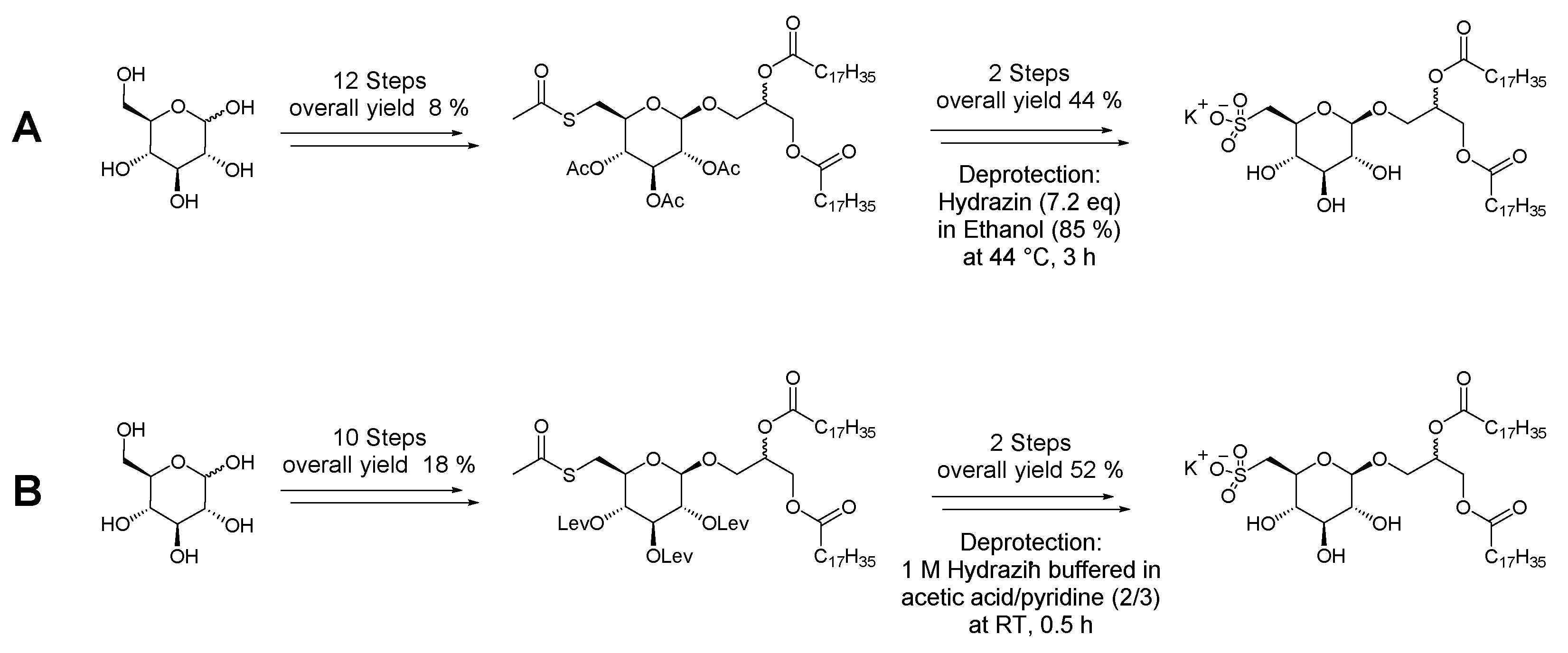
3. Materials and Methods
All reagents were purchased from commercial suppliers (Sigma-Aldrich (Darmstadt, Germany), Honeywell (Seelze, Germany), Acros (Schwerte, Germany) and used without further purification. Solvents (DCM, DMF, pyridine, MeOH, toluene) were purchased as anhydrous over molecular sieves from Acros Organics. Unless otherwise stated, all reactions were performed in dried glassware with magnetic stirring and under nitrogen atmosphere. All reactions were monitored with thin-layer chromatography (TLC) using an aluminum TLC plate (silica gel 60, F
254, Merck KGaA, Darmstadt, Germany). Components on TLC plates were visualized with cerium-based staining (4.5 g ammonium molybdate tetrahydrate, 0.18 g anhydrous cerium sulfate, 2.5 mL sulfuric acid, and 90 mL H
2O) or staining with copper (5 g anhydrous copper sulfate, 10 mL o-phosphoric acid 85%, and 90 mL H
2O) followed by heating. Purification of products was carried out with silica gel 60 (particle size 0.040–0.063 mm, Merck KGaA, Darmstadt, Germany) and freshly distilled solvents were used as eluents. Yields refer to chromatographically purified and spectroscopically pure compounds. NMR spectra were recorded on Bruker instruments (Fourier HD 300; Avance I 400 DRX 500, or Avance III 600 at room temperature. Chemical shift δ is reported in ppm with the solvent resonance signal as the internal standard: chloroform-d1: 7.26 (
1H NMR), 77.16 (
13C NMR), methanol-d4: 3.31 (
1H NMR), 49.00 (
13C NMR). Coupling constant J is given in Hertz (Hz). Multiplicities are classified as follows: s = singlet, d = doublet, t = triplet, q = quartet, and combinations thereof; m = multiplet or br = broad signal. Two-dimensional NMR experiments (COSY, HSQC, HMBC) were used for the assignment of all NMR signals. The approximately diastereomeric ratio was determined by taking
1H and
13C NMR data into account. High-resolution mass spectra were recorded on an Agilent 6224 ESI-TOF coupled with an Agilent HPLC 1200 Series. The spectra can be found in the
supplementary materials.
General procedure for preparation of levulinic acid anhydride: A solution of dicyclohexylcarbodiimide (0.5 equiv.) in dry dichloromethane (15 mL) was cooled to 0 °C. Levulinic acid (1 equiv.) dissolved in dry DCM (10 mL) was added dropwise. The reaction mixture was stirred for 1 h with the formation of a colorless precipitate. The precipitate was removed by filtration and the solvent evaporated in vacuo to result in a colorless syrup. The resulting product was used without further purification.
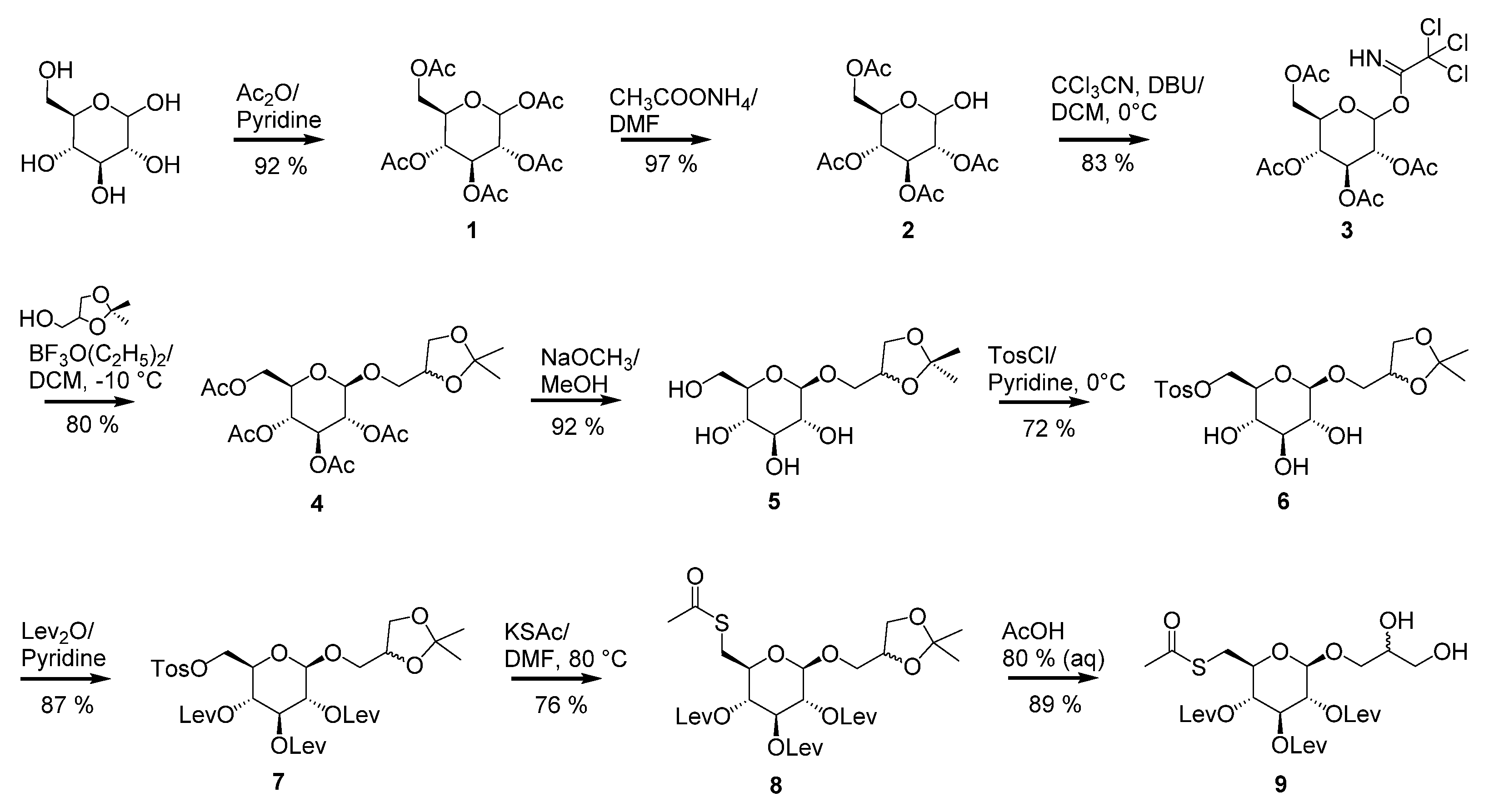
1,2,3,4,6-Penta-O-acetyl-d-glucopyranose (1): d-glucose (29.6 g, 0.164 mol) was mixed with 200 mL of anhydrous pyridine. While stirring with a magnetic stirrer, acetic anhydride (240 mL, 2.54 mol) was slowly added. This mixture was kept at room temperature under stirring for 20 h. When the reaction was complete, the solution was mixed with ice and stirred until all ice melted, whereupon a colorless solid precipitated. The mixture was filtered, and the filtered product was washed with ice-cold water to remove pyridine residues. The filtered product was recrystallized in a solution of water/methanol (v/v = 50/50) and gave compound 1 as a colorless solid (59.6 g, 92%). 1H NMR (600 MHz, CDCl3): δ 6.33 (d, J = 3.7 Hz, 1H, H-1β), 5.47 (dd, J = 9, J < 1.0 Hz, 1H, H-3), 5.14 (t, J = 9.8 Hz, 1H, H-4), 5.10 (dd, J = 10.3, 3.7 Hz, 1H, H-2), 4.29–4.24 (m, 1H, H-6a), 4.18–4.05 (m, 2H, H-5, H-6b), 2.18, 2.09, 2.04, 2.02, 2.01 (all s, 15H, 5 × COCH3). 13C NMR (150 MHz, CDCl3): δ 171.0, 170.32, 170.28, 169.8, (4 × C=O), 89.2 (C-1α), 69.98 (C-3), 69.97 (C-5) 69.3 (C-2) 68.05 (C-4), 61.6 (C-6), 21.0, 20.83, 20.80, 20.7, 20.6 (5 × C(O)CH3).
2,3,4,6-Tetra-O-acetyl-d-glucopyranose (2): Ammonium acetate (19.8 g, 0.257 mol) was added to a solution of compound 1 (50.0 g, 0.128 mol) in dry N,N-dimethylformamide (100 mL). The solution was stirred at room temperature overnight (30 h). After completion of the reaction (monitored by TLC), the solution was evaporated in vacuo. The residue was purified by silica gel column chromatography (ethyl acetate) to give compound 2 (43.3 g, 97% yield) as an amber-colored syrup. The product was present as an anomeric mixture in a ratio of (α:β) 3:1. 1H NMR (500 MHz, CDCl3): main signals α-anomer: δ 5.52 (dd, J = 10.2, 9.4 Hz, 1H, H-3), 5.45 (d, J = 3.6 Hz, 1H, H-1β), 5.07 (t, J = 9.7 Hz, 1H, H-4). 4.89 (dd, J = 10.2, 3.6 Hz, 1H, H-2), 4.73 (d, J = 8.0 Hz, 1H, H-1α), 4.26 (td, J = 3.9, 2.0 Hz, 1H, H-5), 4.23—4.20 (m, 1H, H-6a), 4.14–4.09 (m, 1H, H-6b), 2.08, 2.07, 2.02, 2.01 (all s, 12H, 4 × COCH3). 13C NMR (125 MHz, CDCl3): δ 170.8, 170.4, 169.8, 169.5, 168.9 (5 × C=O), 95.7 (C-1 β), 90,3 (C-1 α), 71.2 (C-2), 70.0 (C-3), 68.7 (C-4), 67.4 (C-5), 62,1 (C-6), 20.86, 20.82, 20.80, 20.73 (4 × C(O)CH3) HRESIMS m/z: 371.096 [M + Na]+ (calculated for C14H20O10Na, 371.095).
2,3,4,6-Tetra-O-acetyl-d-glucopyranosyl trichloroacetimidate (3): To a solution of compound 2 (41.9 g, 0.12 mmol) and in dry dichloromethane (120 mL), trichloroacetonitrile (60 mL, 0.6 mol) was added at 0 °C. Then, 1,8-diazabicyclo [5.4.0]undec-7-ene (DBU) (1.8 mL, 12 mmol) was added dropwise into the solution and stirred for 1 h. After the reaction was completed, the reaction mixture evaporated in vacuo. The residue was purified by silica gel column chromatography (ethyl acetate/cyclohexane, v/v, 50/50) to give compound 2 (48.8 g, 83% yield) as an amber-colored syrup. 1H NMR (400 MHz, CDCl3): δ 8.69 (s, 1H, NH), 6.55 (d, J = 3.7 Hz, 1H, H-1β), 5.55 (t, J = 9.9 Hz, 1H, H-3), 5.17 (t, J = 9.9 Hz, 1H, H-4), 5.12 (dd, J = 10.2, 3.7 Hz, 1H, H-2), 4.27 (dd, J = 12.3, 4.1 Hz, 1H, H-6a), 4.20 (ddd, J = 10.3, 4.2, 2.1 Hz, 1H, H-5), 4.17–4.08 (m, 1H, H-6b), 2.07, 2.04, 2.02, 2.01 (all s, 12H, 4 × COCH3). 13C NMR (100 MHz, CDCl3): δ 170.7, 170.1, 170.0, 169.6, (4 × C=O), 160.9 (HN=C), 93.0 (C-1), 70.1 (C-5), 70.0 (C-3), 69.8 (C-2), 67.9 (C-4), 61.5 (C-6), 20.80, 20.80, 20.7, 20.56 (4 × C(O)CH3). HRESIMS m/z: 514.002 [M + Na]+ (calculated for C16H20Cl3NO10Na, 514.005).
1,2-Isopropylidene-3-O-(β-d-2,3,4,6-tetra-O-acetyl-glucopyranosyl)-(R/S)-glycerol (4): To a solution of compound 3 (56.3 g, 0.11 mol) in dry dichloromethane (60 mL), 1,2-O-isopropylidene glycerol (26.0 mL, 0.20 mol) was added and cooled to -10 °C. Then, boron trifluoride etherate (1.6 mL, 13 mmol) was added dropwise into the solution and stirred. After 2 h, more boron trifluoride etherate (1.0 mL, 8.1 mmol) was added and slowly warmed until reaching room temperature. After stirring overnight, the reaction mixture was quenched with triethylamine (2 mL) and mixed with ice and stirred until all ice melted, whereupon a beige-colored solid precipitated. The mixture was filtered, and the filtered product was washed with ice-cold water. The filtered product was recrystallized in a solution of water/methanol (v/v, 50/50) and gave compound 4 as a colorless solid (40.9 g, 80%). The product was present as a diastereomeric mixture in a ratio of 7:3. 1H NMR (600 MHz, CDCl3): δ 5.19 (tt, J = 9.5, 6.9 Hz, 1H, H-3), 5.09–5.03 (m, 1H, H-4), 5.01–4.95 (m, 1H, H-2), 4.59 (d, J = 8.0 Hz, 1H, H-1α), 4.27–4.22 (m, 1H, H-6a), 4.22–4.19 (m, 1H, H-2′), 4.12 (dd, J = 12.3, 2.4 Hz, 1H, H-6b), 4.02 (dd, J = 8.5, 6.4 Hz, 1H, H-3′a), 3.78 (dd, J = 10.3, 5.9 Hz, 1H, H-1′a), 3.71 (dd, J = 8.4, 5.8 Hz, 1H, H-3′b), 3.68 (dq, J = 7.4, 2.5 Hz, 1H, H-5), 3.63 (dd, J = 10.4, 5.7 Hz, 1H, H-1′b), 2.08, 2.04, 2.01, 1.99 (all s, 12H, 4× COCH3), 1.39 (s, 3H, CH3), 1.33 (s, 3H, CH3). 13C NMR (150 MHz, CDCl3): δ 170.8, 170.4, 169.51, 169.47, (4× C=O), 109.7 (C(CH3)2, 100.1 (C-1), 74.5 (C-2′) 72.9 (C-3), 72.0 (C-5), 71.3 (C-2), 70.7 (C-1′), 68.5 (C-4), 66.9 (C-3′), 62.0 (C-6), 26.9 (CH3), 25.5 (CH3), 20.9, 20.8, 20.73, 20.71 (4× C(O)CH3). HRESIMS m/z: 485.159 [M + Na]+ (calculated for C20H30O12Na, 485.163).
1,2-Isopropylidene-3-O-(β-d-glucopyranosyl)-(R/S)-glycerol (5): Sodium methanolate (3.46 g, 64.1 mmol) was added to a suspension of compound 4 (5.00 g, 10.8 mmol) in dry methanol (50 mL). The reaction mixture was stirred at room temperature. After 2 h, the reaction was completed and the solution was homogeneous. The mixture was neutralized with Amberlyst® 15 (Merck KGaA), filtered, and evaporated in vacuo. The residue was purified by silica gel column chromatography (ethyl acetate/methanol (v/v, 70/30) to give compound 5 (2.95 g, 92% yield) as a colorless syrup. The product was present as a diastereomeric mixture in a ratio of 7:3. 1H NMR (500 MHz, MeOD): 4.36–4.31 (m, 1H, H-2′), 4.29 (d, J = 7.7 Hz, 1H, H-1α), 4.08 (dd, J = 8.5, 6.5 Hz, 1H, H-3′a), 3.90 (dd, J = 10.3, 5.3 Hz, 1H, H-1′a), 3.87 (dd, J = 11.9, 1.7 Hz, 1H, H-6a), 3.77 (dd, J = 8.5, 6.1 Hz, 1H, H-3′b), 3.66 (dd, J = 11.9, 5.3 Hz, 1H, H-6b), 3.62 (dd, J = 10.2, 6.2 Hz, 1H, H-1′b), 3.38–3.33 (m, 1H, H-3), 3.29–3.26 (m, 2H, overlapped H-4, H-5), 3.18 (dd, J = 9.1, 7.7 Hz, 1H, H-2), 1.40 (s, 3H, CH3), 1.34 (s, 3H, CH3). 13C NMR (125 MHz, MeOD): δ 110.6 (C(CH3)2, 104.5 (C-1), 78.0 overlapped (C-3, C-5), 76.1 (C-2′), 75.1 (C-2), 71.59 (C-1′), 71.57 (C-4), 67.7 (C-3′), 62.7 (C-6), 27.1 (CH3), 25.6 (CH3), HRESIMS m/z: 317.121 [M + Na]+ (calculated for C12H22O8Na, 317.121).
1,2-Isopropylidene-3-O-[(6-O-(4-tolylsulfonyl))-β-d-glucopyranosyl]-(R/S)-glycerol (6): To a solution of compound 5 (900 mg, 3.06 mmol) in dry pyridine (10 mL), p-toluenesulfonyl chloride (TsCl, 654 mg, 3.43 mmol, 1.1 equiv.) was added at 0 °C and stirred overnight at room temperature. An additional amount of TsCl (100 mg, 0.52 mmol) was added. After 2 h, the reaction was quenched with EtOH; solvents were evaporated in vacuo and the residue was purified by flash column chromatography (ethyl acetate/methanol (v/v, 100:0→90:10)) to give compound 6 (994 mg, 72% yield) as a colorless resin. The product was present as a diastereomeric mixture in a ratio of 7:3. 1H NMR (300 MHz, CDCl3): δ 7.77 (d, J = 8.3 Hz, 2H, Ar-H), 7.31 (d, J = 8.1 Hz, 2H, Ar-H), 4.35–4.15 (m, 4H, overlapped H-2′, H-1, H-6a,b), 4.01 (dd, J = 8.7, 6.6 Hz, 1H, H-3′a), 3.80–3.71 (m, 1H, H1′a), 3.65 (dd, J = 8.5, 5.8 Hz, 1H, H-3′b), 3.56–3.49 (m, 2H, overlapped H-3, H-1′b), 3.45 3.45–3.37 (m, 2H, overlapped H-4, H-5), 3.34 (t, J = 8.5 Hz, 1H, H-2), 2.42 (s, 3H, aromatic methyl), 1.39 (s, 3H, CH3), 1.32 (s, 3H, CH3). 13C NMR (75 MHz, CDCl3): δ 145.1, Ar, (CCH3), 132.4 Ar, (CSO3), 130.0, 128.1 (4× CH, aromatic methynes), 109.9 (C(CH3)2, 95.8 (C-1), 76.1 (C-3), 74.7 (C-2′), 73.6 (C-5), 73,3 (C-2), 71.3 (C-1′), 69.7 (C-4), 69.2 (C-6), 66.6 (C-3′), 27.1 (CH3), 25.6 (CH3), 21.2 (Ar, CCH3), HRESIMS m/z: 471.131 [M + Na]+ (calculated for C19H28O10SNa, 471.130).
1,2-Isopropylidene-3-O-[(2′,3′,4′-tri-O-levulinyl-6′-O-(4-tolylsulfonyl))-β-d-glucosyl]-(R/S)-glycerol: (7): To a solution of compound 6 (990 mg, 2.01 mmol) in dry pyridine (10 mL), levulinic anhydride (3.0 mL, 15 mmol) dissolved in dry pyridine (7 mL) was added dropwise. The reaction was stirred overnight at room temperature. After the reaction was complete, ice water (50 mL) was added. The mixture was extracted with chloroform (3 × 50 mL) and the combined organic layers were extracted with a 10% solution of sodium bicarbonate (2 × 50 mL) first and then with water, dried over Na2SO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography (ethyl acetate/cyclohexane (v/v, 50/50→100/0)) to give 7 (1.42 g, 87%) as a colorless resin. The product was present as a diastereomeric mixture in a ratio of 7:3. 1H NMR (400 MHz, CDCl3): δ 7.79 (d, J = 8.4 Hz, 2H, Ar-H), 7.34 (d, J = 8.2 Hz, 2H, Ar-H), 5.21 (t, J = 9.6 Hz, 1H, H-3), 4.90 (m, 2H, overlapped H-2, H-4), 4.51 (d, J = 7.9 Hz, 1H, H-1α), 4.24–4.18 (m, 1H, H-2′), 4.15 (dd, J = 11.6, 2.8 Hz, 1H, H-6a), 4.04 (dd, J = 11.6, 6.5 Hz, 1H, H-6b), 4.00 (d, J = 6.5 Hz, 1H, H-3′a), 3.80–3.69 (m, 2H, overlapped H-5, H-1′a), 3.66 (dd, J = 8.5, 5.9 Hz, 1H, H-3′b), 3.57 (dd, J = 10.5, 5.4 Hz, 1H, H-1′b), 2.82–2.66 (m, 6H, 3 × CH2OCO), 2.64–2.46 (m, 6H, 3 × CH2COCH3), 2.44 (s, 3H, aromatic methyl), 2.15 (s, 3H), 2.15 (s, 3H), 2.13 (s, 3H), (3 × CH3CO), 1.39 (s, 3H, CH3), 1.33 (s, 3H, CH3). 13C NMR (100 MHz, CDCl3): δ 206.37, 206.34, 206.31 (3 × CH3CO), 172.1, 171.7, 171.3 (3 × CH2OCO), 145.2, (Ar, CCH3), 132.8 (Ar, CSO3), 130.0, 128.2 (4 × CH, aromatic protons), 109.7 (C(CH3)2, 100.8 (C-1), 74.5 (C-2′), 72.0 (overlapped C-3, C-5), 71.01 (C-2), 70.7 (C-1′), 68.6 (C-4), 68.0 (C-6), 66.9 ( C-3′) 37.90, 37.84, 37.80 (3 × CH2COCH3), 29.86, 29.79, 29.77, (3 × CH3CO), 27.91, 27.88, 27.83 (3 × CH2OCO), 26.9 (CH3), 25,5 (CH3), 21.8 (Ar, CCH3), HRESIMS m/z: 765.239 [M + Na]+ (calculated for C34H46O16SNa, 765.240).
1,2-Isopropylidene-3-O-[(2′,3′,4′-tri-O-levulinyl-6′-thioacetyl)-β-d-glucosyl]-(R/S)-glycerol: (8) Compound 7 (1.40 g, 1.88 mmol) was dissolved in dry dimethylformamide (10 mL) and potassium thioacetate (646 mg, 5.66 mmol) was added. The reaction was stirred at 80 °C. After 2 h, the reaction mixture was cooled until reaching room temperature and further stirred overnight. Subsequently, the solvents were evaporated in vacuo and the residue was purified by silica gel column chromatography (ethyl acetate) to give compound 8 (926 mg, 76% yield) as a colorless resin. The product was present as a diastereomeric mixture in a ratio of 7:3. 1H NMR (400 MHz, CDCl3): δ 5.20 (t, J = 9.7 Hz, 1H, H-3), 5.00–4.88 (m, 2H, overlapped H-2, H-4), 4.52 (d, J = 8.0 Hz, 1H, H-1α), 4.23 (p, J = 5.9 Hz, 1H, H-2′), 4.03 (dd, J = 8.4, 6.4 Hz, 1H, H-3′a), 3.85–3.68 (m, 2H, overlapped H-1′a, H-3′b), 3.63 (dd, J = 10.4, 5.7 Hz, 1H, H-1′b), 3.60–3.53 (m, 1H, H-5), 3.28 (dd, J = 14.3, 3.0 Hz, 1H, H-6a), 3.01–2.92 (m, 1H, H-6b), 2.81–2.68 (m, 6H, 3 × CH2OCO), 2.63–2.50 (m, 6H, 3 × CH2COCH3), 2.32 (s, 3H, CH3C(O)S), 2.16 (s, 3H), 2.15 (s, 3H), 2.13 (s, 3H) (3 × CH3CO), 1.39 (s, 3H, CH3), 1.33 (s, 3H, CH3). 13C NMR (100 MHz, CDCl3): δ 206.6, 206.5, 206.4 (3 × CH3CO), 194.8 (CH3C(O)S), 172.1, 171.8, 171.4 (3 × CH2OCO), 109.6 (C(CH3)2, 100.8 (C-1), 74.5 (C-2′), 73.3 (C-5), 72.1 (C-3), 71.3 (C-4), 70.8 (C-2), 70.5 (C-1′) 67.0 (C-3′), 37.93, 37.87, 37.84 (3 × CH2COCH3), 30.5 (CH3C(O)S), 30.2 (C-6), 29.9, 29.8, 29.79 (3 × CH3CO), 28.02, 27.95, 27.93 (3 × CH2OCO), 26.9 (CH3), 25.5 (CH3). HRESIMS m/z: 669.219 [M + Na]+ (calculated for C29H42O14SNa, 669.219).
3-O-[(2′,3′,4′-tri-O-levulinyl-6′-thioacetyl)-β-d-glucosyl]-(R/S)-glycerol (9): A solution of compound 8 (876 mg, 1.35 mmol) in 80% aq acetic acid (10 mL) was stirred at 45 °C for 2 h. The mixture was evaporated in vacuo and co-evaporated with toluene three times. The residue was purified by silica gel column chromatography (ethyl acetate/methanol (v/v, 100/0→70/30)) to give 9 (730 mg, 89%) as a colorless resin. The product was present as a diastereomeric mixture in a ratio of 7:3. 1H NMR (300 MHz, CDCl3): δ 5.22 (t, J = 9.7 Hz, 1H, H-3), 5.01–4.89 (m, 2H, overlapped H-2, H-4), 4.48 (d, J = 8.0 Hz, 1H, H-1α), 3.96–3.80 (m, 2H, overlapped H-2′, H-1′a), 3.72–3.50 (m, 4H, overlapped H-1′b, H-3′a, H-3′b, H-5), 3.29 (dd, J = 14.3, 2.7 Hz, 1H, H-6a), 2.96 (dd, J = 14.3, 7.2 Hz, 1H, H-6b), 2.86–2.67 (m, 6H, 3 × CH2OCO), 2.64–2.47 (m, 6H, 3 × CH2COCH3), 2.33 (s, 3H, CH3C(O)S), 2.18 (s, 3H), 2.16 (s, 3H), 2.13 (s, 3H) (3 × CH3CO). 13C NMR (75 MHz, CDCl3): δ 208.1, 206.7, 206.5 (3 × CH3CO), 195.0 (CH3C(O)S), 172.0, 171.8, 171.6 (3 × CH2OCO), 101.4 (C-1), 73.2 (C-5), 72.2 (C-1′), 72.0 (C-3), 71.3 (C-4), 70.7 (C-2), 70.6 (C-2′), 63.4 (C-3′), 38.0, 37.9, 37.8 (3 × CH2COCH3), 30.6 (CH3C(O)S), 30.1 (C-6), 29.9, 29.81, 29.80 (3 × CH3CO), 28.0, 27.9, 27.8 (3 × CH2OCO). HRESIMS m/z: 629.188 [M + Na]+ (calculated for C26H38O14SNa, 629.188).
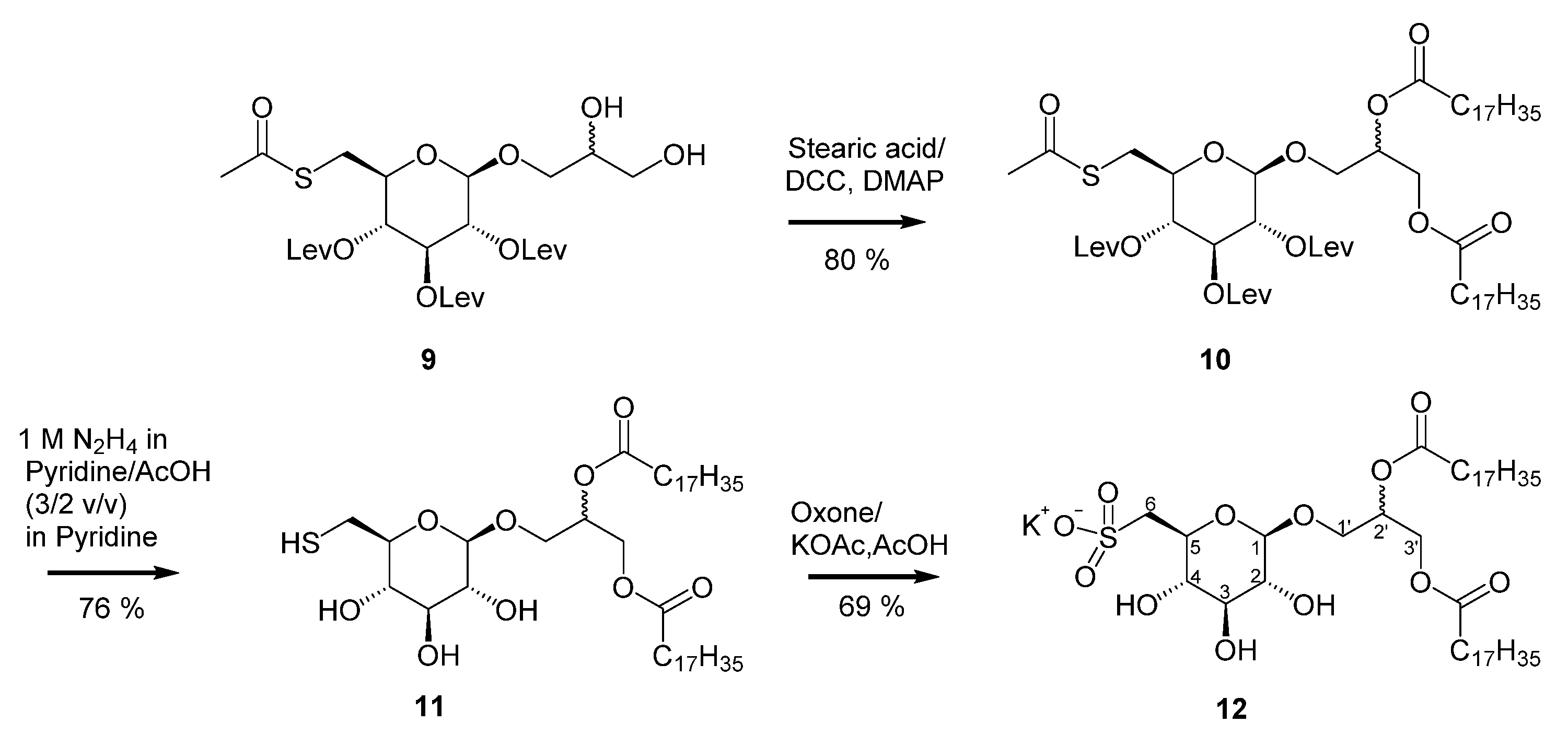
1,2-Distearoyl-3-O-[(2′,3′,4′-tri-O-levulinyl-6′-thioacetyl)-β-d-glucosyl]-(R/S)-glycerol (10): To a solution of compound 9 (700 mg, 1.15 mmol) in dry dichloromethane (15.0 mL), stearic acid (854 mg, 3.00 mmol), dicyclohexylcarbodiimide (608 mg, 2.90 mol), and 4-(Dimethylamino)-pyridine (67 mg, 0.5 mmol) were successively added. The reaction mixture was stirred overnight for 24 h at room temperature. The precipitate was removed by filtration and the solvent evaporated in vacuo. The residue was purified by silica gel column chromatography eluted with ethyl acetate/cyclohexane, v/v, 50/50 to give the product 10 (1.05 g, 80%) as a colorless waxy solid. The product was present as a diastereomeric mixture in a ratio of 7:3. 1H NMR (400 MHz, CDCl3): δ 5.23–5.13 (m, 2H, overlapped H-3, H-2′), 4.98–4.91 (m, 2H, overlapped H-2, H-4), 4.45 (d, J = 8.0 Hz, 1H, H-1α), 4.28 (dd, J = 12.0, 3.7 Hz, 1H, H-3′a), 4.07 (dd, J = 12.0, 6.4 Hz, 1H, H-3′b), 3.89 (dd, J = 10.9, 5.3 Hz, 1H, H-1′a), 3.65 (dd, J = 10.9, 5.4 Hz, 1H, H-1′b), 3.57 (ddd, J = 10.0, 7.2, 2.9 Hz, 1H, H-5), 3.28 (dd, J = 14.3, 2.9 Hz, 1H, H-6a), 2.96 (dd, J = 14.3, 7.2 Hz, 1H, H-6b), 2.87–2.67 (m, 6H, 3 × CH2OCO), 2.66–2.47 (m, 6H, 3 × CH2COCH3), 2.33 (s, 3H, CH3C(O)S), 2.29 (dd, J = 9.0, 6.3 Hz, 4H, α-methylene of stearoyl), 2.16 (s, 3H), 2.15 (s, 3H), 2.13 (s, 3H) (3 × CH3CO), 1.63–1.54 (m, 4H, β-methylene of stearoyl), 1.18 (br s, 56H, aliphatic methylenes of stearoyl), 0.81 (t, J = 7.2, 6.4 Hz, 6H, 2 × terminal CH3 of stearoyl). 13C NMR (100 MHz, CDCl3): δ 206.7, 206.5, 206.4 (3 × CH3CO), 194.7 (CH3C(O)S), 173.5, 173.2 (2 × C(O)O of stearoyl), 172.1, 171.8, 171.3 (3 × CH2OCO), 100.7 (C-1), 73.3 (C-5), 72.0 (C-3), 71.1 (C-4), 70.6 (C-2), 69.9 (C-2′), 67.6 (C-1′), 62.4 (C-3′), 37.94, 37.87, 37.84 (3 × CH2COCH3), 34.4, 34.2 (2 × αC -methylene), 30.6 (CH3C(O)S), 30.6 (C-6), 30.9, 29.98, 29.94 (overlapped 3 × CH3CO), 32.1, 29.9–29.2, 22.8 (aliphatic methylenes, in part overlapped), 28.0, 27.9, 27.8 28.0, 27.9, 27.8 (3 × CH2OCO), 25.0 (2 × βC-methylene of stearoyl), 14.3 (2 × terminal CH3 of stearoyl). HRESIMS m/z: 1183.757 [M + HCOO−]− (calculated for C63H108O18−, 1183.718).
1,2-Distearoyl-3-O-[(6′-thiol)-β-d-glucosyl]-(R/S)-glycerol (11): To a solution of compound 10 (500 mg, 0.44 mmol) in dry pyridine (5 mL), 1 M hydrazine monohydrate in pyridine/glacial acetic acid (v/v, 3/2) (10 mL) was added. The reaction mixture was stirred for 30 min at room temperature. After the reaction was completed, ice water (50 mL) was added. The mixture was extracted with chloroform (3 × 50 mL) and the combined organic layers were extracted with a 10% solution of sodium bicarbonate (2 × 50 mL) first and then with water, dried over Na2SO4, and concentrated in vacuo. The residue was purified by silica gel column chromatography (chloroform/methanol, v/v, 97/3→92/8) to give compound 11 (270 mg, 76%) as a colorless solid. The product was present as a diastereomeric mixture in a ratio of 7:3. 1H NMR (300 MHz, CDCl3): δ 5.34–5.16 (m, 1H, H-2′), 4.42 (dd, J = 12.0, 3.9 Hz, 1H, H-3′a), 4.29 (d, J = 7.7 Hz, 1H, H-1α), 4.14 (dd, J = 12.0, 6.2 Hz, 1H, H-3′b), 3.98 (dd, J = 11.0, 5.5 Hz, 1H, H-1′a), 3.70 (dd, J = 11.0, 5.9 Hz, 1H, H-1′b), 3.58–3.45 (m, 2H, overlapped H-3, H-4), 3.41–3.34 (m, 2H, overlapped H-2, H-5), 2.97 (ddd, J = 14.1, 9.5, 2.7 Hz, 1H, H-6a), 2.74 (dt, J = 14.3, 7.4 Hz, 1H, H-6b), 2.40–2.23 (m, 4H, α-methylene of stearoyl), 1.66–1.52 (m, 4H, β-methylene of stearoyl), 1.37–1.19 (m, 56H, aliphatic methylenes of stearoyl), 0.88 (t, J = 6.5 Hz, 6H, 2 × terminal CH3 of stearoyl). 13C NMR (75 MHz, CDCl3): δ 173.9, 173.5 (2 × C(O)O of stearoyl), 103.1 (C-1), 76.33 (C-3), 76.30 (C-5), 73.8 (C-2), 72.2 (C-4), 70.10 (C-2′), 68.0 (C-1′), 62.5 (C-3′), 34.5, 34.3 (2 × αC-methylene), 32.1, 29.9–29.2, 22.8 (aliphatic methylenes, overlapped), 26.4 (C-6), 25.1, 25.0 (2 × βC-methylene), 14.3 (2 × terminal CH3 of stearoyl), HRESIMS m/z: 801.580 [M − H]- (calculated for C45H85O9S−, 801.592).
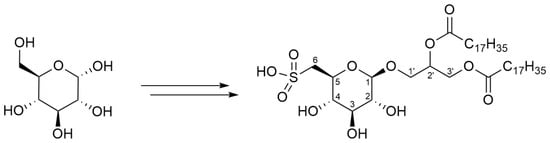
1,2-Distearoyl-3-O-[β-d-sulfoquinovosyl]-(R/S)-glycerol-potassium salt (12): To a solution of compound 11 (170 mg, 0.211 mmol) in glacial acetic acid (5 mL), potassium acetate (550 mg, 5.60 mmol) and oxone (2 KHSO5, KHSO4, K2SO4) (560 mg, 1.82 mmol, 4 equiv. KHSO5) were added. The resultant mixture was stirred at room temperature for 24 h. After the reaction was complete, the mixture was evaporated in vacuo and the residue purified by flash chromatography (chloroform/methanol/water, v/v/v, 92/8/0→65/25/4) to give compound 12 (120 mg 69%), as a colorless solid. The product was present as a diastereomeric mixture in a ratio of 7:3. 1H NMR (300 MHz, CDCl3): δ 5.30–5.21 (m, 1H, H-2′), 4.43 (dd, J = 12.1, 3.1 Hz, 1H, H-3′a), 4.28 (d, J = 7.7 Hz, 1H, H-1α), 4.14 (dd, J = 12.1, 6.8 Hz, 1H, H-3′b), 4.01 (dd, J = 11.0, 5.3 Hz, 1H, H-1′a), 3.77–3.66 (m, 2H, overlapped H-1′b, H-5), 3.39–3.36 (m, 1H, H-3), 3.36–3.33 (m, 1H, H-6a), 3.26–3.21 (m, 1H, H-2), 3.20–3.16 (m, 1H, H-4), 3.04 (dd, J = 14.4, 7.3 Hz, 1H, H-6b), 2.30 (td, J = 7.5, 5.0 Hz, 4H, α-methylene of stearoyl), 1.65–1.52 (m, 4H, β-methylene of stearoyl), 1.24 (br s, 56H, aliphatic methylenes of stearoyl), 0.86 (t, J = 7.2, 6.6 Hz, 6H, 2 × terminal CH3 of stearoyl). 13C NMR (75 MHz, CDCl3): δ 174.5, 174.2 (2 × C(O)O of stearoyl), 103.5 (C-1), 76.4 (C-3), 73.9 (C-2), 73.6 (C-4), 72.4 (C-5), 70.7 (C-2′), 68.2 (C-1′), 63.2 (C-3′), 54.0 (C-6), 34.6, 34.4 (2 × αC-methylene), 32.2, 30.0–29.4, 22.8 (aliphatic methylenes, in part overlapped), 25.2 (2 × βC-methylene of stearoyl), 14.3 (2 × terminal CH3 of stearoyl). HRESIMS m/z: 849.562 [M − H]− (calculated for C45H85O12S−, 849.577).




{kind=link}
{kind=link}
{kind=link}
{kind=link}


