
Total Synthesis of Pagoamide A


Abstract
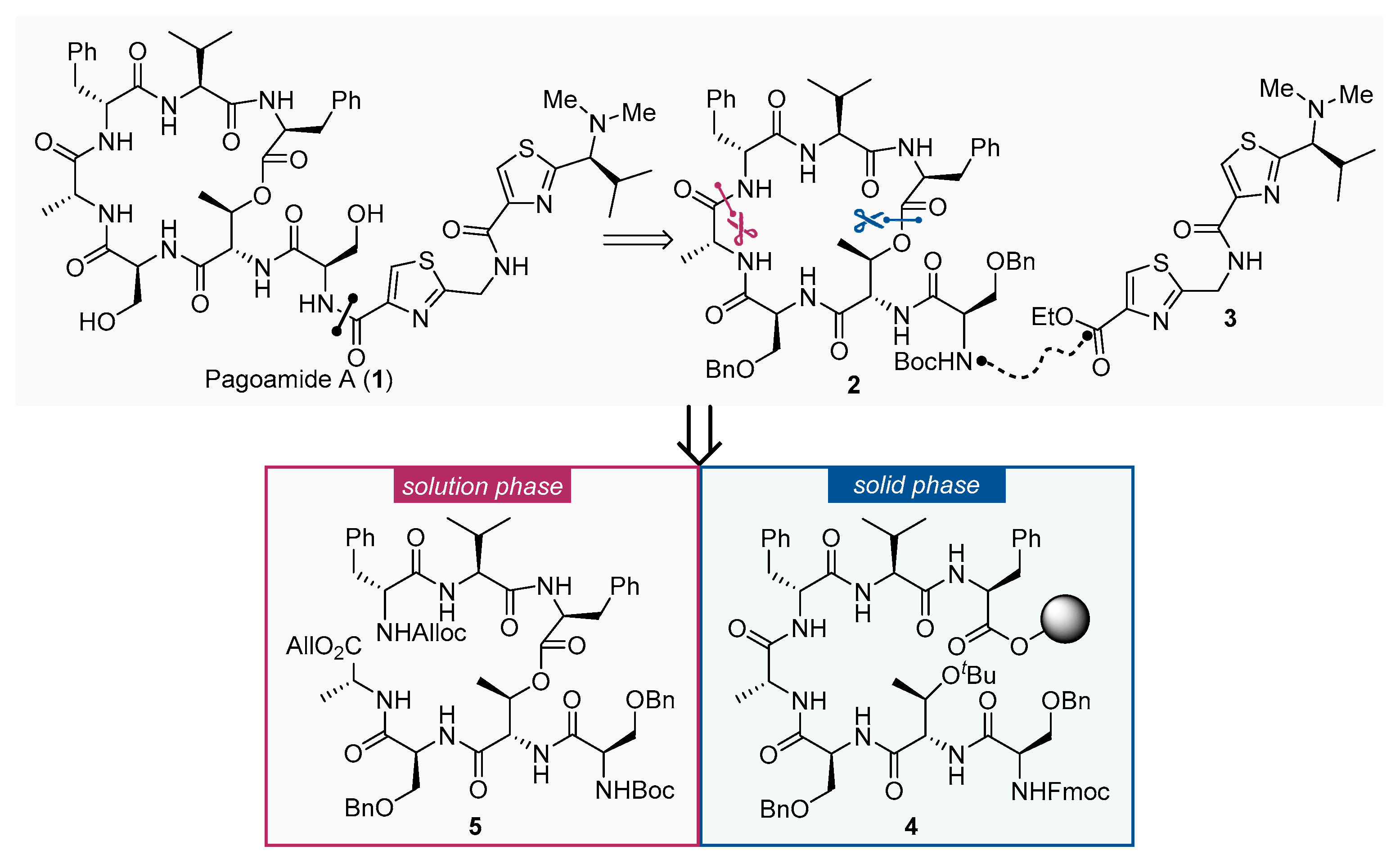
:1. Introduction
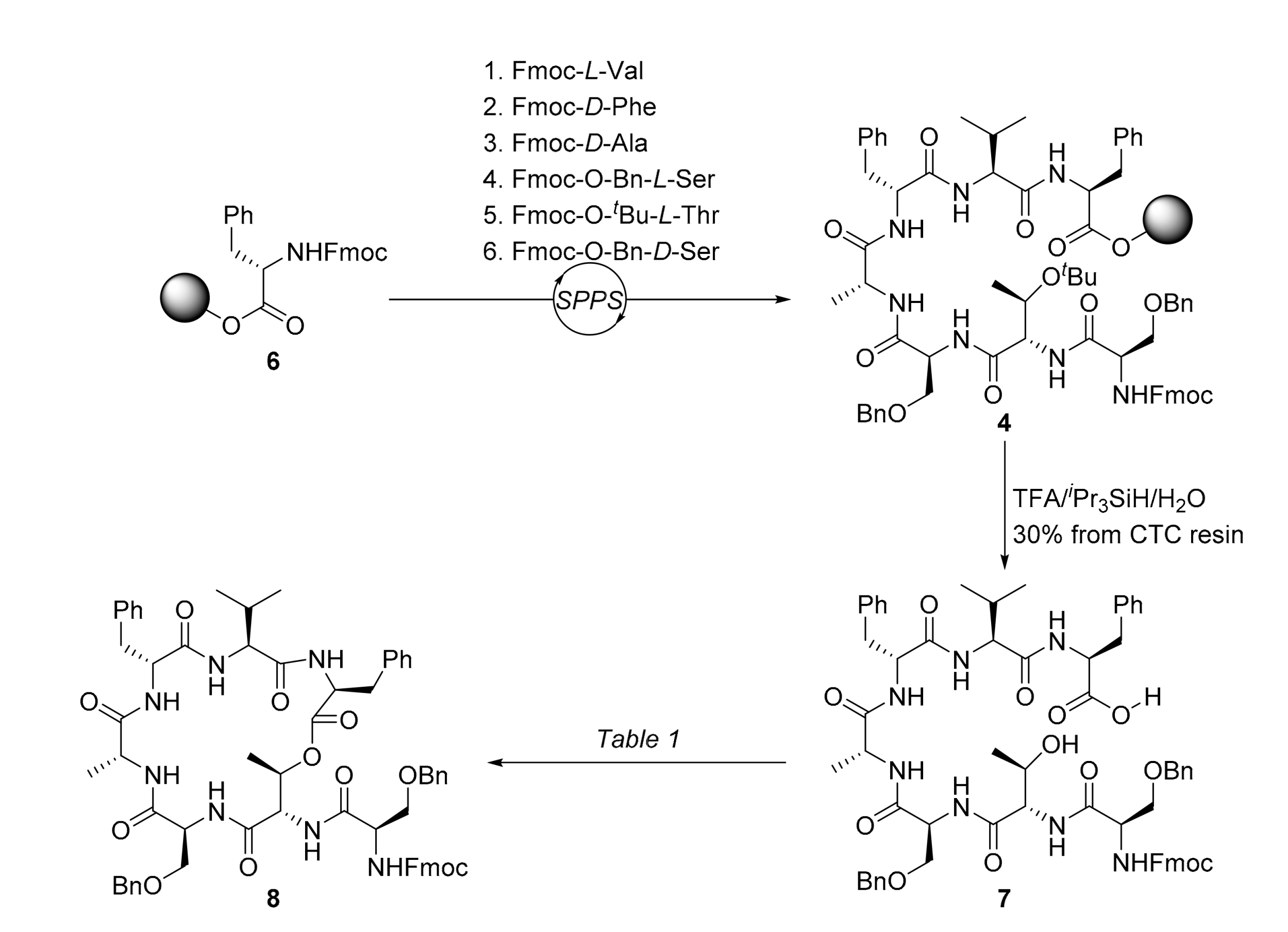
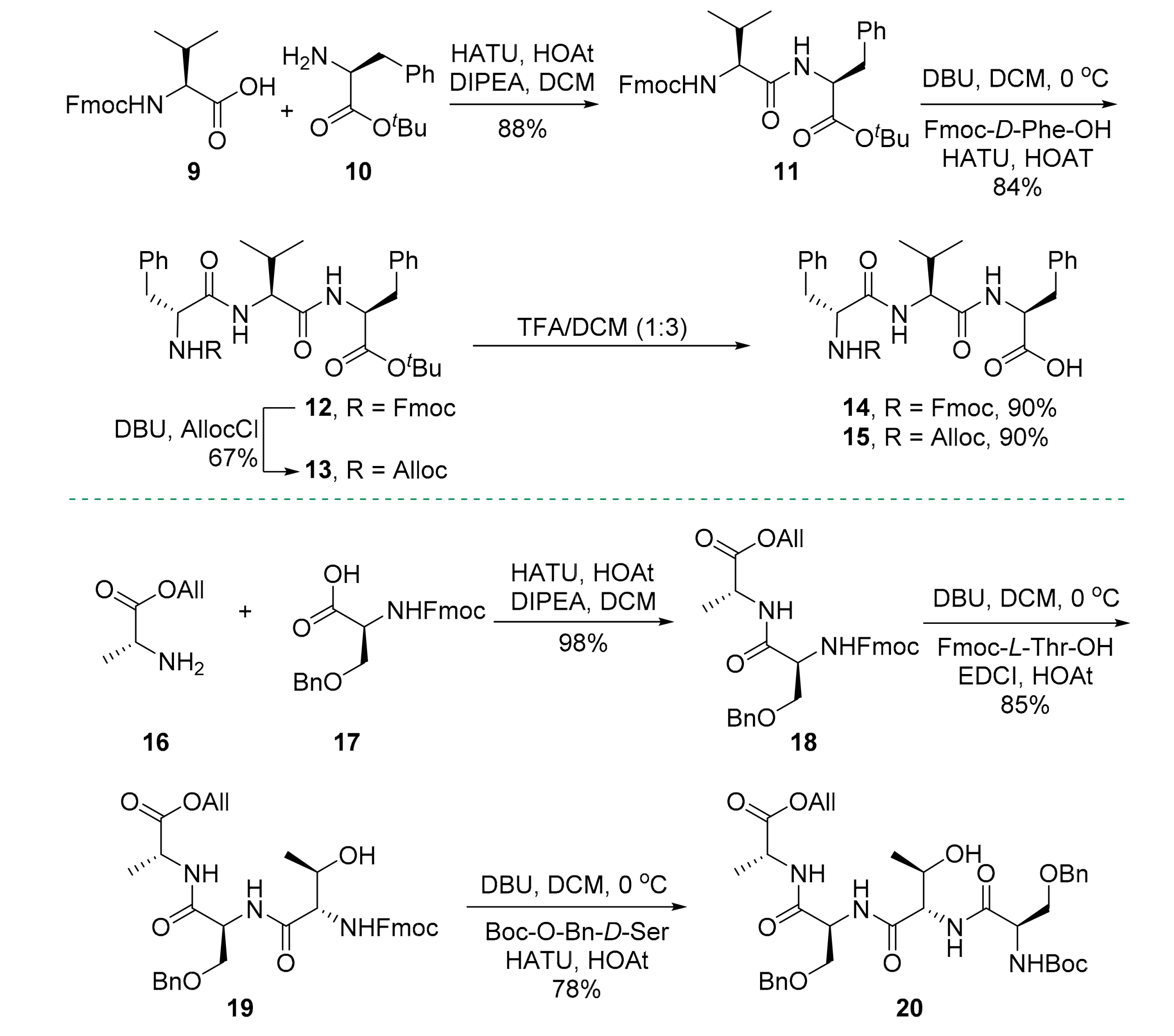
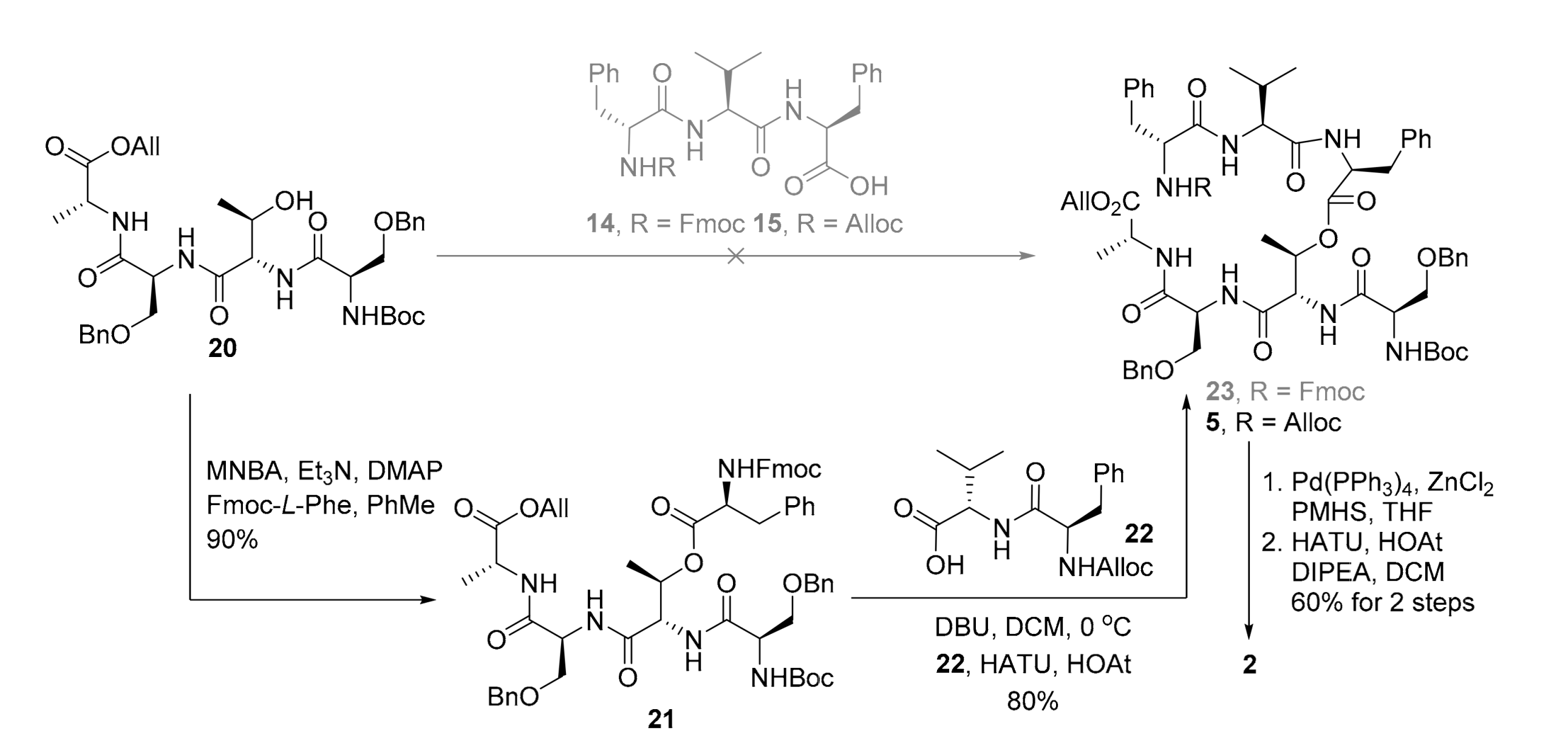
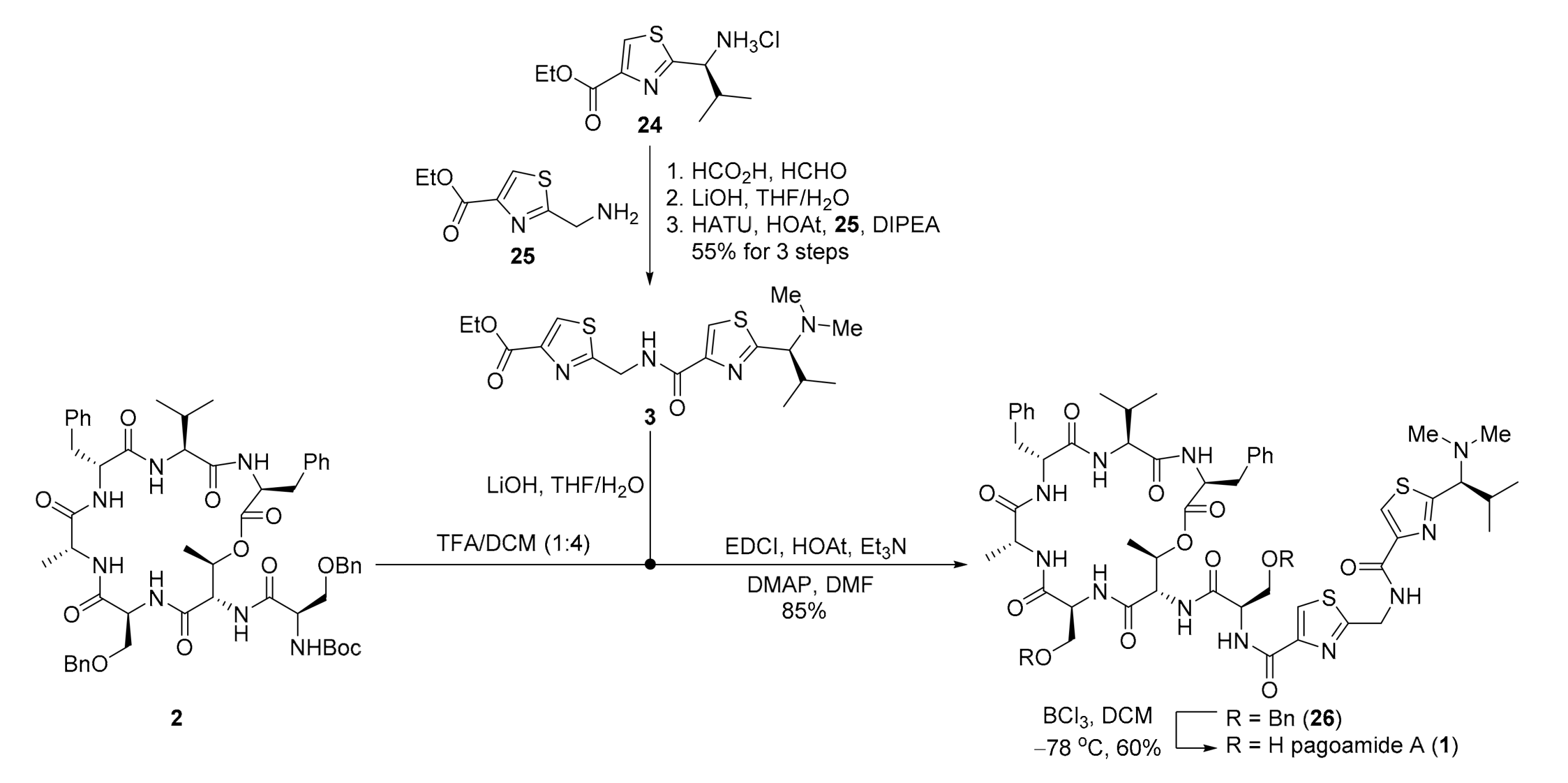
2. Results
3. Materials and Methods
3.1. General Information
3.2. General Experimental Procedures
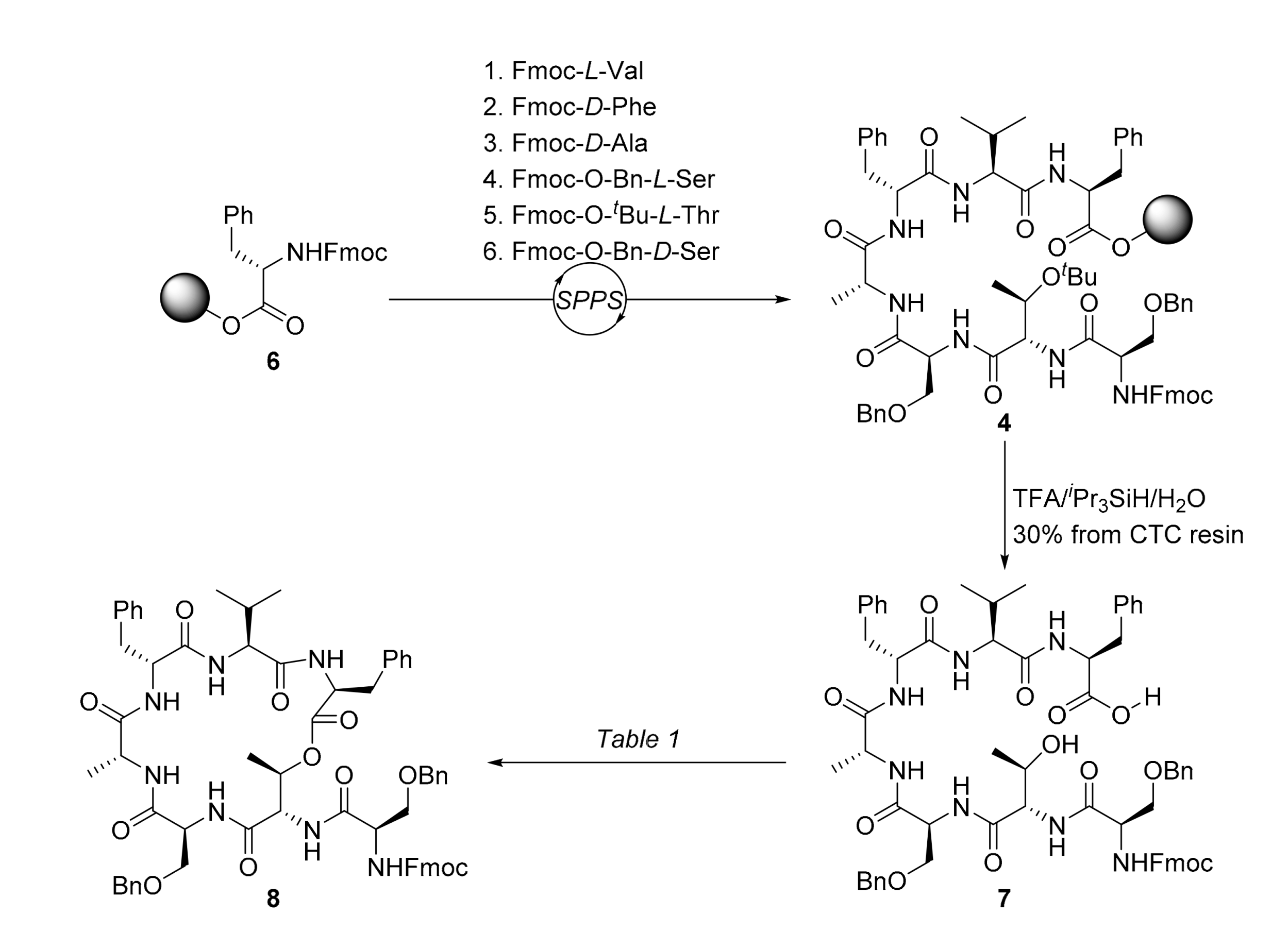
3.2.1. N-N-(((9H-fluoren-9-yl)methoxy)carbonyl)-O-benzyl-d-seryl-l-threonyl-O-benzyl-l-seryl-d-alanyl-d-phenylalanyl-l-valyl-l-phenylalanine (7)
- (A)
- Fmoc-based Solid-phase Peptide Synthesis (SPPS)
- (B)
- Removal of the C-terminal protecting group
3.2.2. (9H-Fluoren-9-yl)methyl ((R)-3-(benzyloxy)-1-(((3S,6S,9R,12R,15S,18S,19R)-3,9-dibenzyl-15-((benzyloxy)methyl)-6-isopropyl-12,19-dimethyl-2,5,8,11,14,17-hexaoxo-1-oxa-4,7,10,13,16-pentaazacyclononadecan-18-yl)amino)-1-oxopropan-2-yl)carbamate (8)
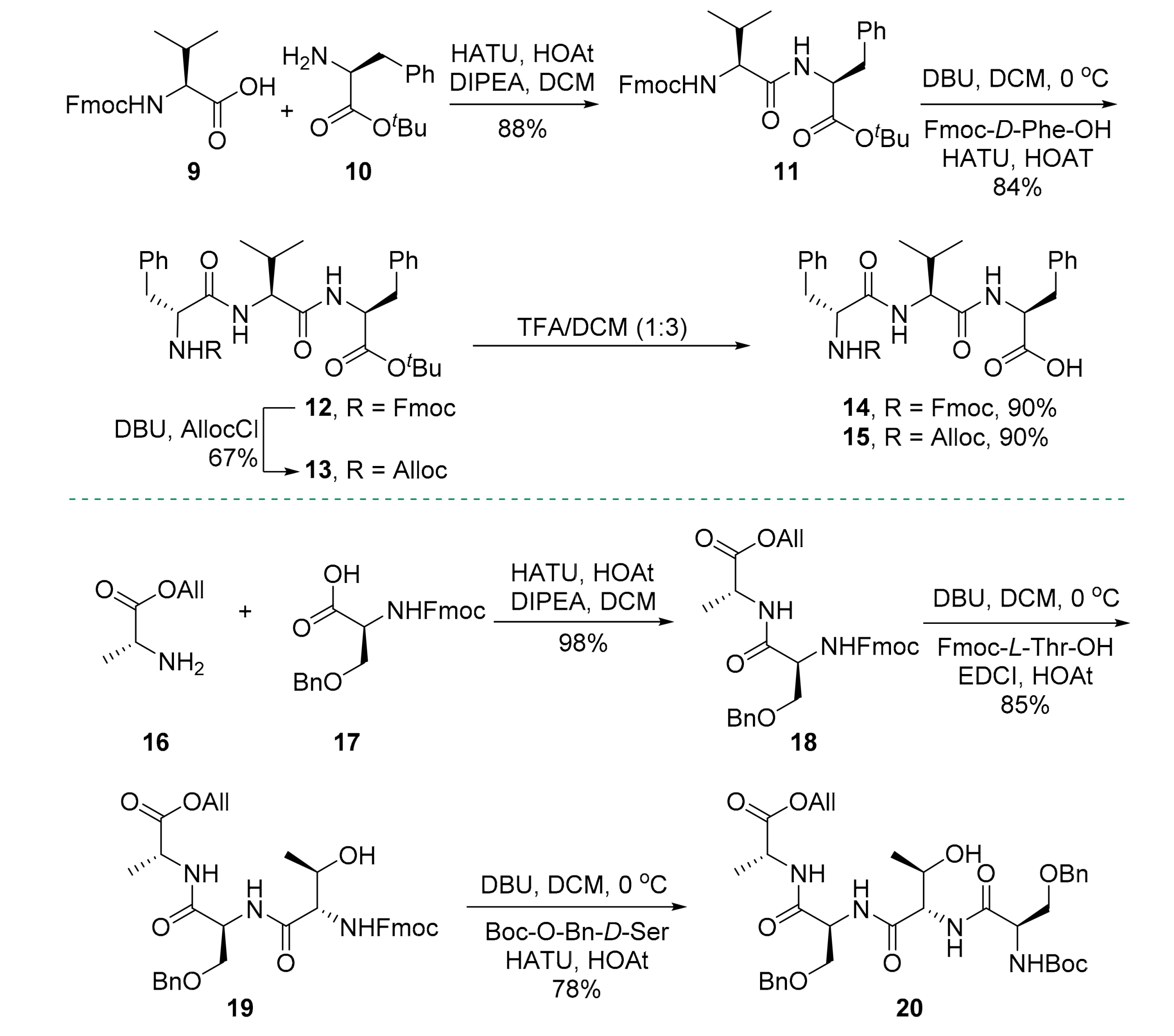
3.2.3. tert-Butyl (((9H-fluoren-9-yl)methoxy)carbonyl)-l-valyl-l-phenylalaninate (11)
3.2.4. tert-Butyl (((9H-fluoren-9-yl)methoxy)carbonyl)-d-phenylalanyl-l-valyl-l-phenylalaninate (12)
3.2.5. tert-Butyl ((allyloxy)carbonyl)-d-phenylalanyl-l-valyl-l-phenylalaninate (13)
3.2.6. (((9H-Fluoren-9-yl)methoxy)carbonyl)-d-phenylalanyl-l-valyl-l-phenylalanine (14)
3.2.7. ((Allyloxy)carbonyl)-d-phenylalanyl-l-valyl-l-phenylalanine (15)
3.2.8. Allyl N-(((9H-fluoren-9-yl)methoxy)carbonyl)-O-benzyl-l-seryl-d-alaninate (18)
3.2.9. Allyl N-((((9H-fluoren-9-yl)methoxy)carbonyl)-l-threonyl)-O-benzyl-l-seryl-d-alaninate (19)
3.2.10. Allyl O-benzyl-N-O-benzyl-N-(tert-butoxycarbonyl)-d-seryl-l-threonyl-l-seryl-d-alaninate (20)
3.2.11. (2.R,3S)-4-(((S)-1-(((R)-1-(Allyloxy)-1-oxopropan-2-yl)amino)-3-(benzyloxy)-1-oxopropan-2-yl)amino)-3-((R)-3-(benzyloxy)-2-((tert-butoxycarbonyl)amino)propanamido)-4-oxobutan-2-yl (((9H-fluoren-9-yl)methoxy)carbonyl)-l-phenylalaninate (21)
3.2.12. ((Allyloxy)carbonyl)-d-phenylalanyl-l-valine (22)
- Step A: To a solution of Alloc-d-Phe (7.5 g, 30.3 mmol, 1.0 eq.), l-valine methyl ester hydrochloride (10.2 g, 60.6 mmol, 2.0 eq.), HOAt (8.2 g, 60.6 mmol, 2.0 eq.) and DIPEA (20 mL, 121.2 mmol, 4.0 eq.) in dry DCM (200 mL, 0.15 M) under an argon atmosphere, was added EDCI (11.6 mg, 60.6 mmol, 2.0 eq.) at 0 °C. The mixture was allowed to stir for 12 h at room temperature and then concentrated in vacuo furnishing a solid residue. The solid residue was redissolved in EtOAc (5 mL) and quenched with 4% citric acid aqueous solution. The aqueous layer was extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with saturated aqueous solution of NaHCO3 (100 mL), brine (100 mL), dried over anhydrous Na2SO4, filtered and concentrated in vacuo. Purification of the crude product was performed by flash chromatography on silica gel (hexanes/EtOAc = 3/1) to afford S1 (9.9 g, 90%) as a white solid. TLC: Rf = 0.5 (Hexanes/EtOAc = 2/1), UV & PMA stain. = +8.7 (c 2.2, CHCl3). 1H-NMR (300 MHz, CDCl3) δ 7.33–7.21 (m, 2H), 7.25–7.14 (m, 3H), 6.61 (d, J = 8.7 Hz, 1H), 5.95–5.72 (m, 1H), 5.62 (d, J = 8.1 Hz, 1H), 5.31–5.15 (m, 1H), 5.21–5.10 (m, 1H), 4.61–4.53 (m, 1H), 4.51 (t, J = 1.4 Hz, 1H), 4.49 (t, J = 1.5 Hz, 1H), 4.45 (dd, J = 8.6, 5.0 Hz, 1H), 3.67 (s, 3H), 3.21–2.94 (m, 2H), 2.19–1.85 (m, 1H), 0.74 (t, J = 6.8 Hz, 6H). 13C-NMR (75 MHz, CDCl3) δ 172.1, 171.1, 155.9, 136.5, 132.6, 129.3, 128.7, 127.0, 117.8, 65.9, 57.3, 56.2, 52.2, 38.8, 31.1, 18.8, 17.7. HRMS (ESI) calculated for C19H26N2O5Na+ [M + Na]+ 385.1734, found 385.1730.
- Step B: To a solution of S1 (5.0 g, 13.8 mmol, 1.0 eq.) in THF/H2O (20 mL/10 mL, 0.46 M) was added LiOH⋅H2O (1.7 g, 41.4 mmol, 3.0 eq.) at 0 °C. after being stirred for 2 h at room temperature, the organic solvents were evaporated. The reaction mixture was diluted with water (10 mL), acidified to pH = 2 with HCl (1.0 M in water), and extracted with EtOAc (3 × 20 mL). The combined organic layers were washed with saturated aqueous solution brine (20 mL), dried over anhydrous Na2SO4, filtered and concentrated in vacuo. Purification of the crude product was performed by flash chromatography on silica gel (Hexanes/EtOAc = 3/1) to afford 22 (4.4 g, 91%) as a white solid. TLC: Rf = 0.6 (hexanes/EtOAc = 1/1), UV & PMA stain. = +9.6 (c 7.3, CHCl3). 1H-NMR (500 MHz, CDCl3) δ 9.08 (s, 1H), 7.34–7.23 (m, 2H), 7.24–7.15 (m, 3H), 7.08 (t, J = 11.0 Hz, 1H), 6.12 (t, J = 9.1 Hz, 1H), 5.82 (ddt, J = 16.4, 10.9, 5.3 Hz, 1H), 5.26–5.18 (m, 1H), 5.16 (d, J = 10.4 Hz, 1H), 4.80–4.70 (m, 1H), 4.50–4.46 (m, 3H), 3.14–2.89 (m, 2H), 2.13–1.94 (m, 1H), 0.79 (d, J = 6.8 Hz, 3H), 0.74 (d, J = 6.7 Hz, 3H). 13C-NMR (125 MHz, CDCl3) δ 174.1, 171.8, 156.4, 136.4, 132.4, 129.4, 128.7, 127.0, 117.9, 66.1, 57.4, 56.1, 39.3, 31.1, 18.6, 17.7. HRMS (ESI) calculated for C18H24N2O5Na+ [M + Na]+ 371.1577, found 371.1577.
3.2.13. (2R,3S)-4- (((S)-1- (((R)-1- (allyloxy)-1-oxopropan-2-yl)amino)-3- (benzyloxy)-1-oxopropan-2-yl)amino)-3- ((R)-3-(benzyloxy)-2- ((tert-butoxycarbonyl)amino)propanamido)-4-oxobutan-2-yl ((allyloxy)carbonyl)-d-phenylalanyl-l-valyl-l-phenylalaninate (5)
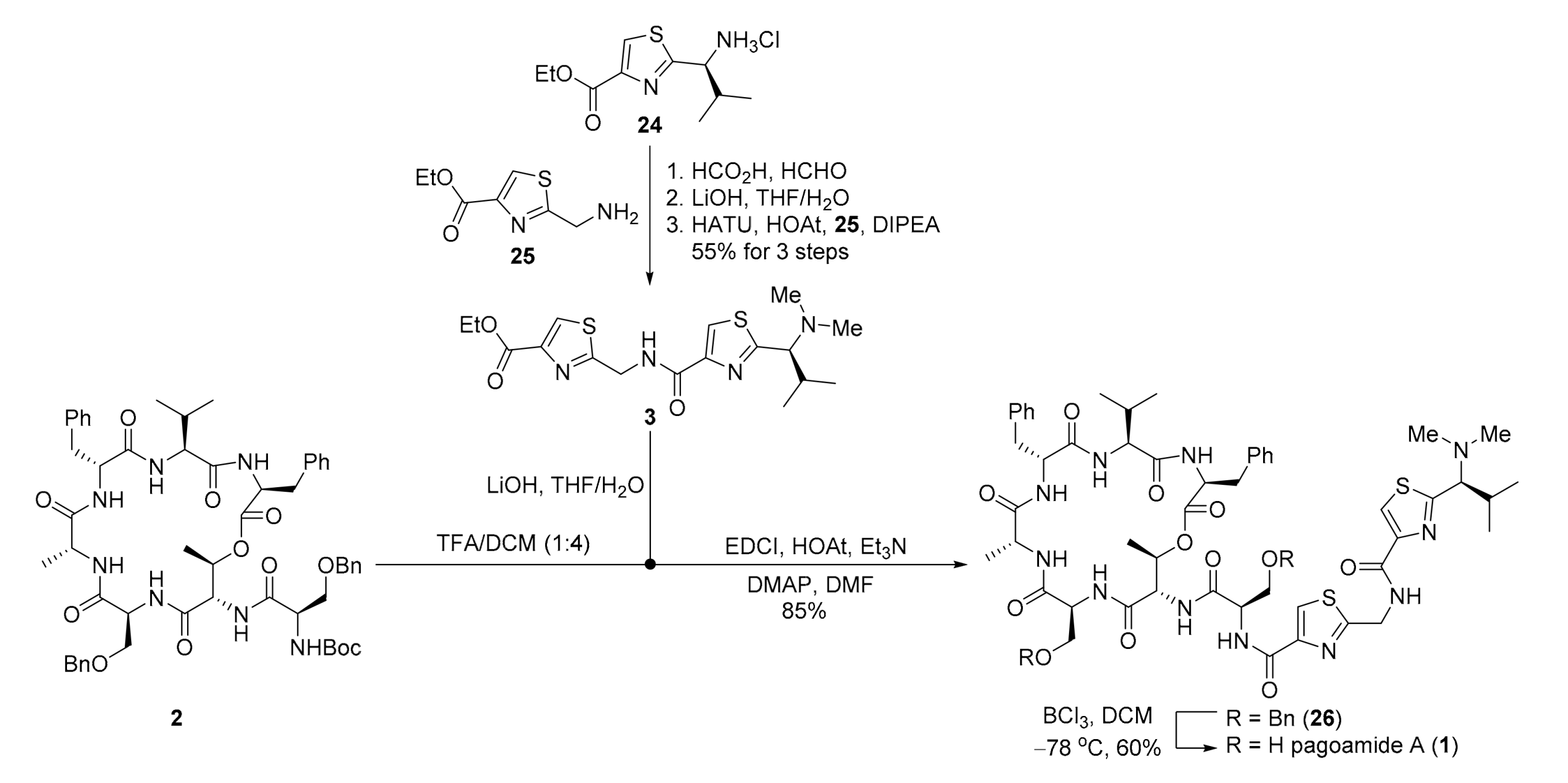
3.2.14. Ethyl (S)-2-((2-(1-(dimethylamino)-2-methylpropyl)thiazole-4-carboxamido)methyl)thiazole-4-carboxylate (3)
- Step A: A solution of 24 [22] (5.0 g, 18.9 mmol, 1.0 eq.) and formaldehyde solution (37% in H2O, 10.3 mL, 375 mmol) in formic acid (10 mL, 1.89 M) was stirred for 12 h at 80 °C. After cooling to room temperature, concentrated HCl (3 mL) was slowly added to the reaction mixture. All volatiles were removed in vacuo, and the resulting residue was extracted with Et2O (3 × 20 mL). The aqueous layer was added saturated aqueous solution of NaHCO3 (20 mL) and extracted with Et2O (3 × 20 mL). The combined organic layers were washed with brine (20 mL), dried over anhydrous Na2SO4, filtered and concentrated in vacuo providing S2 (3.15 g, 65%) as a colorless oil. TLC: Rf = 0.5 (hexanes/EtOAc = 1/1), UV & PMA stain. = +1.6 (c 1.0, MeOH). 1H-NMR (300 MHz, CDCl3) δ 8.03 (s, 1H), 5.19 (s, 1H), 4.28 (q, J = 7.1 Hz, 2H), 3.51 (d, J = 8.9 Hz, 1H), 2.11 (s, 6H), 1.26 (t, J = 7.1 Hz, 3H), 0.89 (d, J = 6.6 Hz, 3H), 0.69 (d, J = 6.6 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 169.6, 161.3, 146.2, 127.1, 72.9, 61.1, 41.6, 30.1, 20.0, 18.9, 14.2. HRMS (ESI) calculated for C12H20N2O2SNa+ [M + Na]+ 279.1138, found 279.1140.
- Step B: To a solution of S2 (3.15 g, 12.3 mmol, 1.0 eq.) in THF/H2O (10 mL/5 mL, 0.8 M) was added LiOH⋅H2O (1.55 g, 36.9 mmol, 3.0 eq.) at 0 °C. After being stirred for 2 h at room temperature, the reaction mixture was quenched with concentrated HCl (5 mL) and all solvents were removed in vacuo providing crude product S3 which was used directly in the next step without further purification.Step C: To a solution of the crude S3 in DCM (20 mL, 0.6 M) at 0 °C was added DIPEA (10.1 mL, 61.5 mmol, 5.0 eq.), followed by consecutive addition of 25 [23] (4.6 g, 24.6 mmol, 2.0 eq.), HOAt (3.4 g, 24.6 mmol, 2.0 eq.) and HATU (9.4 g, 24.6 mmol, 2.0 eq.). The mixture was allowed to stir for 12 h at room temperature and then concentrated in vacuo furnishing a solid residue. Purification of the crude product was performed by flash chromatography on silica gel (MeOH/DCM = 1/40) to afford 3 (3.8 g, 84% for 2 steps) as a colorless oil. TLC: Rf = 0.4 (MeOH/DCM = 1/20), UV & PMA stain. = +0.5 (c 0.75, CHCl3). 1H-NMR (400 MHz, CDCl3) δ 8.10 (s, 1H), 8.07 (s, 1H), 4.93 (d, J = 6.5 Hz, 2H), 4.37 (q, J = 7.1 Hz, 2H), 3.38 (d, J = 9.2 Hz, 1H), 2.75 (s, 2H), 2.17 (s, 6H), 1.35 (t, J = 7.1 Hz, 3H), 0.98 (d, J = 6.7 Hz, 3H), 0.76 (d, J = 6.6 Hz, 3H). 13C-NMR (100 MHz, CDCl3) δ 169.3, 168.5, 161.5, 161.3, 148.4, 146.8, 128.4, 123.7, 73.0, 61.6, 41.8, 40.8, 38.6, 30.1, 20.2, 19.3, 14.4. HRMS (ESI) calculated for C17H24N4O3S2Na+ [M + Na]+ 419.1182, found 419.1187.
3.2.15. tert-Butyl ((R)-3-(benzyloxy)-1- (3S,6S,9R,12R,15S,18S,19R)-3,9-dibenzyl-15-((benzyloxy)methyl)-6-isopropyl-12,19-dimethyl-2,5,8,11,14,17-hexaoxo-1-oxa-4,7,10,13,16-pentaazacyclononadecan-18-yl)amino)-1-oxopropan-2-yl)carbamate (2))
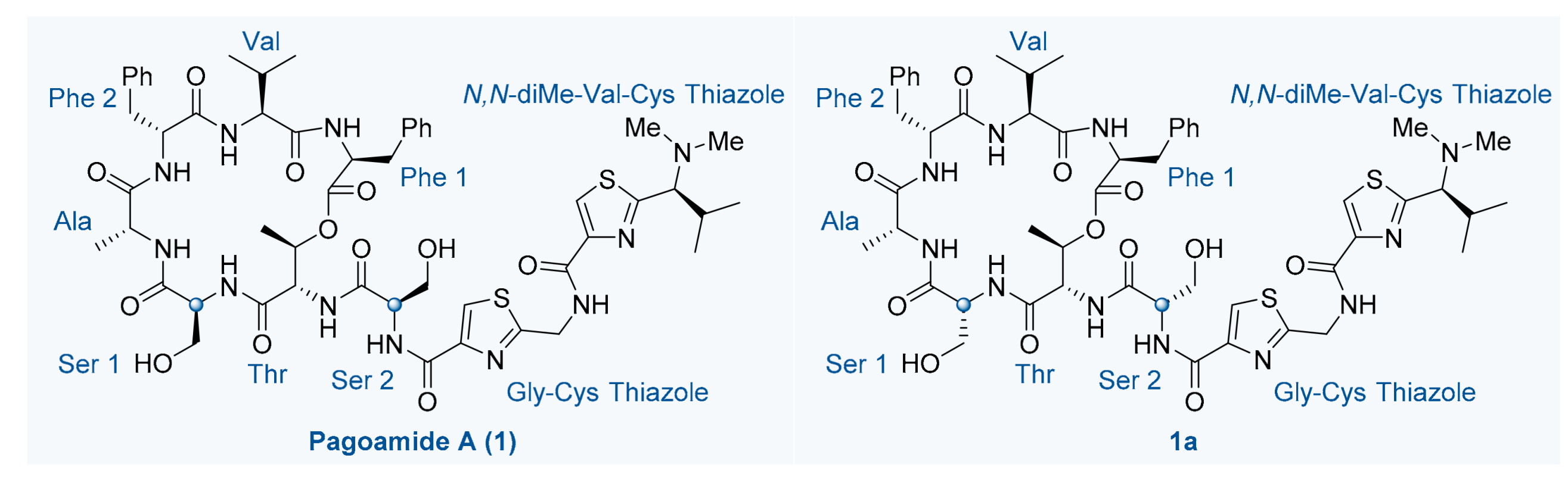
3.2.16. Pagoamide A (1)
- Step A: To a solution of 2 (180 mg, 0.18 mmol, 1.0 eq.) in DCM (3 mL, 0.05 M) was added TFA (1 mL) dropwise at 0 °C. After being stirred at room temperature for 3 h, the reaction mixture was concentrated in vacuo to afford the crude amine 3, which was used directly in the next step without further purification. To a solution of 3 (285.0 mg, 0.72 mmol, 4.0 eq.) in THF/H2O (4 mL/2 mL, 0.12 M) was added LiOH⋅H2O (92.0 mg, 2.2 mmol, 12.0 eq.) at 0 °C. After being stirred for 3 h at room temperature, the reaction mixture was quenched with concentrated HCl (1 mL), and all solvent was removed in vacuo providing the crude acid which was used directly in the next step without further purification. To a solution of the above crude amine (0.18 mmol, 1.0 eq.) and the crude acid (0.72 mmol, 4.0 eq.) in DMF (5 mL, 0.09 M) were added HOAt (98 mg, 0.72 mmol, 4.0 eq.), Et3N (0.2 mL, 1.4 mmol, 8.0 eq.), DMAP (11.0 mg, 0.04 mmol, 0.5 eq.) and EDCI (138.0 mg, 0.72 mmol, 4.0 eq.) under argon atmosphere at 0 °C. The mixture was allowed to stir for 36 h at room temperature and then concentrated in vacuo furnishing a solid residue. The solid residue was redissolved in EtOAc (10 mL) and quenched with saturated aqueous solution of NaHCO3 (10 mL). The aqueous layer was extracted with EtOAc (3 × 10 mL). The combined organic layers were washed with brine (10 mL), dried over anhydrous Na2SO4, filtered and concentrated in vacuo. Purification of the crude product was performed by flash chromatography on silica gel (MeOH/DCM = 1/40-1/20) to afford 26 (194 mg, 85%) as a white solid. TLC: Rf = 0.4 (MeOH/DCM = 1/10), UV & PMA stain. = +9.7 (c 1.0, CHCl3). 1H-NMR (500 MHz, CDCl3) δ 8.07 (dd, J = 7.6, 2.2 Hz, 1H), 8.05–8.02 (m, 2H), 7.98 (d, J = 9.4 Hz, 1H), 7.94 (d, J = 8.8 Hz, 1H), 7.92 (s, 1H), 7.33–7.27 (m, 6H), 7.27–7.23 (m, 5H), 7.23–7.19 (m, 3H), 7.18–7.13 (m, 5H), 7.10 (t, J = 7.5 Hz, 2H), 7.01 (t, J = 7.3 Hz, 1H), 6.89 (s, 1H), 6.85 (d, J = 10.0 Hz, 1H), 5.66 (qd, J = 6.3, 2.7 Hz, 1H), 5.41–5.33 (m, 1H), 5.25 (ddd, J = 11.7, 9.9, 4.5 Hz, 1H), 5.11 (dd, J = 9.6, 2.7 Hz, 1H), 4.82 (dd, J = 16.1, 6.4 Hz, 1H), 4.74 (ddd, J = 16.1, 8.0, 6.3 Hz, 1H), 4.69–4.58 (m, 2H), 4.52 (d, J = 12.4 Hz, 2H), 4.43 (d, J = 12.0 Hz, 1H), 4.16 (dd, J = 6.7, 3.8 Hz, 1H), 4.12 (dd, J = 7.3, 5.1 Hz, 1H), 4.09 (dd, J = 7.3, 3.9 Hz, 1H), 4.06 (ddd, J = 9.9, 4.3, 1.8 Hz, 1H), 3.86 (dd, J = 10.0, 5.0 Hz, 1H), 3.61 (d, J = 7.0 Hz, 12H), 3.46 (d, J = 9.0 Hz, 1H), 3.41–3.32 (m, 2H), 3.28–3.21 (m, 1H), 3.03 (dd, J = 14.2, 11.7 Hz, 1H), 2.29–2.21 (m, 1H), 2.22 (s, 6H), 2.13–2.02 (m, 1H), 1.20 (d, J = 7.3 Hz, 3H), 1.15 (d, J = 6.2 Hz, 3H), 1.02 (d, J = 6.6 Hz, 3H), 0.82 (d, J = 6.6 Hz, 3H), 0.75 (d, J = 6.9 Hz, 3H), 0.32 (d, J = 6.9 Hz, 3H). 13C-NMR (125 MHz, CDCl3) δ 173.9, 173.5, 172.0, 171.3, 171.1, 170.8, 169.8, 168.1, 161.7, 160.7, 149.5, 148.4, 137.9, 137.8, 137.8, 137.1, 129.5, 129.2, 128.7, 128.5, 128.5, 128.2, 128.0, 127.8, 127.7, 126.5, 126.2, 124.3, 124.0, 73.5, 73.2, 73.2, 70.6, 70.4, 68.1, 59.8, 55.8, 55.5, 53.3, 52.6, 52.4, 51.4, 41.8, 41.0, 36.5, 33.8, 30.2, 29.8, 29.6, 20.3, 19.7, 19.4, 16.9, 16.7, 16.3. HRMS (ESI) calculated for C65H80N11O12S2+ [M + Na]+ 1270.5424, found 1270.5429.
- Step B: To a solution of 26 (32 mg, 0.025 mmol, 1.0 eq.) in anhydrous DCM (3 mL, 0.008 M) was added BCl3 (0.25 mL, 0.25 mmol, 10.0 eq.,1 M in DCM) at −78 °C under argon atmosphere. The reaction mixture was stirred at the same temperature for 4 h. Then, MeOH was added while the temperature was still at −78 °C, and the mixture was allowed to reach room temperature. The solvent was removed in vacuo furnishing a solid residue. The solid residue redissolved in DCM (5 mL) and saturated aqueous solution of NaHCO3 (10 mL) was added. The aqueous layer was extracted with DCM (3 × 10 mL). The combined organic layers were washed with brine (10 mL), dried over anhydrous Na2SO4, filtered and concentrated in vacuo. Purification of the crude product was performed by flash chromatography on silica gel (MeOH/DCM = 1/40-1/20) to afford pagoamide A (1) (16.3 mg, 60%) as a white solid. TLC: Rf = 0.2 (MeOH/DCM = 1/10), UV & PMA stain. = +8.0 (c 0.1, MeOH). 1H-NMR (500 MHz, DMSO-d6) δ 9.24 (t, J = 6.3 Hz, 1H), 9.14–8.99 (m, 1H), 8.36 (d, J = 5.7 Hz, 1H), 8.31 (s, 1H), 8.20 (d, J = 9.0 Hz, 1H), 8.09 (s, 1H), 8.06 (d, J = 8.0 Hz, 1H), 7.74 (d, J = 9.2 Hz, 1H), 7.38 (d, J = 7.6 Hz, 1H), 7.22 (d, J = 4.4 Hz, 4H), 7.20 (d, J = 7.2 Hz, 2H), 7.19–7.13 (m, 3H), 7.16–7.10 (m, 1H), 6.85 (d, J = 9.5 Hz, 1H), 5.70–5.61 (m, 1H), 4.91–4.85 (m, 1H), 4.85 (dt, J = 10.2, 2.8 Hz, 2H), 4.74 (d, J = 6.0 Hz, 2H), 4.60 (td, J = 9.8, 4.9 Hz, 1H), 4.09–4.00 (m, 1H), 3.94 (dt, J = 10.4, 4.8 Hz, 1H), 3.87 (dd, J = 7.8, 4.5 Hz, 1H), 3.75 (dt, J = 10.2, 4.6 Hz, 1H), 3.64–3.54 (m, 3H), 3.27–3.17 (m, 2H), 3.17–3.10 (m, 2H), 2.31–2.22 (m, 1H), 2.17 (s, 6H), 1.93–1.81 (m, 1H), 1.15 (d, J = 7.3 Hz, 3H), 1.12 (d, J = 6.2 Hz, 3H), 1.00 (d, J = 6.6 Hz, 3H), 0.75 (d, J = 6.6 Hz, 3H), 0.59 (d, J = 6.9 Hz, 3H), 0.28 (d, J = 6.9 Hz, 3H). 13C-NMR (125 MHz, DMSO-d6) δ 173.5, 172.5, 171.3, 170.4, 170.2, 170.0, 169.9, 169.1, 168.4, 161.2, 159.7, 148.8, 148.5, 138.0, 137.8, 129.2, 129.1, 128.1, 128.0, 126.2, 126.1, 124.4, 124.3, 71.9, 71.3, 62.3, 60.7, 58.7, 57.3, 54.6, 54.1, 52.9, 52.4, 50.1, 41.3, 40.8, 36.1, 34.2, 29.5, 29.3, 20.1, 19.4, 19.0, 16.6, 16.4, 15.9. HRMS (ESI) calculated for C51H67N11O12S2Na+ [M + Na]+ 1112.4304, found 1112.4300.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Li, Y.; Yu, H.B.; Zhang, Y.; Leao, T.; Glukhov, E.; Pierce, M.L.; Zhang, C.; Kim, H.; Mao, H.H.; Fang, F.; et al. Pagoamide A, a Cyclic Depsipeptide Isolated from a Cultured Marine Chlorophyte, Derbesia sp. Using MS/MS-Based Molecular Networking. J. Nat. Prod. 2020, 83, 617–625. [Google Scholar] [CrossRef]
- Zhao, M.; Xiao, Y.; Otsuka, S.; Nakao, Y.; Guo, Y.; Ye, T. Total Synthesis and Biological Evaluation of Kakeromamide A and Its Analogues. Front. Chem. 2020, 8, 410. [Google Scholar] [CrossRef]
- Guo, Y.; Zhou, J.; Gao, B.; Zhao, M.; Yan, J.L.; Xu, Z.; Choi, S.; Ye, T. Total Synthesis of Hoiamide A Using an Evans-Tishchenko Reaction As a Key Step. Org. Lett. 2019, 21, 5471–5474. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Liu, Y.; Wang, Z.; Xing, X.; Maguire, R.A.; Luesch, H.; Zhang, H.; Xu, Z.; Ye, T. Total Synthesis and Biological Evaluation of Grassypeptolide A. Chem. Eur. J. 2013, 19, 6774–6784. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xu, Z.; Ye, T. Total Synthesis of Hoiamide C. Org. Lett. 2011, 13, 2506–2509. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Liu, Y.; Wang, Z.; Xing, X.; Xu, Z.; Ye, T. Total Synthesis of Grassypeptolide. Chem. Commun. 2010, 46, 7486–7488. [Google Scholar] [CrossRef]
- Gao, X.; Liu, Y.; Kwong, S.; Xu, Z.; Ye, T. Total Synthesis and Stereochemical Reassignment of Bisebromoamide. Org. Lett. 2010, 12, 3018–3021. [Google Scholar] [CrossRef]
- Ren, Q.; Dai, L.; Zhang, H.; Tan, W.; Xu, Z.; Ye, T. Total Synthesis of Largazole. Synlett 2008, 2008, 2379–2383. [Google Scholar]
- Pan, H.; Xu, Z.; Chen, Z.; Ye, T. Total Synthesis of Lyngbyabellin A. Lett. Org. Chem. 2005, 2, 699–702. [Google Scholar]
- Zhao, J.C.; Yu, S.M.; Liu, Y.; Yao, Z.J. Biomimetic Synthesis of ent-(-)-Azonazine and Stereochemical Reassignment of Natural Product. Org. Lett. 2013, 15, 4300–4303. [Google Scholar] [CrossRef]
- Wu, Q.X.; Crews, M.S.; Draskovic, M.; Sohn, J.; Johnson, T.A.; Tenney, K.; Valeriote, F.A.; Yao, X.J.; Bjeldanes, L.F.; Crews, P. Azonazine, a Novel Dipeptide from a Hawaiian Marine Sediment-Derived Fungus, Aspergillus insulicola. Org. Lett. 2010, 12, 4458–4461. [Google Scholar] [CrossRef] [Green Version]
- Kashinath, K.; Jachak, G.R.; Athawale, P.R.; Marelli, U.K.; Gonnade, R.G.; Reddy, D.S. Total Synthesis of the Marine Natural Product Solomonamide B Necessitates Stereochemical Revision. Org. Lett. 2016, 18, 3178–3181. [Google Scholar] [CrossRef] [PubMed]
- Festa, C.; Marino, S.D.; Sepe, V.; Auria, M.V.; Bifulco, G.; Debitus, C.; Bucci, M.; Vellecco, V.; Zampella, A. Solomonamides A and B, New Anti-inflammatory Peptides from Theonella swinhoe. Org. Lett. 2005, 2, 699–702. [Google Scholar]
- Rodriquez, M.; Terracciano, S.; Cini, E.; Settembrini, G.; Bruno, I.; Bifulco, G.; Taddei, M.; Gomez-Paloma, L. Total Synthesis, NMR Solution Structure, and Binding Model of the Potent Histone Deacetylase Inhibitor FR235222. Angew. Chem. Int. Ed. 2006, 45, 423–427. [Google Scholar] [CrossRef]
- Shiina, I.; Kubota, M.; Oshiumi, H.; Hashizume, M. An Effective Use of Benzoic Anhydride and Its Derivatives for the Synthesis of Carboxylic Esters and Lactones: A Powerful and Convenient Mixed Anhydride Method Promoted by Basic Catalysts. J. Org. Chem. 2004, 69, 1822–1830. [Google Scholar] [CrossRef]
- Inanaga, J.; Hirata, K.; Saeki, H.; Katsuki, T.; Yamaguchi, M. A Rapid Esterification by Means of Mixed Anhydride and Its Application to Large-Ring Lactonization. Bull. Chem. Soc. Jpn. 1979, 52, 1989–1993. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Xu, J.-C. 1-Ethyl 2-Halopyridinium Salts, Highly Efficient Coupling Reagents for Hindered Peptide Synthesis both in Solution and the Solid-Phase. Tetrahedron 2000, 56, 8119–8131. [Google Scholar] [CrossRef]
- Boden, E.P.; Keck, G.E. Proton-Transfer Steps in Steglich Esterification: A very Practical New Method for Macrolactonization. J. Org. Chem. 1985, 50, 2394–2395. [Google Scholar] [CrossRef]
- Beutner, G.L.; Young, I.S.; Davies, M.L.; Hickey, M.R.; Park, H.; Stevens, J.M.; Ye, Q. TCFH-NMI: Direct Access to N-Acyl Imidazoliums for Challenging Amide Bond Formations. Org. Lett. 2018, 20, 4218–4222. [Google Scholar] [CrossRef] [Green Version]
- Isley, N.A.; Endo, Y.; Wu, Z.C.; Covington, B.C.; Bushin, L.B.; Seyedsayamdost, M.R.; Boger, D.L. Total Synthesis and Stereochemical Assignment of Streptide. J. Am. Chem. Soc. 2019, 141, 17361–17369. [Google Scholar] [CrossRef]
- Chandrasekhar, S.; Raji Reddy, C.; Jagadeeshwar Rao, R. Facile and Selective Cleavage of Allyl Ethers, Amines and Esters Using Polymethylhydrosiloxane-ZnCl2/Pd(PPh3)4. Tetrahedron 2001, 57, 3435–3438. [Google Scholar] [CrossRef]
- Aguilar, E.; Meyers, A.I. Reinvestigation of a Modified Hantzsch Thiazole Synthesis. Tetrahedron Lett. 1994, 35, 2473–2476. [Google Scholar] [CrossRef]
- Peña, S.; Scarone, L.; Manta, E.; Serra, G. First Total Synthesis of Aerucyclamide B. Tetrahedron Lett. 2013, 54, 2806–2808. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Conditions | Yield |
|---|---|---|
| 1 | MNBA, DMAP, DCM, r.t. | 20% |
| 2 | MNBA, DMAP, DCM/THF (1:1), r.t. | 10% |
| 3 | MNBA, DMAP, CH3CN, r.t. | 5% |
| 4 | TCBC, Et3N, DCM; then DMAP, PhMe, r.t. | 5% |
| 5 | TCBC, Et3N, PhMe; then DMAP, PhMe, r.t. | trace |
| 6 | BEP, DMAP, DCM/DMF (10:1), r.t. to 40 °C | 5% |
| 7 | EDCI, DMAP, DCM/DMF (10:1), 60 °C | 5% |
| 8 | DCC, DMAP, DMAP⋅HCl, CHCl3, 65 °C | 5% |
| 9 | TCFH, NMI, CH3CN, r.t. to 40 °C | trace |
| 10 | TCFH, NMI, DMF, r.t. to 40 °C | trace |
| No. | Natural (δ1) | Sample 1 (δ2) | δ = δ1−δ2 | No. | Natural (δ1) | Sample 1 (δ2) | δ = δ1−δ2 |
|---|---|---|---|---|---|---|---|
| 1 | 170.4 | 170.3 | 0.1 | 29 | 60.7 | 60.7 | 0 |
| 2 | 52.4 | 52.4 | 0 | 30 | 169.1 | 169 | 0.1 |
| 3 | 36.1 | 36.1 | 0 | 31 | 54.1 | 54.1 | 0 |
| 4 | 137.9 | 137.8 | 0.1 | 32 | 71.3 | 71.4 | −0.1 |
| 5,9 | 128.1 | 128.1 | 0 | 33 | 16 | 15.9 | 0.1 |
| 6,8 | 129.2 | 129.2 | 0 | 34 | 169.9 | 169.9 | 0 |
| 7 | 126.2 | 126.2 | 0 | 35 | 54.5 | 54.6 | −0.1 |
| 10 | 170 | 170 | 0 | 36 | 62.4 | 62.3 | 0.1 |
| 11 | 58.7 | 58.7 | 0 | 37 | 159.7 | 159.8 | −0.1 |
| 12 | 29.3 | 29.3 | 0 | 38 | 148.8 | 148.8 | 0 |
| 13 | 19.1 | 19 | 0.1 | 39 | 124.2 | 124.3 | −0.1 |
| 14 | 16.4 | 16.3 | 0.1 | 40 | 170.3 | 170.2 | 0.1 |
| 15 | 172.5 | 172.5 | 0 | 41 | 40.8 | 40.8 | 0 |
| 16 | 53 | 53 | 0 | 42 | 161.1 | 161.2 | −0.1 |
| 17 | 34.1 | 34.1 | 0 | 43 | 148.5 | 148.4 | 0.1 |
| 18 | 138.1 | 138 | 0.1 | 44 | 124.4 | 124.4 | 0 |
| 19,23 | 128 | 128 | 0 | 45 | 168.4 | 168.4 | 0 |
| 20,22 | 129.1 | 129.1 | 0 | 46 | 71.9 | 71.9 | 0 |
| 21 | 126.1 | 126.1 | 0 | 47 | 29.5 | 29.5 | 0 |
| 24 | 173.5 | 173.5 | 0 | 48 | 19.4 | 19.3 | 0.1 |
| 25 | 50.1 | 50.1 | 0 | 49 | 20.1 | 20.1 | 0 |
| 26 | 16.7 | 16.6 | 0.1 | 50 | 41.3 | 41.3 | 0 |
| 27 | 171.4 | 171.3 | 0.1 | 51 | 41.3 | 41.3 | 0 |
| 28 | 57.4 | 57.5 | −0.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, F.; Yu, J.; Meng, J.; Guo, Y.; Ye, T. Total Synthesis of Pagoamide A. Molecules 2021, 26, 4224. https://doi.org/10.3390/molecules26144224
Wu F, Yu J, Meng J, Guo Y, Ye T. Total Synthesis of Pagoamide A. Molecules. 2021; 26(14):4224. https://doi.org/10.3390/molecules26144224
Chicago/Turabian StyleWu, Fusong, Jie Yu, Jiawei Meng, Yian Guo, and Tao Ye. 2021. "Total Synthesis of Pagoamide A" Molecules 26, no. 14: 4224. https://doi.org/10.3390/molecules26144224
APA StyleWu, F., Yu, J., Meng, J., Guo, Y., & Ye, T. (2021). Total Synthesis of Pagoamide A. Molecules, 26(14), 4224. https://doi.org/10.3390/molecules26144224