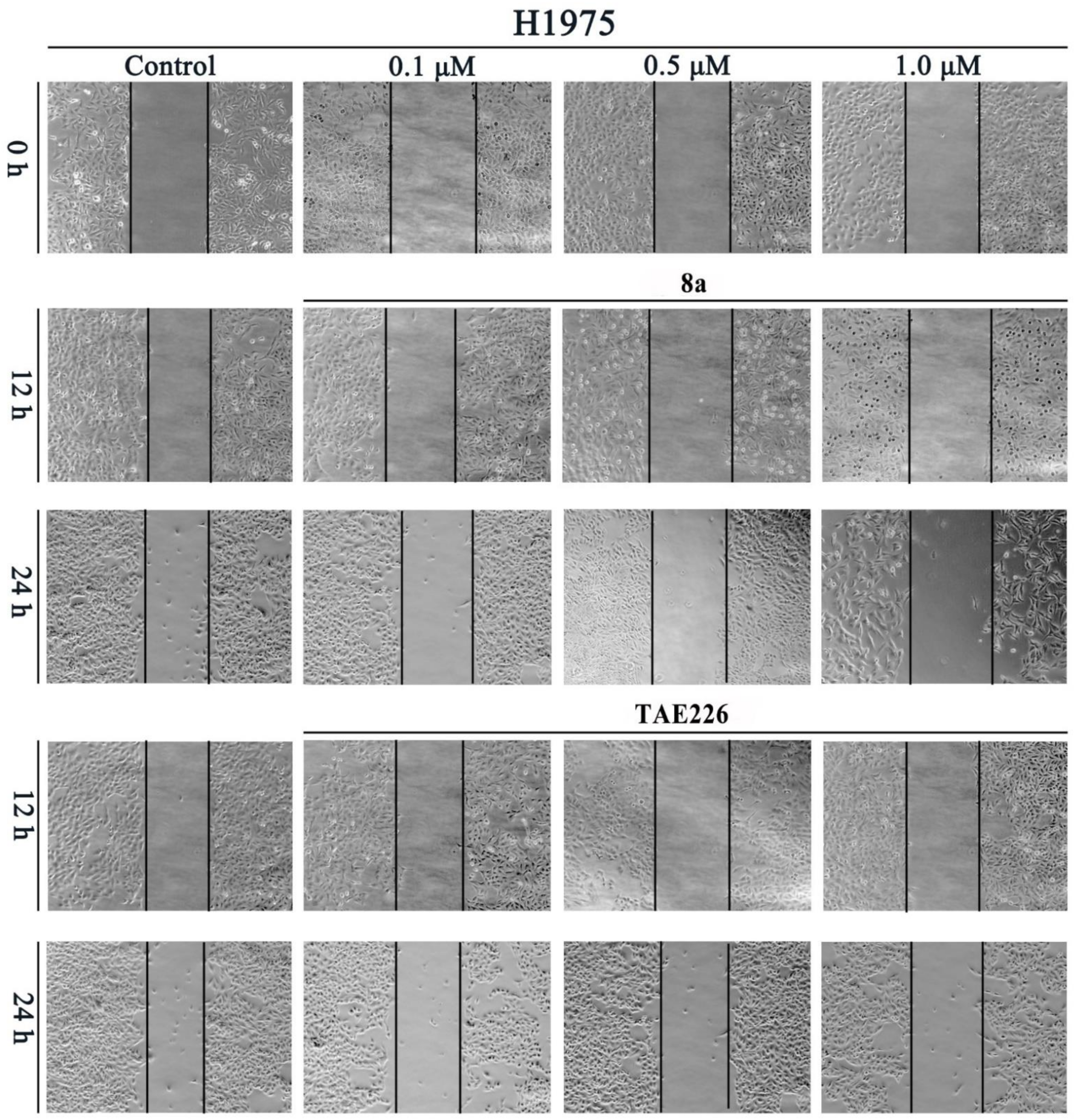
2.7. Cell Migration Assay
2.8. Molecular Docking Study
3. Materials and Methods
3.1. Chemistry
3.1.1. Methyl 2-((2,5-Dichloropyrimidin-4-yl)amino)benzoate (5)
3.1.2. Methyl 2-((5-Chloro-2-((4-(morpholinomethyl)phenyl)amino)pyrimidin-4-yl)amino)benzoate (6)
3.1.3. 2-((5-Chloro-2-((4-(morpholinomethyl)phenyl)amino)pyrimidin-4-yl)amino)benzoic acid (7)
3.1.4. General Procedure for the Preparation of 8a–8i
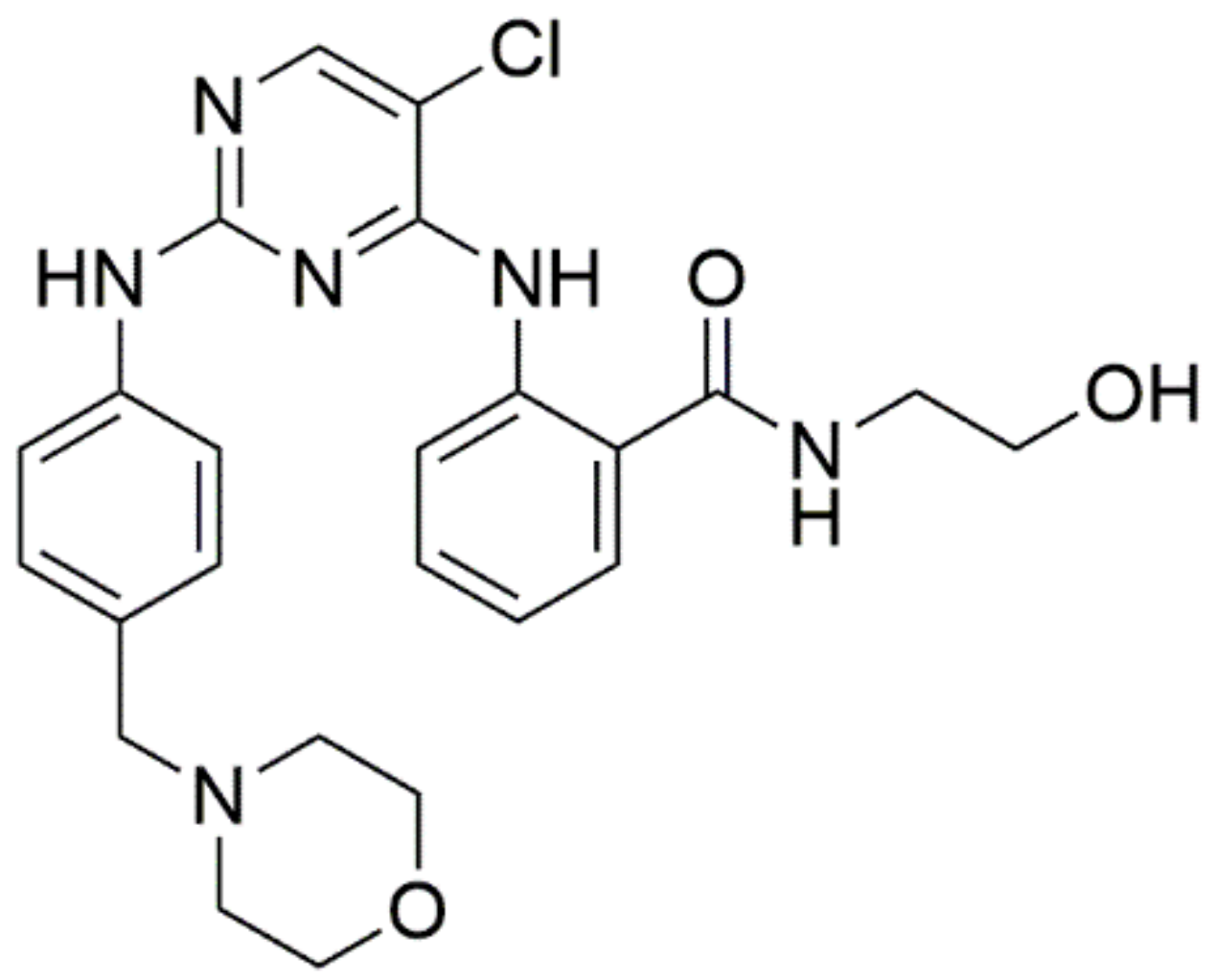
2-((5-Chloro-2-((4-(morpholinomethyl)phenyl)amino)pyrimidin-4-yl)amino)-N-(2-hydroxyethyl)benzamide (8a)
2-((5-Chloro-2-((4-(morpholinomethyl)phenyl)amino)pyrimidin-4-yl)amino)-N-(3-hydroxypropyl)benzamide (8b)
2-((5-Chloro-2-((4-(morpholinomethyl)phenyl)amino)pyrimidin-4-yl)amino)-N-(2-hydroxypropyl)benzamide (8c)
2-((5-Chloro-2-((4-(morpholinomethyl)phenyl)amino)pyrimidin-4-yl)amino)-N-(2-hydroxyethyl)-N-methylbenzamide (8d)
2-((5-Chloro-2-((4-(morpholinomethyl)phenyl)amino)pyrimidin-4-yl)amino)phenyl)(4-hydroxypiperidin-1-yl)methanone (8e)
2-((5-Chloro-2-((4-(morpholinomethyl)phenyl)amino)pyrimidin-4-yl)amino)phenyl)(4-(hydroxymethyl)piperidin-1-yl)methanone (8f)
2-((5-Chloro-2-((4-(morpholinomethyl)phenyl)amino)pyrimidin-4-yl)amino)-N-(6-hydroxyhexyl)benzamide (8g)
2-((5-Chloro-2-((4-(morpholinomethyl)phenyl)amino)pyrimidin-4-yl)amino)-N-(2,3-dihydroxypropyl)benzamide (8h)
2-((5-Chloro-2-((4-(morpholinomethyl)phenyl)amino)pyrimidin-4-yl)amino)-N-(1-hydroxy-3-methylpentan-2-yl)benzamide (8i)
3.1.5. General Procedure for the Preparation of 9a–9g
4-(2-(2-((5-Chloro-2-((4-(morpholinomethyl)phenyl)amino)pyrimidin-4-yl)amino)benzamido)ethoxy)-3-(phenylsulfonyl)-1,2,5-oxadiazole 2-oxide (9a)
4-(3-(2-((5-Chloro-2-((4-(morpholinomethyl)phenyl)amino)pyrimidin-4-yl)amino)benzamido)propoxy)-3-(phenylsulfonyl)-1,2,5-oxadiazole 2-oxide (9b)
4-((1-(2-((5-Chloro-2-((4-(morpholinomethyl)phenyl)amino)pyrimidin-4-yl)amino)benzamido)propan-2-yl)oxy)-3-(phenylsulfonyl)-1,2,5-oxadiazole 2-oxide (9c)
4-(2-(2-((5-Chloro-2-((4-(morpholinomethyl)phenyl)amino)pyrimidin-4-yl)amino)-N-methylbenzamido)ethoxy)-3-(phenylsulfonyl)-1,2,5-oxadiazole 2-oxide (9d)
4-((1-(2-((5-Chloro-2-((4-(morpholinomethyl)phenyl)amino)pyrimidin-4-yl)amino)benzoyl)piperidin-4-yl)oxy)-3-(phenylsulfonyl)-1,2,5-oxadiazole 2-oxide (9e)
4-((1-(2-((5-Chloro-2-((4-(morpholinomethyl)phenyl)amino)pyrimidin-4-yl)amino)benzoyl)piperidin-4-yl)methoxy)-3-(phenylsulfonyl)-1,2,5-oxadiazole 2-oxide (9f)
4-((6-(2-((5-Chloro-2-((4-(morpholinomethyl)phenyl)amino)pyrimidin-4-yl)amino)benzamido)hexyl)oxy)-3-(phenylsulfonyl)-1,2,5-oxadiazole 2-oxide (9g)
3.2. In Vitro Kinase Enzymatic Assay
3.3. Cell Viability Determination
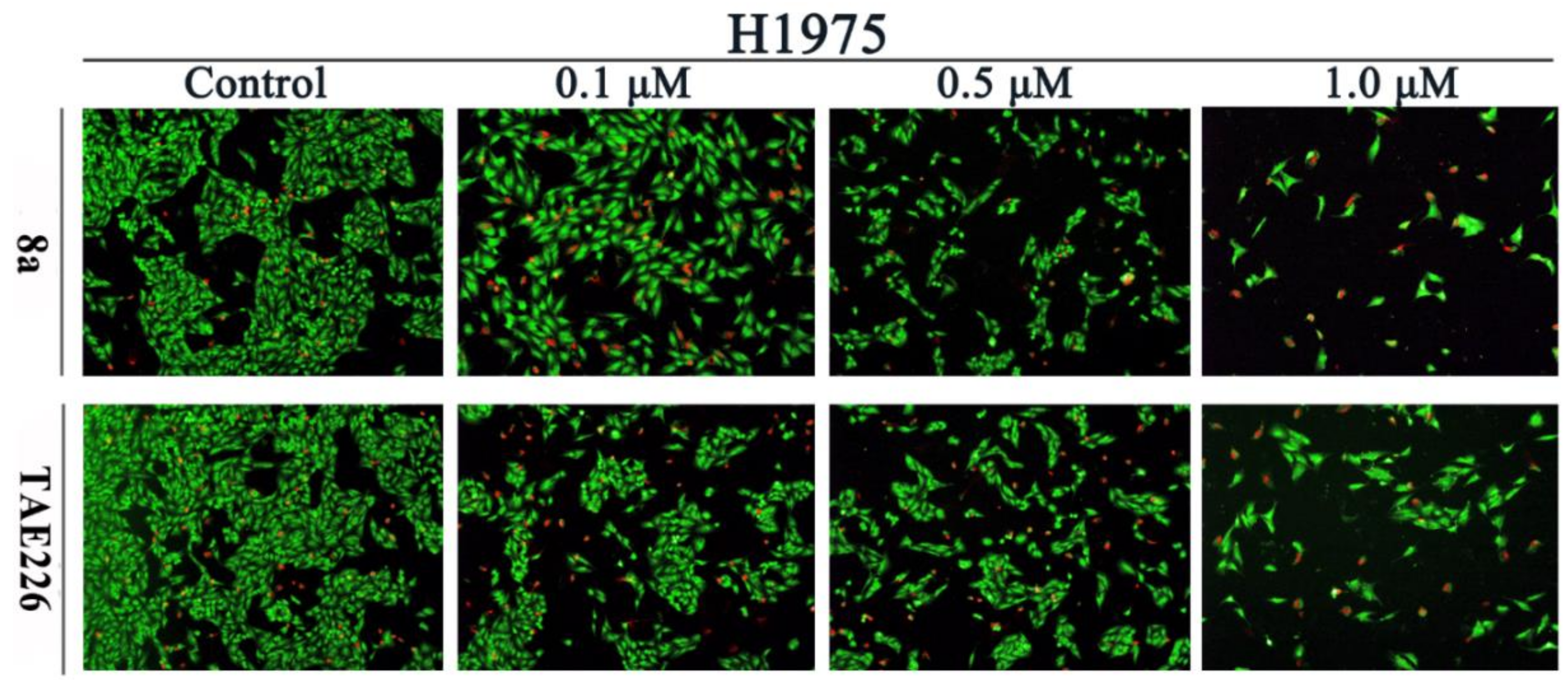
3.4. Acridine Orange/Ethidium Bromide (AO/EB) Staining Assay
3.5. Flow Cytometric Analysis
3.6. Scratch Assay
3.7. Molecular Docking Study
3.8. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Murphree, A.L.; Villablanca, J.G.; Deegan, W.F., 3rd; Sato, J.K.; Malogolowkin, M.; Fisher, A.; Parker, R.; Reed, E.; Gomer, C.J. Chemotherapy plus local treatment in the management of intraocular retinoblastoma. Arch. Ophthalmol. 1996, 114, 1348–1356. [Google Scholar] [CrossRef]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef] [PubMed]
- Hoelder, S.; Clarke, P.A.; Workman, P. Discovery of small molecule cancer drugs: Successes, challenges and opportunities. Mol. Oncol 2012, 6, 155–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.; Dehart, J.P.; Murphy, J.M.; Lim, S.T. Understanding the roles of FAK in cancer: Inhibitors, genetic models, and new insights. J. Histochem. Cytochem. 2015, 63, 114–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cance, W.G.; Harris, J.E.; Iacocca, M.V.; Roche, E.; Yang, X.; Chang, J.; Simkins, S.; Xu, L. Immunohistochemical analyses of focal adhesion kinase expression in benign and malignant human breast and colon tissues: Correlation with preinvasive and invasive phenotypes. Clin. Cancer Res. 2000, 6, 2417–2423. [Google Scholar] [PubMed]
- Owens, L.V.; Xu, L.; Dent, G.A.; Yang, X.; Sturge, G.C.; Craven, R.J.; Cance, W.G. Focal adhesion kinase as a marker of invasive potential in differentiated human thyroid cancer. Ann. Surg Oncol. 1996, 3, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, L.; Hauck, W.; Aprikian, A.G.; Begin, L.R.; Chapdelaine, A.; Chevalier, S. Focal adhesion kinase (pp125FAK) expression, activation and association with paxillin and p50CSK in human metastatic prostate carcinoma. Int. J. Cancer 1996, 68, 164–171. [Google Scholar] [CrossRef]
- Natarajan, M.; Hecker, T.P.; Gladson, C.L. FAK signaling in anaplastic astrocytoma and glioblastoma tumors. Cancer J. 2003, 9, 126–133. [Google Scholar] [CrossRef]
- Sood, A.K.; Coffin, J.E.; Schneider, G.B.; Fletcher, M.S.; DeYoung, B.R.; Gruman, L.M.; Gershenson, D.M.; Schaller, M.D.; Hendrix, M.J. Biological significance of focal adhesion kinase in ovarian cancer: Role in migration and invasion. Am. J. Pathol. 2004, 165, 1087–1095. [Google Scholar] [CrossRef]
- Yuan, Z.; Zheng, Q.; Fan, J.; Ai, K.X.; Chen, J.; Huang, X.Y. Expression and prognostic significance of focal adhesion kinase in hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 2010, 136, 1489–1496. [Google Scholar] [CrossRef]
- Park, J.H.; Lee, B.L.; Yoon, J.; Kim, J.; Kim, M.A.; Yang, H.K.; Kim, W.H. Focal adhesion kinase (FAK) gene amplification and its clinical implications in gastric cancer. Hum. Pathol. 2010, 41, 1664–1673. [Google Scholar] [CrossRef]
- Carelli, S.; Zadra, G.; Vaira, V.; Falleni, M.; Bottiglieri, L.; Nosotti, M.; Di Giulio, A.M.; Gorio, A.; Bosari, S. Up-regulation of focal adhesion kinase in non-small cell lung cancer. Lung Cancer 2006, 53, 263–271. [Google Scholar] [CrossRef]
- Diaz Osterman, C.J.; Ozmadenci, D.; Kleinschmidt, E.G.; Taylor, K.N.; Barrie, A.M.; Jiang, S.; Bean, L.M.; Sulzmaier, F.J.; Jean, C.; Tancioni, I.; et al. FAK activity sustains intrinsic and acquired ovarian cancer resistance to platinum chemotherapy. eLife 2019, 8, e47327. [Google Scholar] [CrossRef]
- Tavora, B.; Reynolds, L.E.; Batista, S.; Demircioglu, F.; Fernandez, I.; Lechertier, T.; Lees, D.M.; Wong, P.P.; Alexopoulou, A.; Elia, G.; et al. Endothelial-cell FAK targeting sensitizes tumours to DNA-damaging therapy. Nature 2014, 514, 112–116. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Hegde, S.; Knolhoff, B.L.; Zhu, Y.; Herndon, J.M.; Meyer, M.A.; Nywening, T.M.; Hawkins, W.G.; Shapiro, I.M.; Weaver, D.T.; et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat. Med. 2016, 22, 851–860. [Google Scholar] [CrossRef]
- Serrels, A.; Lund, T.; Serrels, B.; Byron, A.; McPherson, R.C.; von Kriegsheim, A.; Gomez-Cuadrado, L.; Canel, M.; Muir, M.; Ring, J.E.; et al. Nuclear FAK controls chemokine transcription, Tregs, and evasion of anti-tumor immunity. Cell 2015, 163, 160–173. [Google Scholar] [CrossRef] [Green Version]
- Kostourou, V.; Lechertier, T.; Reynolds, L.E.; Lees, D.M.; Baker, M.; Jones, D.T.; Tavora, B.; Ramjaun, A.R.; Birdsey, G.M.; Robinson, S.D.; et al. FAK-heterozygous mice display enhanced tumour angiogenesis. Nat. Commun. 2013, 4, 2020. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Sun, H. Progress in the Development of Small Molecular Inhibitors of Focal Adhesion Kinase (FAK). J. Med. Chem. 2020, 63, 14382–14403. [Google Scholar] [CrossRef]
- Zhang, J.; He, D.H.; Zajac-Kaye, M.; Hochwald, S.N. A small molecule FAK kinase inhibitor, GSK2256098, inhibits growth and survival of pancreatic ductal adenocarcinoma cells. Cell Cycle 2014, 13, 3143–3149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, W.G.; Ung, E.; Whalen, P.; Cooper, B.; Hulford, C.; Autry, C.; Richter, D.; Emerson, E.; Lin, J.; Kath, J.; et al. Antitumor activity and pharmacology of a selective focal adhesion kinase inhibitor, PF-562,271. Cancer Res. 2008, 68, 1935–1944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, G.R.; Cheng, M.; Learn, K.S.; Wagner, J.; Gingrich, D.E.; Lisko, J.G.; Curry, M.; Mesaros, E.F.; Ghose, A.K.; Quail, M.R.; et al. Discovery of Clinical Candidate CEP-37440, a Selective Inhibitor of Focal Adhesion Kinase (FAK) and Anaplastic Lymphoma Kinase (ALK). J. Med. Chem. 2016, 59, 7478–7496. [Google Scholar] [CrossRef] [PubMed]
- Tiede, S.; Meyer-Schaller, N.; Kalathur, R.K.R.; Ivanek, R.; Fagiani, E.; Schmassmann, P.; Stillhard, P.; Hafliger, S.; Kraut, N.; Schweifer, N.; et al. The FAK inhibitor BI 853520 exerts anti-tumor effects in breast cancer. Oncogenesis 2018, 7, 73. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.M.; Lee, B.Y.; Castillo, L.; Spielman, C.; Grogan, J.; Yeung, N.K.; Kench, J.G.; Stricker, P.D.; Haynes, A.M.; Centenera, M.M.; et al. Effect of FAK inhibitor VS-6063 (defactinib) on docetaxel efficacy in prostate cancer. Prostate 2018, 78, 308–317. [Google Scholar] [CrossRef]
- Walsh, C.; Tanjoni, I.; Uryu, S.; Tomar, A.; Nam, J.O.; Luo, H.; Phillips, A.; Patel, N.; Kwok, C.; McMahon, G.; et al. Oral delivery of PND-1186 FAK inhibitor decreases tumor growth and spontaneous breast to lung metastasis in pre-clinical models. Cancer Biol. Ther. 2010, 9, 778–790. [Google Scholar] [CrossRef] [Green Version]
- Kurio, N.; Shimo, T.; Fukazawa, T.; Takaoka, M.; Okui, T.; Hassan, N.M.; Honami, T.; Hatakeyama, S.; Ikeda, M.; Naomoto, Y.; et al. Anti-tumor effect in human breast cancer by TAE226, a dual inhibitor for FAK and IGF-IR in vitro and in vivo. Exp. Cell Res. 2011, 317, 1134–1146. [Google Scholar] [CrossRef]
- Wang, R.; Yu, S.; Zhao, X.; Chen, Y.; Yang, B.; Wu, T.; Hao, C.; Zhao, D.; Cheng, M. Design, synthesis, biological evaluation and molecular docking study of novel thieno[3,2-d]pyrimidine derivatives as potent FAK inhibitors. Eur. J. Med. Chem. 2020, 188, 112024. [Google Scholar] [CrossRef]
- Lietha, D.; Eck, M.J. Crystal structures of the FAK kinase in complex with TAE226 and related bis-anilino pyrimidine inhibitors reveal a helical DFG conformation. PLoS ONE 2008, 3, e3800. [Google Scholar] [CrossRef] [Green Version]
- Huang, Z.J.; Fu, J.J.; Zhang, Y.H. Nitric Oxide Donor-Based Cancer Therapy: Advances and Prospects. J. Med. Chem. 2017, 60, 7617–7635. [Google Scholar] [CrossRef]
- Cross, D.A.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014, 4, 1046–1061. [Google Scholar] [CrossRef] [Green Version]
- Murugesan, D.; Mital, A.; Kaiser, M.; Shackleford, D.M.; Morizzi, J.; Katneni, K.; Campbell, M.; Hudson, A.; Charman, S.A.; Yeates, C.; et al. Discovery and structure-activity relationships of pyrrolone antimalarials. J. Med. Chem. 2013, 56, 2975–2990. [Google Scholar] [CrossRef]
- Farrar, W.V. The 3, 4-bisarenesulphonylfuroxans. J. Chem. Soc. 1964, 904–906. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | R | FAK, IC50 (μM) a | Antiproliferative Activity IC50 (μM) a | SI Values b | ||
|---|---|---|---|---|---|---|
| H1975 | A431 | HBE | ||||
| 8a | 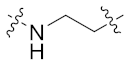 | 0.047 ± 0.006 | 0.044 ± 0.011 | 0.119 ± 0.036 | 1.039 ± 0.266 | 24 |
| 8b | 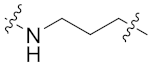 | 0.061 ± 0.009 | 0.075 ± 0.011 | 0.207 ± 0.036 | 1.153 ± 0.283 | 15 |
| 8c | 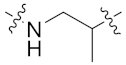 | 0.030 ± 0.007 | 0.119 ± 0.047 | 0.158 ± 0.027 | 4.007 ± 0.231 | 34 |
| 8d | 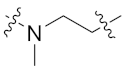 | 0.040 ± 0.011 | 0.293 ± 0.050 | 0.161 ± 0.082 | 3.825 ± 0.500 | 13 |
| 8e |  | 0.129 ± 0.037 | 1.414 ± 0.132 | 0.238 ± 0.098 | 0.435 ± 0.045 | 0.3 |
| 8f | 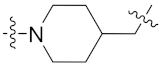 | 0.173 ± 0.023 | 2.221 ± 0.261 | 0.195 ± 0.097 | 0.862 ± 0.125 | 0.4 |
| 8g | 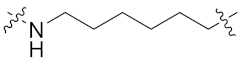 | 0.198 ± 0.032 | 0.287 ± 0.061 | 0.507 ± 0.019 | 1.645 ± 0.813 | 6 |
| 8h | 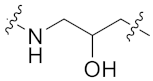 | 0.072 ± 0.007 | 0.219 ± 0.082 | 0.047 ± 0.006 | 0.688 ± 0.121 | 3 |
| 8i | 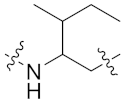 | 0.126 ± 0.021 | 0.815 ± 0.061 | 1.245 ± 0.909 | >20 | >24 |
| 9a |  | 0.149 ± 0.033 | 0.047 ± 0.009 | 0.182 ± 0.032 | 0.916 ± 0.092 | 19 |
| 9b | 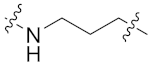 | 0.338 ± 0.038 | 0.164 ± 0.051 | 0.138 ± 0.031 | 2.078 ± 0.366 | 13 |
| 9c |  | 0.089 ± 0.014 | 0.567 ± 0.086 | 1.074 ± 0.154 | 2.418 ± 0.546 | 4 |
| 9d | 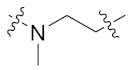 | 0.066 ± 0.014 | 1.094 ± 0.137 | 2.738 ± 0.206 | 4.552 ± 0.533 | 4 |
| 9e | 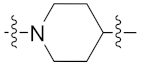 | >1 | 1.724 ± 0.295 | 2.414 ± 0.587 | 0.763 ± 0.100 | 0.4 |
| 9f |  | 0.300 ± 0.009 | 1.570 ± 0.265 | 1.739 ± 0.305 | 3.761 ± 1.604 | 2 |
| 9g | 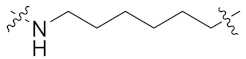 | >1 | 0.592 ± 0.073 | 0.617 ± 0.040 | 3.009 ± 0.090 | 5 |
| TAE226 | 0.0034 ± 0.0005 | 0.141 ± 0.041 | 0.392 ± 0.053 | >20 | >142 | |
| Osimertinib | ND | 0.116 ± 0.032 | 0.988 ± 0.119 | 1.208 ± 0.177 | 10 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, C.; Shen, K.; Wang, S.; Wang, Z.; Su, F.; Wu, X.; Hu, X.; Li, M.; Han, J.; Wu, L. Discovery of Novel 2,4-Dianilinopyrimidine Derivatives Containing 4-(Morpholinomethyl)phenyl and N-Substituted Benzamides as Potential FAK Inhibitors and Anticancer Agents. Molecules 2021, 26, 4187. https://doi.org/10.3390/molecules26144187
Han C, Shen K, Wang S, Wang Z, Su F, Wu X, Hu X, Li M, Han J, Wu L. Discovery of Novel 2,4-Dianilinopyrimidine Derivatives Containing 4-(Morpholinomethyl)phenyl and N-Substituted Benzamides as Potential FAK Inhibitors and Anticancer Agents. Molecules. 2021; 26(14):4187. https://doi.org/10.3390/molecules26144187
Chicago/Turabian StyleHan, Chun, Kemin Shen, Shijun Wang, Zhijun Wang, Feng Su, Xi Wu, Xiaoqin Hu, Mengyao Li, Jing Han, and Lintao Wu. 2021. "Discovery of Novel 2,4-Dianilinopyrimidine Derivatives Containing 4-(Morpholinomethyl)phenyl and N-Substituted Benzamides as Potential FAK Inhibitors and Anticancer Agents" Molecules 26, no. 14: 4187. https://doi.org/10.3390/molecules26144187
APA StyleHan, C., Shen, K., Wang, S., Wang, Z., Su, F., Wu, X., Hu, X., Li, M., Han, J., & Wu, L. (2021). Discovery of Novel 2,4-Dianilinopyrimidine Derivatives Containing 4-(Morpholinomethyl)phenyl and N-Substituted Benzamides as Potential FAK Inhibitors and Anticancer Agents. Molecules, 26(14), 4187. https://doi.org/10.3390/molecules26144187