Abstract
We performed an X-ray crystallographic study of complexes of protein kinase PIM-1 with three inhibitors comprising an adenosine mimetic moiety, a linker, and a peptide-mimetic (d-Arg)6 fragment. Guided by the structural models, simplified chemical structures with a reduced number of polar groups and chiral centers were designed. The developed inhibitors retained low-nanomolar potency and possessed remarkable selectivity toward the PIM kinases. The new inhibitors were derivatized with biotin or fluorescent dye Cy5 and then applied for the detection of PIM kinases in biochemical solutions and in complex biological samples. The sandwich assay utilizing a PIM-2-selective detection antibody featured a low limit of quantification (44 pg of active recombinant PIM-2). Fluorescent probes were efficiently taken up by U2OS cells and showed a high extent of co-localization with PIM-1 fused with a fluorescent protein. Overall, the developed inhibitors and derivatives represent versatile chemical tools for studying PIM function in cellular systems in normal and disease physiology.
1. Introduction
The PIM (proviral integration site for Moloney murine leukemia virus) family of protein kinases (PKs) includes three constitutively active serine/threonine kinases (PIM-1, PIM-2, and PIM-3) that regulate key biological processes, including cell survival, proliferation, differentiation, and apoptosis [1,2]. Elevated expression levels of PIM kinases have been observed in hematologic malignancies such as myelomas and non-Hodgkin lymphomas [3,4]. PIM kinases play important roles in the development and progression of other types of cancer (e.g., non-small-cell lung cancer, pancreatic and prostate cancers, gastric, hepatocellular and squamous cell carcinomas, liposarcoma, glioblastoma) [2,5]. These findings suggest that PIM kinases are potential cancer drug targets and biomarkers [6]. Recent success in the development of PIM-selective inhibitors with low picomolar inhibitory potency [4] has intensified clinical testing of these inhibitors for the treatment of hematologic cancers.
Protein kinases (PKs) have been the most important targets for cancer drug development in this century. More than 60 small-molecule PK inhibitors have reached the drug market [7]. Most of these inhibitors follow Lipinski’s rules for oral drugs [8] and bind to the ATP-binding pocket or flanking regions of PKs. However, all PKs as well as other proteins of the purinome (a total of 3266 proteins encoded in the human genome) bind purine and its derivatives (e.g., adenine, the heteroaromatic moiety of ATP) [9]. Therefore, ATP-mimicking PK inhibitors bear a high off-target risk.
In recent years, the bisubstrate approach has gained popularity for the construction of potent and selective inhibitors of PKs [10,11,12]. Bisubstrate inhibitors consist of two conjugated fragments, each targeting the binding site of a particular substrate. This approach facilitates the formation of additional interactions between the inhibitor and less-conserved protein substrate-binding site of the kinase. Thus, the two important biochemical characteristics of an efficient inhibitor, affinity and selectivity, can be improved.
Previously, we have used the bisubstrate approach for the development of high-affinity bisubstrate inhibitors, ARC inhibitors (ARC: adenosine–arginine conjugate) possessing values of the dissociation constant (KD) in the picomolar range for several PKs: catalytic subunit α of protein kinase A (PKAcα), PIM-1, Rho-associated protein kinase 2 (ROCK2), and Haspin [13,14,15,16]. In ARCs, an adenosine analogue heteroaromatic moiety and substrate peptide mimetic d-Arg-rich fragment are covalently tethered by a linker. These conjugates are proteolytically stable and efficiently penetrate the plasma membrane of mammalian cells [17,18]. The latter property of ARCs is based upon the transport peptide fragment (d-Arg)6 incorporated into several ARC inhibitors [17]. In case of basophilic PKs, the (d-Arg)6 fragment associates with the substrate protein-binding domain of the target PK and thereby substantially contributes to the binding of ARCs to the PK. On the other hand, oligo-(d-Arg) peptides comprising six or more d-Arg residues are considered as classical transport peptides that efficiently penetrate the cell plasma membrane. Unfortunately, (d-Arg)6 peptide is highly charged and can develop specific and non-specific interactions with certain biomolecules (e.g., endoprotease furin (KD = 106 nM and 1.3 nM for (d-Arg)6-NH2 and (d-Arg)9-NH2, respectively) [19], nucleic acids [20], components of cell nucleoli [21]) as well as labware surfaces [22].
Here, we report co-crystal structures of ARC/PIM-1 complexes. Based on the newly developed ARC-type inhibitor BPTP-Ahx-(d-Arg)6-d-Lys-NH2 (ARC-3126; BPTP-7-bromo-2-(methylene)pyrido[4,5]thieno[3,2-d]pyrimidin-4-one, Ahx-6-aminohexanoic acid), new compounds with a reduced number of d-Arg residues were constructed in order to decrease the risk of non-specific interactions in biochemical experiments. The affinities of the novel compounds were assessed along with their selectivity profiles. An ARC-affinity surface was designed for capturing PIM-2, which was quantitatively detected with a specific monoclonal antibody. Finally, fluorescent probes derived from the newly developed compounds were examined in live U2OS cells to assess their cell plasma membrane-penetrative properties and intracellular localization.
2. Results and Discussion
2.1. Thermal Shift Assay of ARC/PIM Complexes
The study was started by choosing a set of structurally diverse ARCs, which had previously revealed low nanomolar or subnanomolar KD values toward their reported PK targets (PKAcα, ROCK2 etc.) [13,23]. For establishing affinity toward different PKs of the PIM family, a thermal shift assay was used that measured the stabilization of the 3D structure of the PK upon its binding to the compound under study [24]. As control compounds, an ATP-binding site PIM inhibitor SGI-1776, as well as ARC-3119 and ARC-3125 (previously tested with PIM kinases) were utilized.
The results of measurements are summarized in Table 1. All the characterized compounds exhibited ΔTm values of over 5 °C with all PIM isoforms (according to the literature, ΔTm values over 5 °C typically point to nanomolar affinity of inhibitors [24]). SGI-1776 featured a ΔTm value of 9.5 °C with PIM-1, which is comparable with the previously reported data (ΔTm value of 9.7 °C was reported for a very similar structural analogue of SGI-1776 [25]).

Table 1.
Values of thermal shift of screened compounds in complex with PIM kinases.
All tested ARCs featuring large ΔTm values contained at least six d-Arg residues, whereas the binding of compounds incorporating TIBI ((4,5,6,7-tetraiodo-1H-benzimidazol-1-yl)acetic acid moiety) and BBTP (8-bromo-2-(methylene)[1]benzothieno[3,2-d]pyrimidin-4-one moiety) as adenosine analogue moieties led to higher stability of the protein structure. In our previous study, ARCs with AMTH and PIPY bound with high affinity to a large set of basophilic PKs [13,23], whereas the BBTP moiety conferred a PIM-selective inhibitor [14] and the TIBI moiety conferred a promiscuous CK2 inhibitor [26]. The TIBI-comprising conjugate ARC-3125 revealed high ΔTm values (12.7–14.7 °C) due to the strong binding of both its moieties [26] to the substrate pockets of PIM. Moreover, ARC-3125 stabilized PIM kinases significantly more compared to previously studied PKs DYRK2 and CLK2 that showed ΔTm values of only 3.3 °C and 4.0 °C, respectively [15].
2.2. Co-Crystal Structures of ARC-1411, ARC-1415, and ARC-3126 with PIM-1
Based on the results of thermal shift assay, we selected the candidates for co-crystallization with PIM-1: ARC-1411, ARC-3119, and their derivatives ARC-1415 and ARC-3126. Adenosine analogue moieties of ARC-1415 and ARC-3126 were structurally different (PIPY vs. BPTP, respectively), and these two compounds also comprised different linkers (α,ω-nonanedioic acid vs. 6-aminohexanoic acid). The only structural difference between ARC-1415 and ARC-1411 was the lack of a C-terminal d-Lys residue in ARC-1415 (Figure 1).
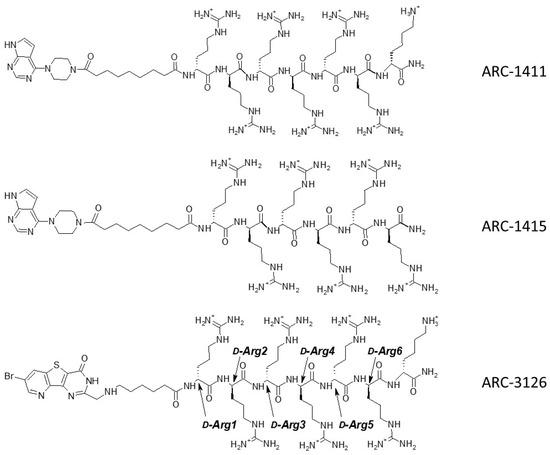
Figure 1.
Structures of ARC inhibitors co-crystallized with PIM-1.
Unfortunately, the co-crystallization of ARC-3119 with PIM-1 was not successful; co-crystals were obtained for PIM-1 complexes with ARC-1411, ARC-1415, and ARC-3126. The corresponding crystal structures were solved and refined at 1.9 Å resolution (Table S1) and the coordinates were deposited in the protein databank (PDB) (ID 7OOV for ARC-1411, 7OOW for ARC-1415, and 7OOX for ARC-3126). All crystal structures contained one protein molecule in the asymmetric unit, in which the adenosine analogue moiety of the inhibitors was positioned in the ATP-binding site of PIM-1 and the peptide mimetic moiety bound on the surface of the substrate protein site (Figure 2). The electron densities for the ARCs in the obtained co-crystals are shown in Supplementary Figure S1. The peptide mimetic moiety of ARC-1411 was resolved in the complex with PIM-1 to a larger extent compared to the previously reported complex with PKAcα [13]. However, some of the side chains of the peptidic portion of the inhibitor that were oriented toward the solvent showed weak electron density. The same applied to the ARC-1415 complex, where first d-Arg (d-Arg1; Figure 1) and a small portion of the linker as well as the side chain of d-Arg5 were poorly resolved; again, these moieties made few interactions with the PIM-1 catalytic domain. In contrast, in the ARC-3126 complex, the linker and the side chain of d-Arg1 were well resolved, but the terminal arginine and lysine residues showed only weak density in the 2Fo-Fc electron density maps (Supplementary Figure S1). As the side chains of the terminal arginine and lysine made few interactions, the peptide portion of these bisubstrate inhibitors could be truncated at d-Arg5 without losing affinity for the target.
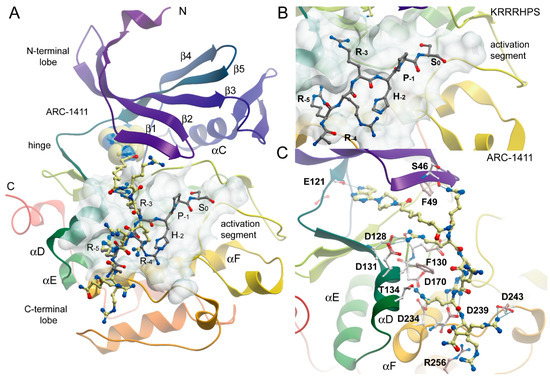
Figure 2.
Binding mode of ARC-1411 in PIM-1 and comparison with a PIM substrate complex. (A) Overall structure of the PIM-1 catalytic domain in complex with ARC-1411 (PDB-ID: 7OOV). The main secondary structure elements are labeled. ARC-1411 is shown in a ball and stick representation, and the ATP mimetic fragment of the inhibitor is additionally highlighted in transparent CPK. The binding site of ARC-1411 is shown as a semi-transparent surface. The structural model has been superimposed with a PIM-1 substrate complex with the consensus peptide KRRRHPS (PDB-ID: 2BIL). The peptide is also shown in ball and stick representation with gray carbon atoms. (B) Details of the interaction of the PIM-1 complex with the consensus peptide. (C) Details of the interactions of ARC-1411 with PIM-1.
The structure of the ARC-1411 complex with PIM-1 revealed a canonical type-1 inhibitor interaction of its pyrrolo-pyrimidine hinge binding moiety, forming the anticipated hydrogen bond of the pyrrolo nitrogen with the backbone carbonyl of the hinge residue E121. Due to the insertion of a proline residue in the hinge of PIM kinases, the pyrimidine moiety did not engage in additional hinge backbone hydrogen bonds. Comparison with a complex of the PIM-1 kinase with a consensus peptide (PDB-ID: 2BIL) revealed high conservation of key interactions with consensus substrate residues (Figure 2). As a basophilic kinase of the CAMK family, PIM kinases recognize basic residues, in particular arginines at positions -3 and -5 of the phosphorylation site, which are required for high affinity interaction [27] (Figure 2B,C).
These key interactions were closely mimicked by arginine moieties in ARC-1411. Remarkably, the first arginine moiety connected via an aliphatic linker to the ATP mimetic hinge binding motif engages with the glycine-rich loop (GRL) residue S46. The GRL itself assumed a folded conformation, which is often observed in PIM-1 inhibitor complexes where the phenylalanine (F49) is positioned at the tip of the GRL and flipped into the ATP binding pocket [28]. The second arginine in ARC-1411 mimicked the interaction of the arginine in the key substrate position -3, which pointed into an acidic binding pocket lined by the αD residues D128 and D131. The following arginine in ARC-1411 formed polar interactions with D239 that were not present in the substrate complex, most likely increasing the affinity of ARC-1411 (Figure 2C). However, similar to the substrate residue R-4, the next arginine moiety in ARC-1411 pointed toward the solvent, but the following arginine mimicked again the substrate arginine in position -5, which is required for high-affinity substrate interactions. At this position, the arginine side chains formed an intricate network of polar interactions with residues located in αD (T134), αF (D234), and D170. The terminal arginines in ARC-1411 formed a number of long-range salt bridge (D243) and stacking interactions (R256). In summary, the well-resolved ARC-1411 complex with PIM-1 revealed the presence of key interactions that are critical for high-affinity substrate binding as well as additional interactions that explain the high affinity of this bisubstrate inhibitor for PIM kinases. Similar interactions were also conserved in the closely related ARC-1415 complex (Figure 3A).
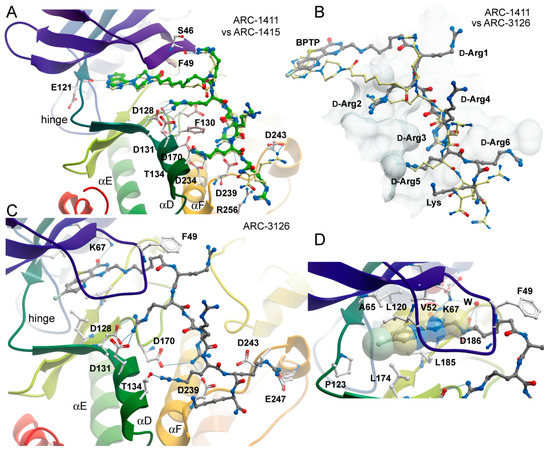
Figure 3.
Binding modes of ARC-1415 and ARC-3126. (A) Comparison of the binding modes of ARC-1411 and ARC-1415. The figure shows a superimposition of both structures. ARC-1411 carbon atoms are colored in yellow, and ARC-1415 carbon atoms are colored in green. Main interacting residues of PIM-1 are labeled. (B) Comparison of the binding modes of ARC-1411 and ARC-3126. Both structures have been superimposed, but for clarity, only the inhibitors are shown on the surface representation of PIM-1. ARC-1411 carbon atoms are colored in yellow and ARC-3126 carbon atoms are colored in gray. The residues in ARC-3126 are labeled. (C) Details of the interactions formed by ARC-3126. The main interacting residues are labeled. ARC-3126 carbon atoms are colored in gray. (D) Details of the interactions of the ATP-mimetic BPTP moiety and the linker in the ARC-3126 complex. W stands for water molecule.
In contrast, ARC-3126 utilized a different exit vector based on its diverse ATP-competitive moiety BPTP (Figure 3C,D). Remarkably, ARC-3126 did not form canonical hydrogen bonds with the hinge backbone but was anchored to the backpocket via a polar interaction with the conserved VAIK lysine (K67) similar to other kinases that harbor a large hydrophobic residue immediately N-terminal to the DFG motif [29], which is a binding mode that has been associated with favorable selectivity profiles (Figure 3D). The non-classical hinge-binding mode of BPTP in the ARC-3126/PIM-1 complex, similar to those of pyrimidinones and pyridazines [25,30], is a distinctive element for its selectivity, and thus for the selectivity of ARC-3126 over the promiscuous inhibitor ARC-3125. Consistently, ARC incorporating a pyrimidinone derivative as an ATP mimetic moiety possessed higher affinity toward PIM kinases than to other basophilic PKs in a previously reported selectivity panel [14], whereas ARC-1411 and ARC-1415 were reported to inhibit a wide list of basophilic PKs [13]. Attachment of the linker to the peptidic portion of ARC-3126 at the 2 position of the pyrimidin-4-one ring resulted in reorientation of the GRL, forcing F49 into an extended active-like conformation (Figure 3C,D).
In the ARC-1411 and ARC-1415 co-crystals, the d-Arg1 interacted with the GRL; in the case of ARC-3126, the interaction with GRL was formed by the carboxyl group of the 6-aminohexanoic acid linker (Figure 3A–C). Except for d-Arg1, the peptide mimetic moieties of ARC-1411, ARC-1415, and ARC-3126 formed similar polar interactions with PIM-1. Remarkably, despite the different exit vectors, the main substrate mimetic interactions with d-Arg2, d-Arg3, and d-Arg5 were highly conserved and superimposed well with the peptidic portion in ARC-1411. d-Arg2 interacted with PIM-1 residues D128 and D131, d-Arg3 with D239, and d-Arg5 with F130, D170, and T134. Additionally, d-Arg5 of ARC-1411 and ARC-1415 made contacts with D234 and G238. On the other hand, the C-terminus of the inhibitor showed a high degree of structural diversity, and d-Arg4 and d-Arg6 did not interact with the protein (Figure 3A–C).
PIM-1 has a strong preference for substrates with basic residues, particularly Arg, at positions −5 and −3 from the phosphorylatable serine or threonine residue [25,27]; for instance, the PIM-1 substrate peptide Pimtide (ARKRRRHPSGPPTA) possesses very high affinity to the kinase (KD value of 58 nM [27]). According to the previously reported crystal structure (PDB-ID: 2BZK, [27]), Arg residues at −5 and −3 positions in Pimtide interact with the same PIM-1 residues as ARC-1411, ARC-1415, and ARC-3126. Therefore, ARC inhibitors that were the focus of this study have the capacity of preventing interactions involving PIM kinases and their substrate proteins, similarly to a structurally different inhibitor that also reveals multiple interactions with residues of the substrate-binding site [31].
2.3. Design and Biochemical Characterization of New Inhibitors
Guided by the 3D structure of the PIM-1 bound lead compound ARC-3126 and taking into consideration the revealed cues regarding the putative binding mode and selectivity features of the compound, a new set of ARC compounds was designed. As the crystal structure analysis revealed that d-Arg1, d-Arg4, and d-Arg6 residues of ARC-3126 did not contribute to the binding in the ARC-3126/PIM-1 complex, these structural elements were omitted from the new ARC inhibitors to make the compounds structurally smaller, simpler, and less charged. To conserve the positioning of d-Arg2, d-Arg3, and d-Arg5, longer linker chains were used in inhibitors, and the d-Arg4 residue was replaced by Gly. The C-terminal d-Lys residue was kept for potential labeling of inhibitors via the amino group of its side chain. Potentially, these new structures were expected to reveal lower non-specific binding to components of biological solutions, as well as to become less prone to binding to nucleic acids in the cellular context [17].
The affinities of the compounds toward the PIM family PKs were determined in a binding/displacement assay with measurement of fluorescence anisotropy (FA) and time-gated luminescence intensity (TGLI) (Table 2). For the initial assessment of the selectivity of compounds, PKAcα was used as the reference basophilic kinase [10]. As a control compound, the commercially available ATP-competitive PIM inhibitor AZD1208 was used.
Table 2.
Structures of tested ARC inhibitors and their affinities toward PIM kinases and PKAcα.
The six d-Arg residues-comprising lead compound ARC-3126 possessed a KD value of 1.8 nM toward PIM-1 and, in comparison, a KD value of over 18 μM toward PKAcα. Compound 1 (incorporating an 8-aminooctanoic acid (Aoc) linker) bound to PIM-1 with a KD value of 11.4 nM but was more than 50-fold less selective than ARC-3126 based on comparison of their respective affinities toward PIM-1 and PKAcα (Table 2). This may reflect the fact that compound 1 did not interact with all the substrate binding hotspots of PIM-1 compared to ARC-3126. Upon replacement of the adenosine analogue moiety BPTP (compound 1) with BBTP (compound 2) that has been reported to have high affinity and selectivity to PIM kinases [30], the selectivity to PIM-1 was increased 11-fold.
PKAcα binds with high affinity to ARCs that comprise a flexible linker between the adenosine analogue and peptide mimetic moieties [13]. Aoc is a highly flexible linker in compounds 1 and 2. Therefore, rigidified linkers comprising a 1,2,3-triazole fragment were tested in compounds 3 and 4. The 1,2,3-triazole moiety is resistant to cellular proteolytic degradation and capable of forming hydrogen bonds. When [4-(aminomethyl)-1H-1,2,3-triazol-1-yl]acetic acid (Amt) was used as the linker, the affinity and selectivity of the resulting compound 3 was comparable to those of compound 2 (Table 2). The use of another linker, 5-(1H-1,2,3-triazol-4-yl)pentanoic acid (Tap) in compound 4 led to over 5-fold higher selectivity as compared to compound 3.
Furthermore, it has been shown that ARC inhibitors harboring a hydroxyproline (Hyp)-incorporating linker possess good PIM selectivity [14]. Therefore, 2 was modified by the insertion of Hyp between BBTP and Aoc. This modification resulted in compound 5 possessing over 4-fold lower selectivity toward PIM-1 than compound 2, suggesting less suitable positioning of the peptide mimetic moiety of 5 in complex with PIM-1.
Fluorescent probes are important tools in biochemical research due to the high sensitivity of fluorescence-based assays, applicability of the probes for high-throughput measurements, and amenableness of the assays for automation [34]. The labeling of compounds 2 and 4 with the fluorescent cyanine dye Cy5 via the side-chain amino group of the C-terminal d-Lys residues yielded fluorescent probes 6 and 7, featuring nanomolar affinity toward PIM-1 (KD = 1.7 nM and 21.9 nM, respectively). The KD value of 6 demonstrated that attachment of the dye did not affect much the binding of the inhibitor to the kinase and pointed to the potential applicability of the probe for screening of PIM inhibitors. Importantly, the affinities of 6 and 7 to PKAcα were much lower, falling in the sub-micromolar and micromolar ranges (Table 2). The analogue of 7 with 5-TAMRA dye (compound 8) revealed similar affinity to PIM-1 and PKAcα as 7.
The binding properties of the designed peptide mimetic moiety of compounds 1–5 (PIM peptide) to PIM-1 were also tested. It revealed only weak affinity, possessing a KD value of over 10 µM (Table 2). Finally, the tested compounds revealed almost the same affinity and selectivity to PIM-3 compared to PIM-1, while they demonstrated lower affinity to PIM-2 (except for ARC-3126, which showed similar affinity toward all PIM kinases; Table 2).
2.4. Kinome-Wide Selectivity Profiling
To evaluate the kinome-wide selectivity of novel ARCs, compounds 1, 2, and 8 as well as the lead compounds ARC-3125 and ARC-3126 were tested against a panel of 140 PKs in the MRC PPU International Centre for Kinase Profiling in Dundee.
The residual activities of PKs in the presence of 1 µM final concentration of compounds are summarized in Table S3. The profiling confirmed the promiscuous behavior of TIBI-comprising ARC-3125 with a Gini coefficient of 0.508 and a hit rate of 0.62 (the latter value indicated that 62% of the tested PKs had residual activity below 50%). The PKs with residual activity below 20% belonged to different groups of the kinome [35]. As expected, high inhibitory potency was revealed toward the PIM family (CAMK group), yet the residual activity of another PK of the CMGC group CDK1/cyclin A, a STE group member MAP4K3, a tyrosine kinase-like group member IRAK1, as well as the tyrosine kinase IGF1R were all below 25% residual activity at 1 µM inhibitor concentration.
The selectivity of ARC-3126 was highlighted by a Gini coefficient value of 0.73 and a hit rate of 0.09 (Table S3). Notably, PIM-1 had zero residual activity, while PIM-2 and PIM-3 revealed residual activity of 10% and 13%, respectively (Table S3). The major off-target of ARC-3126 was MAPKAP-K2 kinase with a residual activity of 13%; the other members of the MAPKAP family were not inhibited. The observed inhibition of CK2 in the kinase panel was most likely an assay artefact caused by the interaction of Arg-rich ARCs with the aspartic acid-rich substrate peptide of CK2.
On the other hand, compound 1 comprising three d-Arg residues showed a remarkably narrow selectivity profile compared to ARC-3126, with a Gini coefficient value of 0.887 and hit rate of 0.022 (Table S3). Compound 1 strongly inhibited kinases of the PIM family (residual activities of 9% and 17% for PIM-1 and PIM-3, respectively) except for PIM-2, which unexpectedly retained 72% of residual activity. Other basophilic kinases in the panel had high residual activities above 81%.
Despite the higher affinity of compound 2 toward PIM kinases compared to compound 1 (Table 2), the selectivity profile of 2 was slightly broader, as revealed by its Gini coefficient value of 0.824 and a hit rate of 0.079 (Table S3). Still, these parameters indicated that the selectivity profile of 2 was narrower than that of our previously reported selective CK2 inhibitor ARC-772 (Gini coefficient of 0.752, [36]), or that of the clinically used Bcr-Abl inhibitor imatinib (Gini coefficient of 0.76, [37]). Expectedly, compound 2 strongly inhibited PKs of the PIM family (residual activity in the range of 4–7% for all isoforms). The major off-target PKs of 2 compared to 1 were DYRK1α (residual activity of 5% vs. 74%), CLK2 (24% vs. 97%), and RSK1 (31% vs. 95%).
The radiometric assay format used for the kinase panel also enabled the screening of fluorescent molecules. 5-TAMRA-labeled compound 8 displayed a Gini coefficient of 0.682 and hit rate of 0.066 (Table S3); in this respect, 2 and 8 demonstrated a similar level of selectivity and possessed similar ratios of the Gini coefficient over hit rate (around 10). Accordingly, 8 inhibited the PIM family to the same extent as 2. The examples of kinases inhibited less potently by 8 as compared to 2 included DYRK1α (residual activity of 37% vs. 5%), RSK1 (74% vs. 31%), and TAO1 (80% vs. 49%) kinases. Contrarily, some kinases were inhibited more potently by 8 as compared to 2, such as PKBβ (residual activity of 54% vs. 101%), TBK1 (46% vs. 113%), and CAMK1 (49% vs. 81%).
2.5. Surface Sandwich Assay for the Measurement of PIM-2 Concentration
PIM-2 is largely expressed in both leukemia and solid tumors. It plays an important role in promoting cell survival and preventing apoptosis [38]. Therefore, new analytical methods are needed to assess its expression level and activity in cells, cell lysates, and tissue extracts.
Previously, we have worked out a method for PIM-2 analysis and screening of its inhibitors in cell lysates [39], yet that method required the utilization of fluorescently tagged PIM-2 protein, and it could not be used for measurement of subnanomolar concentrations of the native kinase. Thus, we aimed at the development of a more sensitive and more robust method for the quantitative detection of PIM-2 with the aid of new ARC-probes.
To our knowledge, there are few reports on protein analysis assays based on the formation of a three-component complex [40] on solid support that uses the combination of a small-molecule inhibitor and specific antibody as capture/detection reagents [41]. Usually, larger molecules (proteins, nucleic acids, aptamers, etc.) are used for establishing such assays [42]. For example, PIM-2 can be analyzed with conventional enzyme-linked immunosorbent assays (ELISA). On the market, there are various sandwich-type ELISA kits from several suppliers. On the other hand, a classical ELISA assay requires the application of two orthogonal antibodies of the analyte, which are not always easy to obtain [43]. In this study, we have substituted an ARC-inhibitor-based capture molecule for the capture antibody. ARC inhibitors are synthetic mid-sized organic molecules that feature several advantages as compared to an antibody, such as shorter generation time, lower costs of manufacturing, the lack of batch-to-batch variability, higher modifiability, and better thermal stability. Another distinctive feature of the ARC-utilizing assay that might be physiologically relevant is that only the active kinase is captured, whereas the non-active (e.g., denatured) PIM-2 is washed out.
For the surface assay, ARC-2073, a biotinylated derivative of compound 2 was synthesized (Table S2). ARC-2073 revealed the KD value of 10 ± 2 nM in solution toward PIM-2, which was comparable to that of compound 2 (Table 2). Following the immobilization of ARC-2073 onto a streptavidin surface, PIM-2 was captured from a solution containing the recombinant protein. Thereafter, the captured PIM-2 was detected using rabbit anti-PIM-2 antibody D1D2 and visualized with an europium chelate-labeled secondary goat antibody against rabbit immunoglobulin G (G0506) (Figure 4A). The obtained calibration graph is depicted in Figure 4B. The lowest quantifiable concentration of PIM-2 was 25 ± 1 pM (quantification range from 25 to 625 pM), pointing to the high sensitivity of the assay, affording the measurement (LoQ) of 1.3 fmol (44 pg) of PIM-2 in the sample (50 μL). Thus, this sensitivity is advantageous versus the PIM-2 ELISA kit from ELK Biotechnology (product #ELK6492), which has a quantification limit of 1.8 fmol.
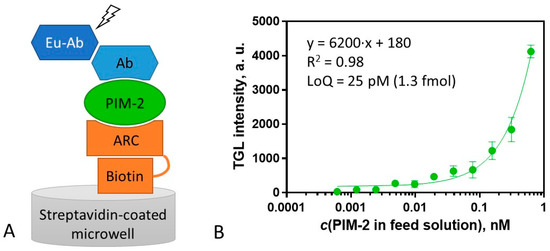
Figure 4.
Scheme and calibration curve of the assay for measurement of concentration of PIM-2. (A) Scheme of the assay format for PIM-2 measurement. PIM-2 is bound to the ARC-2073 functionalized surface (step 1), the solution of the primary antibody (Ab) is added (step 2), and Eu chelate-labeled secondary antibody (Eu-Ab) is added (step 3). (B) Calibration curve for PIM-2 analysis (immobilized ARC-2073 (4 pmol), detection with D1D2 (84 fmol) and G0506 (110 fmol)). TGL intensity was measured with excitation at 330 (60) nm; emission was detected at 590 (50) nm (integration from 80 to 1000 μs). All measurements were performed in triplicate, and the variance was estimated as SEM.
To assess the selectivity of the assay, the ARC-2073-coated surface was treated with solutions of PIM-1, PIM-3, and DYRK1α (an off-target PK of ARC-2073). These kinases were not detected with the pair of detection reagents D1D2/G0506 (Figure S2), confirming that D1D2 is specifically binding to PIM-2. In essence, the developed assay can be used for screening antibodies orthogonal to ARC-2073. On the other hand, the developed method can be potentially adapted for the quantitative detection of PIM-1 or PIM-3 by switching to the appropriate primary antibodies.
According to the literature, D1D2 has been used for the specific detection of PIM-2 in cell lysates [44]. Unfortunately, our assay did not reliably work in complex biological solutions such as crude cell lysates (Figure S3), which was probably due to matrix effects (presence of other biological molecules) on the stability of ARC-2073/PIM-2 complex in such solutions. We are currently working on reversing the assay format to capture PIM-2 with a surface-immobilized antibody to overcome the aforementioned issues.
2.6. Cell Plasma Membrane-Penetrative Properties of Dye-Labeled ARCs
Fluorescence sensors have gained great importance in biomedical research. Fluorescent probes based on selective inhibitors can be helpful for mapping the localization, concentration, and activity of target proteins in cells [45].
Several fluorescent probes based on inhibitors binding to the ATP-binding site have been constructed for PKs of the PIM family. First, a fluorescent dansyl derivative of a pyrrolo[2,3-a]carbazole-based PIM-1 inhibitor (fluorescent probe PCC-13) was reported to penetrate the plasma membrane and locate to the cytoplasm in live cells [46]. Later, a red-emitting fluorescent probe NB-BF targeting PIM-1 kinase was developed for the imaging of cancer cells [47]. NB-BF was prepared by conjugating 5-bromobenzofuran-2-carboxylic acid (a PIM-1 inhibitor possessing an IC50 value of 8.5 μM) with a red-emitting fluorophore Nile blue via hexanediamine linker. Unfortunately, the probe was not characterized in biochemical assays, and therefore, its binding affinity toward PIM-1 and selectivity to other PKs is unknown. The experiments performed in living cells and animals pointed to localization of the probe as well as PIM-1 protein in mitochondria. Recently, a novel near-infrared imaging agent QCAi-Cy7d was constructed by conjugating a small molecule inhibitor of PIM-1 and near-infrared heptamethine cyanine dye [48]. The effective antitumor activity of the novel theranostic agent was attributed to PIM-1 inhibition and mitochondrial targeting.
Fluorescently labeled ARCs comprising multiple d-Arg residues have been previously shown to penetrate the plasma membrane of live cells [17,49]. The uptake efficiency of ARCs depends on the structure of the adenosine analogue fragment, the number of d-Arg residues in the conjugate, and the hydrophobicity of the fluorescent dye. While the exact mechanism of the cellular entry of cell-penetrating peptides (CPPs) is still disputable, two main contributing pathways have been proposed—endosomal internalization and direct cell membrane penetration [50,51]. According to the mainstream paradigm, CPPs should comprise at least six guanidinium groups to demonstrate good penetrability of cell plasma membrane [51,52]. Previously, we reported that an ARC-probe comprising two d-Arg residues and a positively charged 5-TAMRA dye did not efficiently internalize into cells [17].
New PIM-targeting ARCs comprise three d-Arg residues; thus, we were interested in whether the derivatization of compounds 2 and 4 with a hydrophobic Cy5 dye, bearing a single positive charge, would facilitate the internalization of compounds 6 and 7 into cells. Due to its optical properties (excitation wavelength above 600 nm), Cy5 dye features reduced background cell autofluorescence in microscopy. As control compounds, compound 8 and ARC-2090 were used: the former compound contains three d-Arg residues and is labeled with TAMRA dye, whereas the latter compound contains six d-Arg residues and is labeled with Cy5 (Table S2).
The ability of fluorescent compounds to cross the plasma membrane of live U2OS cells was examined by following the effects of the incubation time (10–60 min) and concentration of the compounds (1–5 μM). Remarkably, 6 and 7 efficiently internalized into the cells (Figure 5), whereas the fluorescence of cells with 6 was somewhat stronger. In line with previous studies [36], the cellular penetrability was strongly concentration-dependent, with uptake at 2.5 μM or higher concentrations being remarkably more efficient than at 1 μM concentration. At 5 μM probe concentration and after 60 min incubation, the concentration of the compound in cells was so high that dark vesicles could be seen in brightfield images of 6 (Figure S4). In contrast, 8 featured much lower internalization efficiency and gave a reduced signal-to-noise ratio in microscopy images. The higher background signal was mostly caused by adsorption of the probe to the surface of the plastic well (Figure S5). Compounds 2 and 4 labeled with a sulfonated analogue of Cy5 (sulfo-Cy5) could not penetrate the plasma membrane of U2OS cells even at 5 μM concentration (data not shown), which can be attributed to the negative charges of sulfo-Cy5, leading to repulsion from the negatively charged cell plasma membrane.
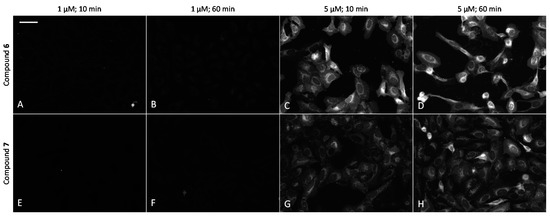
Figure 5.
Comparison of cellular uptake of ARCs labeled with Cy5 dye into live U2OS cells. (A–D) compound 6; (E–H) compound 7. Incubation conditions are listed above the images. Representative images from a single experiment are shown; in all wells, imaging settings were identical (see Materials and Methods section of the main text). Scale bar: 50 μm.
In cells, compounds 6 and 7 showed mainly perinuclear vesicular localization, with some additional cytoplasmic signal. As compared to ARC-2090 incorporating six d-Arg residues, the nuclear staining of compounds incorporating three d-Arg residues was remarkably lower (Figure S6), as it was conceived when the probes were designed.
To examine whether the vesicular staining of novel ARC probes might reflect the intracellular localization of PIM kinases or the compartmentalization is rather related to endosomatic uptake characteristic for many CPPs [53], we proceeded with co-localization studies using high-resolution total internal reflection-fluorescence (TIRF) microscopy. U2OS cells were transiently transfected for 48 h with genetic constructs representing either PIM-1 fused with GFP and PIM-1 fused with TagRFP, or endosomal marker Endo-14 fused with fluorescent protein mCherry [39,54]. As a negative control for co-localization studies, peroxisomal marker (Peroximal Targeting Signal 1) fused with mCherry was used (this marker is sequestered into the well-defined vesicular structures, yet there is no evidence-based reason to associate the intracellular location of PIM-1 with peroxisomes). Thereafter, 1 h incubation with 2.5 μM or 5 μM solutions of compounds 6 or 7 was carried out, followed by live cell imaging. PIM-1 was preferred as a model representative of the PIM family due to its higher ATP KM value of 400 μM compared to ATP KM values of PIM-2 and PIM-3 (4 and 40 µM, respectively) [4]. Therefore, in the cellular environment, the binding of ARCs to PIM-1 is less affected by high intracellular concentrations (1–10 mM) of ATP than the binding to PIM-2 and PIM-3.
We observed that in U2OS cells overexpressing PIM-1 fused with a fluorescent protein, the kinase showed enrichment in the nucleoplasm, which was combined with a somewhat lower cytoplasmic signal, which is consistent with previous reports [44,55,56]. However, under low expression conditions, the signal of PIM-1 was observed in perinuclear vesicles; importantly, in cells with vesicular PIM-1 localization, a high extent of co-localization between ARC probes (either compound 6 or 7) and PIM-1 fusions (either GFP-PIM-1 or PIM-1-TagRFP) was evident (Figure 6A–C and Figure 7A–F; Supplementary Video S1). Both ARC and labeled proteins were confined to the well-defined spot objects inside the cell cytoplasm. The co-localization extent was estimated with Manders’ coefficient (assessing overlap of objects segmented from the labeled protein channel inside the objects segmented from the ARC channel). The highest co-localization was observed for ARCs and PIM-1-TagRFP: for compound 6, Manders’ coefficient was 0.72 ± 0.06 (N = 9), and for compound 7, Manders’ coefficient was 0.43 ± 0.16 (N = 4). Additionally, some co-localization between ARC probes and mCherry-Endo-14 could be detected (Figure 7G–I), with the Manders’ coefficient value of 0.20 ± 0.03 (N = 5). Expectedly, the Manders’ coefficient values for the PIM-targeting ARC and peroxisome marker were very low (≤0.02), indicating absence of co-localization.
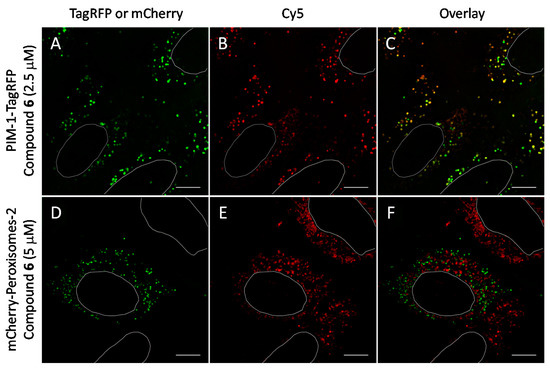
Figure 6.
Co-localization studies of PIM-targeting compound 6 with fluorescent protein-tagged PIM-1 and peroxisomal marker (following 48 h transfection) in live U2OS cells. The PIM-1-mRFP and mCherry-Peroxisomes-2 are shown in green and compound 6 is shown in red. White ovals indicate nuclei borders. Individual channels and overlayed images: (A–C) PIM-1-TagRFP and compound 6 (2.5 μM, 1 h incubation); (D–F) mCherry-Peroxisomes-2 and compound 6 (5 μM, 1 h incubation). Representative images (in cells with low expression of constructs) from two independent experiments are shown. Scale bar: 10 μm.
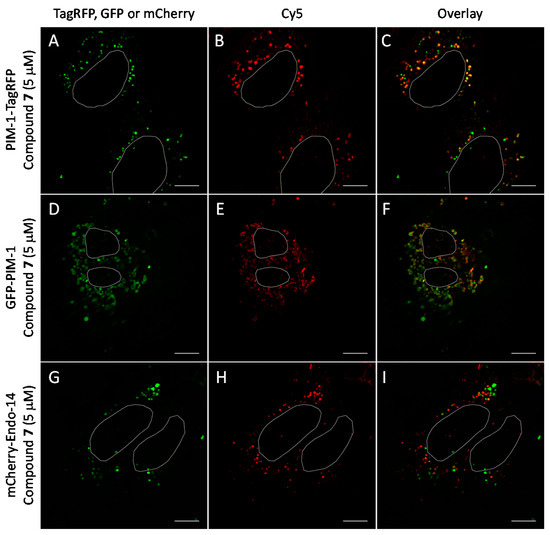
Figure 7.
Co-localization studies of PIM-targeting compound 7 (5 μM, 1 h incubation) with fluorescent protein-tagged PIM-1 and endosomal marker mCherry-Endo-14 in live U2OS cells. The genetically encoded PIM-1 constructs are shown in green and compound 7 is shown in red. Individual channels and overlaid images: (A–C) PIM-1-TagRFP and compound 7; (D–F) GFP-PIM-1 and compound 7; (G–I) mCherry-Endo-14 and compound 6. Representative images (in cells with low expression of constructs) from two independent experiments are shown. White ovals indicate nuclei borders. Scale bar: 10 μm.
3. Materials and Methods
3.1. Materials
Chemicals and resins for synthesis were obtained from Iris Biotech GmbH (Marktredwitz, Germany), Novabiochem (Läufelfingen, Switzerland), AnaSpec (Fremont, CA, USA), ABCR (Karlsruhe, Germany), Acros (Geel, Belgium), Alfa Aesar (Kandel, Germany), Fluka (Buchs, Switzerland; Seelze, Germany), Macherey-Nagel (Bethlehem, PA, USA), and Fisher Scientific (Geel, Belgium; Loughborough, UK; Ward Hill, MA, USA; Hampton, NH, USA). SGI-1776 and AZD1208 were from Axon Medchem (Reston, VA, USA) and Cayman Chemicals (Ann Arbor, MI, USA), respectively. N-hydroxysuccinimide esters of 5-TAMRA and Cy5 were obtained from Sigma-Aldrich (St. Louis, MO, USA) and Lumiprobe GmbH (Hannover, Germany), respectively. ARC-1141, ARC-3125 [15], ARC-668 [57], ARC-684 [23], ARC-3119, ARC-1188 [14], ARC-1411, and ARC-1415 [13] were synthesized as previously described. ARC-583 was previously developed for PKAcα and other PKs [32]. PIM kinases (recombinant human proteins, full sequence) were produced as previously described [27,58]. PKAcα (recombinant human protein, full sequence) was obtained from Biaffin GmbH & Co KG (Kassel, Germany). Rabbit monoclonal anti-PIM-2 antibody, clone D1D2 (2.5 μM) was purchased from Cell Signaling Technology (#4730; Danvers, MA, USA). Europium chelate-labeled goat anti-rabbit IgG (6.7 μM) was obtained from Tokyo Chemical Industry (G0506; Tokyo, Japan). Human bone osteosarcoma cell line U2OS was obtained from The Leibniz Institute DSMZ—German Collection of Microorganisms and Cell Cultures GmbH (Braunschweig, Germany); human prostate adenocarcinoma cell line PC-3 (derived from metastatic site in bone) was from the American Type Culture Collection (Manassas, VA, USA). The solutions and growth medium components for the cell culture were obtained from the following sources: phosphate-buffered saline (PBS), charcoal-purified fetal bovine serum (ccFBS), L-glutamine, Dulbecco’s modified Eagle’s medium (DMEM)/Ham’s F12 medium without phenol red, McCoy’s 5A medium, DMEM high-glucose medium, fetal bovine serum (FBS)—Sigma-Aldrich (Steinheim, Germany); a mixture of penicillin, streptomycin, and amphotericin B—Capricorn (Ebsdorfergrund, Germany); imaging medium components (MEMO and Supplement A)—Cell Guidance Systems (Cambridge, UK). Plasmids of the markers of peroxisomes (mCherry-Peroxisomes-2) and endosomes (mCherry-Endo-14) were a gift from Michael Davidson via Addgene (plasmid #54520 and #55040, respectively). The plasmids encoding GFP-PIM-1 and PIM-1-TagRFP fusion proteins were constructed as previously reported [39]. The transfection reagents FuGENE® HD and TurboFect™ were obtained from Promega Corporation (Madison, WI, USA) and ThermoScientific (R0531; Vilnius, Lithuania), respectively.
3.2. Synthesis of Compounds
BBTP and BPTP were synthesized according to the reported procedure [14]. ARC-3126, compounds 1–5, PIM peptide, and the precursors of ARC-1451 were synthesized on the Rink amide 4-methylbenzhydrylamine resin according to fluorenylmethoxycarbonyl (Fmoc) solid-phase peptide synthesis method as published earlier [14,59]. ARC-1451, ARC-2090, and compounds 6 and 7 were obtained by labeling their precursors with corresponding fluorescent dyes following the published protocol [14]. The detailed synthetic procedures as well as high-resolution mass spectrometry (HRMS) and HPLC purity data are presented in the Supplementary Information (Supplementary Methods and Tables S2 and S4).
After synthesis, the compounds were purified with Shimadzu Prominence LC Solution HPLC system (Columbia, MD, USA) using a Phenomenex Luna C18 reverse-phase column (5 μm, 25 cm × 0.46 cm; Torrance, CA, USA). A Thermo Electron LTQ Orbitrap (Langenselbold, Germany) mass spectrometer was used for the determination of exact masses of synthesized compounds. NMR spectra were recorded on a Bruker Avance-III 700 MHz (16.4 T; Bruker BioSpin GmbH, Rheinstetten, Germany). NanoDrop 2000c spectrophotometer (Thermo Scientific; Wilmington, DE, USA) was used for quantification of the compounds based on the following molar extinction coefficient (ε) values: BBTP (5700 M−1cm−1 at 343 nm), BPTP (5400 M−1cm−1 at 308 nm), 5-TAMRA (80,000 M−1cm−1 at 558 nm), or Cy5 (250,000 M−1cm−1 at 646 nm). The ε value for PIM peptide was calculated according to the literature [60] as 5906 M−1cm−1 at 214 nm.
3.3. Thermal Shift Assay for ARC/PIM Complexes
Thermal shift assay measurements were performed following the previously published protocol [61] using a real-time PCR instrument (Mx3005p RT-PCR, Stratagene; La Jolla, California). The final total concentrations of PIM kinases and ARCs in the experiments were 2 and 12.5 μM, respectively. The measurements were carried out in PCR low-profile microplate wells (ABgene; Portsmouth, NH, USA). The temperature scan was run from 25 to 95 °C at 1 °C/min. GraphPad Prism v. 6.0 (GraphPad Software, Inc.; San Diego, CA, USA) and Microsoft Excel v. 2007 (Microsoft Office; Redmond, WA, USA) were used for data processing and analysis.
3.4. Co-Crystallization, Diffraction Data Collection, and Structure Determination
Final concentrations of PIM-1 and the inhibitors were 0.2 mM and 1 mM, respectively. Crystals of complexes were grown using the sitting-drop vapor-diffusion method at 4 °C (ARC-1411) or 20 °C (ARC-1415, ARC-3126) with a reservoir solution listed in Table S1. Diffraction data were collected at Diamond Light Source, and processed and scaled with XDS [62] and AIMLESS [63], respectively. Structures were solved by molecular replacement using Phaser [64] and the coordinates of crystal structure of PIM-1 (PDB 2J2I; [65,66]) as the search model. Model rebuilding and structure refinement were performed in COOT [67] and REFMAC5 [68], respectively. Data collection and refinement statistics are summarized in Table S1. For preparation of the illustrative materials (Figure 2 and Figure 3), ICM software was used (Molsoft L.L.C.; San Diego, CA, USA).
3.5. Biochemical Binding/Displacement Assays with Measurement of FA or TGLI
Optical FA measurement modules for 5-TAMRA-labeled compounds (ex. 540(20) nm, em. 590(20) nm and 590(20) nm), for Cy5-labeled compounds (ex. 590(50) nm, em. 675(50) nm and 675(50) nm), and optical TGLI measurement modules for ARC-1451 (ex. 330(60) nm, em. 590(50) nm), for ARC-1188 (ex. 330(60) nm, em. 675(50) nm) were used for assays with PHERAstar microplate reader (BMG Labtech; Ortenberg, Germany). TGLI measurements were performed with a delay time of 50 μs and integration of the signal for 150 μs. In each measurement, emission from 200 flashes of excitation radiation was collected. The measurement solutions (20 µL volumes) were prepared in black 384-well low-binding surface microtiter plates (Corning #4514; Kennebunk, ME, USA) and incubated for 20 min at 30 °C before the measurements. The assay buffer contained HEPES hemisodium salt (pH 7.4, 50 mM), NaCl (150 mM), dithiothreitol (5 mM), bovine serum albumin (0.5 mg/mL), and Tween-20 (0.005%). GraphPad Prism v. 5.04 (GraphPad Software, Inc.) was used for data analysis with FA [32] or TGLI [33] readouts. Representative titration curves can be found in Supplementary Figure S7.
3.6. Kinase Selectivity Profiling
Kinase selectivity profiling was performed using the commercial service provided by the MRC PPU International Centre for Kinase Profiling (Dundee, UK) using a radiometric filter-binding assay [69]. Kinases used for analysis were of human origin. The ATP concentration applied for inhibition measurements was at or below the ATP KM value of the particular PK. ARC-3125 and ARC-3126 were screened against 139 PKs, whereas compounds 1, 2, and 8 were screened against 140 PKs. The final total concentration of inhibitors in the selectivity panel was 1 µM. Residual activities were expressed as the percentage of retained activity compared to the control without inhibitor.
From the residual activities, values of the Gini coefficient and hit rate were calculated for each individual compound. The Gini coefficient is a single-value metric whose value for a specific inhibitor depends on the number and origin of the PKs in the panel, but also on concentration of the tested compound [70]. Hit rate is the number of kinases inhibited by over 50%.
3.7. Surface Sandwich Assay for PIM-2 Measurement
The composition of the assay buffer was as follows: HEPES (50 mM), NaCl (150 mM), and Tween 20 (0.005%).
The Pierce streptavidin-coated microtiter plate (#15407; Waltham, MA, USA) was used for the preparation of the ARC-affinity surface. ARC-2073 in 10 mM phosphate buffer (pH = 7.0; 80 nM, 50 μL) was added to the wells and incubated for 60 min at room temperature (RT) under constant shaking at 300 rpm. After incubation, the wells were washed with assay buffer (100 μL) for 10 s at RT under constant shaking at 300 rpm. All the following washing procedures were performed similarly. Thereafter, PIM-2 in assay buffer (50 μL) was added to the ARC affinity wells. The wells were incubated for 60 min at RT under constant shaking at 300 rpm. After another washing procedure, primary antibody D1D2 (1.7 nM, 50 μL) in assay buffer was incubated in the wells for 60 min at RT under constant shaking at 300 rpm. The washing was repeated. Finally, europium chelate-labeled secondary antibody (G0506) (2.2 nM, 50 μL) in assay buffer was incubated in the wells for 60 min at RT under constant shaking at 300 rpm. The wells were washed, dried under a fume hood for 10 min, and TGLI was measured on their surfaces. GraphPad Prism v. 5.04 was used for data analysis. The limit of quantification (LoQ) was calculated based on the standard deviation of the calibration curve (SD) and the slope of the calibration curve (S) using the equation LoQ = 10 × SD/S.
PC-3 cells were cultured in DMEM high-glucose medium supplemented with 10% FBS, 2 mM glutamine, 100 U/mL penicillin, 100 µg/mL streptomycin, and 0.25 µg/mL amphotericin B at 37 °C in humidified atmosphere containing 5% CO2. PC-3 cells were suspended in assay buffer supplemented with EDTA (1 mM), protease inhibitor cocktail (1x), and phenylmethylsulfonyl fluoride (0.5 mM). The lysates were prepared by exposing PC-3 cells to continuous-wave 40-kHz ultrasound at an intensity of 0.4 W/cm2 for 10 s three times in ultrasonic bath at RT with a 30 s interval on ice to avoid overheating [71].
3.8. Cell Culture, Transfection, and Imaging
U2OS cells were cultured in McCoy’s 5A medium supplemented with 10% FBS, 2 mM glutamine, 100 U/mL penicillin, 100 µg/mL streptomycin, and 0.25 µg/mL amphotericin B at 37 °C in humidified atmosphere containing 5% CO2. The imaging was performed at 30 °C in humidified atmosphere containing 5% CO2.
For studies of the general internalization properties of ARCs, the cells were seeded at the density of 12,000–25,000 cells per well onto an Ibidi μ-plate (96-well; Ibidi GmbH, Planegg, Germany). After 24 h incubation in growth medium, the cells were treated for 10 min, 30 min, or 1 h with dilutions of compounds 6, 7, 8, or ARC-2090 (5 μM, 2.5 μM, or 1 μM final concentration) in the phenol red-free DMEM/Ham’s F12 medium supplemented with 2% ccFBS and 2 mM glutamine. Next, cells were washed twice with PBS, and imaging medium (MEMO + Supplement A 1:100) was applied subsequently. Cells were imaged with Cytation 5 wide-field microscope (BioTek; Winooski, VT, USA) using 20× air objective and the following settings: for Cy5, 623 nm LED (intensity: 2) and Cy5 filter cube (685 nm) with 104 ms integration time and detector gain value of 2; for TAMRA, 523 nm LED (intensity: 7) and RFP filter cube (593 nm) with 252 ms integration time and detector gain value of 16; for bright-field, white LED with intensity 6, 186 ms integration time and detector gain value of 15. Four independent experiments were performed.
For the co-localization analysis, the cells were seeded at the density of 30,000 cells per well onto the 8-well CG imaging chambers (Zell Kontakt GmbH; Nörten-Hardenberg, Germany). After 24 h incubation in growth medium, the cells were transfected for 48 h with mixtures comprising 0.8 μg of plasmid (mCherry-Endo-14, GFP-PIM-1, PIM-1-TagRFP, or mCherry-Peroxisomes-2) and 2.4 μL of FuGENE® HD per well. The following treatment of cells with compounds 6 or 7 (5 μM or 2.5 μM final concentrations) was performed as described above. Imaging was carried out with the TIRF microscopy setup as described in [72]. Briefly, widefield epifluorescence and Highly Inclined and Laminated Optical sheet (HILO) imaging [73] was conducted using an inverted microscope built around a Till iMIC body (Till Photonics/FEI; Munich, Germany), equipped with TIRF APON 60× oil (NA 1.49) objective lenses (Olympus Corp., Tokyo, Japan). The samples were sequentially excited with 488 nm (100 mW), 515 nm (80 mW), or 638 nm (150 mW) PhoxX laser diodes (Omicron-Laserage; Rodgau, Germany) combined in the SOLE-6 light engine (Omicron-Laserage). Excitation light was launched into Yanus scan head, which along with a Polytrope galvanometric mirror (Till Photonics/FEI; Munich, Germany) was used to position the laser for widefield epifluorescence or HILO illumination. Excitation and emission light was spectrally separated with imaging filter cubes consisting of a zt 491 rdc beamsplitter (Chroma) and 525/45 BrightLine emitter filter (Semrock; West Henrietta, NY, USA) for proteins labeled with GFP; zt 514/640 rpc dual line beamsplitter (Chroma) and the 577/690–25 nm BrightLine dual-band bandpass emission filter (Semrock) for mCherry or TagRFP-labeled proteins with 515 nm excitation laser or for Cy5-labeled ligands with 638 nm excitation laser. Additionally, the brightfield channel was used for the determination of cell borders and nucleus position. The electron-multiplying charge-coupled device Ultra 897 camera (Andor Technology; Belfast, UK) was mounted to a microscope through a TuCam adapter with 2× magnification (Andor Technology). The camera was cooled down to −100 °C with the assistance of a liquid recirculating chiller Oasis 160 (Solid State Cooling Systems; Wappingers Falls, NY, USA).
All measurements were performed the 8-well CG imaging chambers (Zell Kontakt GmbH), and 3 to 9 randomly selected areas per well were captured as 16-bit OME-TIFF images at EM gain 300 with 100 ms exposure time. In case of ARCs, the maximal laser power of 15% was used; in case of genetically encoded proteins, the imaging settings were varied to enable optimal signal levels (the absence of detectable crosstalk signal in the channels was ensured). Live Acquisition v.2.07 (TILL Photonics/FEI) software autofocus was used to determine the focal plane, and multicolor Z-stacks (100 frames, with 200 nm piezo-focusing increment) were taken. The z-stack was deconvolved with EpiDEMIC plugin [74], the objects were segmented with Spot Detection plugin in an ICY platform [75], and the degree of multichannel co-localizations were estimated by Mander’s coefficients using Colocalization Studio [76]. Two independent experiments were performed.
4. Conclusions
We report the first high-resolution structures from co-crystals of PIM-1 protein and ARC inhibitors. The X-ray crystallographic study confirmed the bisubstrate binding of ARCs to PIM kinases [14], where the adenosine analogue and peptide mimetic moieties bind to the corresponding substrate pockets of PIM-1 (Figure 2). Considering the structural similarity among PIM kinases, ARCs are also bisubstrate inhibitors of PIM-2 and PIM-3 proteins (Table 1).
New ARC inhibitors were constructed by deleting structural elements that lacked interaction with PIM-1 based on the co-crystal structures. These simplified conjugates still retained low-nanomolar affinity and remarkable selectivity toward the kinases of the PIM family.
A biotinylated ARC inhibitor was tested as a kinase capture agent for a surface sandwich assay and used in combination with a PIM-2 detection antibody and secondary antibody labeled with Eu chelate for time-gated measurement of photoluminescence. This highly sensitive approach revealed an LoQ value of 44 pg recombinant PIM-2 protein in a biochemical assay. Further optimization of the assay for its use for analysis of PIM-2 in complex biological matrices is in progress.
ARCs labeled with Cy5 red fluorescent dye were efficiently taken up by live U2OS cells. Partial co-localization of ARC probes with the endosomal marker mCherry-Endo-14 was observed. In U2OS cells transiently transfected with PIM-1 fused with a fluorescent protein, a high extent of co-localization of PIM-1 with fluorescent ARC probes was evident.
Supplementary Materials
The following are available online; Supplementary methods: details of synthesis protocols, Figure S1: 2Fo-Fc electron density maps for the inhibitors in co-crystal structures with PIM-1, Figure S2: specificity of D1D2 antibody to PIM-2 detection, Figure S3: stability of ARC-1451/PIM-2 complex in PC-3 cell lysate, Figure S4: time- and concentration-dependence of the cellular uptake of compound 6 into live U2OS cells, Figure S5: comparison of cellular uptake of compound 8 (labelled with TAMRA) and compound 6 (labelled with Cy5) into live U2OS cells, Figure S6: intracellular localization of compounds labelled with Cy5 and incorporating different number of Arg residues, Figure S7: biochemical characterization of the synthesized compounds and commercially available inhibitors, Table S1: data collection and refinement statistics of PIM-1/ARC co-crystals, Table S2: structural formulae and HRMS data of the new compounds, Table S3: selectivity profiling of ARC inhibitors towards a panel of PKs, Table S4: HPLC purity data for the previously unpublished compounds, Video S1: colocalization studies of PIM-targeting compound 6 with fluorescently tagged PIM-1 vs. peroxisomal marker in live U2OS cells.
Author Contributions
Conceptualization, O.E.N., A.U.; Methodology, E.E., A.C., K.V., S.K. (Stefan Knapp); Validation, K.K., A.N.B.; Formal Analysis, O.E.N., K.K., A.C., S.E.D.-C., S.K. (Sergei Kopanchuk); Investigation, O.E.N., D.L., K.K., S.E.D.-C., S.K. (Sergei Kopanchuk), T.I., R.E., K.V.; Resources, A.C., A.N.B., S.K. (Sergei Kopanchuk), S.K. (Stefan Knapp), A.U.; Writing—Original Draft Preparation, O.E.N.; Writing—Review and Editing, D.L., K.K., A.C., A.N.B., S.K. (Sergei Kopanchuk), K.V., S.K. (Stefan Knapp), A.U.; Visualization, O.E.N., D.L., S.K. (Sergei Kopanchuk), S.K. (Stefan Knapp); Supervision, A.N.B., S.K. (Stefan Knapp), A.U.; Project Administration, A.U.; Funding Acquisition, A.C., A.N.B., S.K. (Sergei Kopanchuk), S.K. (Stefan Knapp), A.U. All authors have read and agreed to the published version of the manuscript.
Funding
The work was supported by the Estonian Research Council grants PRG454 and PSG230. O.E.N. was supported by the UT ASTRA project Graduate School “Functional materials and technologies” receiving funding from the European Regional Development Fund (OEN). S.E.D.-C. was supported by a NDM Prize Studentship, which was funded by the Medical Research Council and the Nuffield Department of Medicine, University of Oxford. S.Kn. and A.C. are grateful for support by the SGC, a registered charity (no. 1097737) that receives funds from AbbVie, BayerAG, Boehringer Ingelheim, the Canada Foundation for Innovation, Eshelman Institute for Innovation, Genentech, Genome Canada through Ontario Genomics Institute (OGI-196), EU/EFPIA/OICR/McGill/KTH/Diamond, Innovative Medicines Initiative 2 Joint Undertaking (EUbOPEN grant 875510), Janssen, Merck KGaA, Merck & Co, Pfizer, the São Paulo Research Foundation-FAPESP, Takeda and Wellcome (106169/ZZ14/Z). S.Kn. is also grateful for support by the German translational cancer network (DKTK) as well as the Frankfurt Cancer Institute (FCI).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The crystallographic data are available from https://www.rcsb.org/ (PDB codes: 7OOV for PIM-1-ARC-1411, 7OOW for PIM-1-ARC-1415, 7OOX for PIM-1-ARC-3126). Other data are available upon reasonable request to the corresponding author.
Acknowledgments
The authors would like to thank Diamond Light Source for beamtime (proposal mx8421), as well as the staff of beamlines IO2 and IO3 for assistance with crystal testing and data collection.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds 1–8 are available from the authors.
References
- Narlik-Grassow, M.; Blanco-Aparicio, C.; Carnero, A. The PIM Family of Serine/Threonine Kinases in Cancer. Med. Res. Rev. 2014, 34, 136–159. [Google Scholar] [CrossRef] [PubMed]
- Jinesh, G.G.; Mokkapati, S.; Zhu, K.; Morales, E.E. Pim kinase isoforms: Devils defending cancer cells from therapeutic and immune attacks. Apoptosis 2016, 21, 1203–1213. [Google Scholar] [CrossRef] [PubMed]
- Keane, N.A.; Reidy, M.; Natoni, A.; Raab, M.S.; O’Dwyer, M. Targeting the Pim kinases in multiple myeloma. Blood Cancer J. 2015, 5, e325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, P.D.; Langowski, J.L.; Wang, Y.; Chen, M.; Castillo, J.; Fanton, C.; Ison, M.; Zavorotinskaya, T.; Dai, Y.; Lu, J.; et al. Pan-PIM Kinase Inhibition Provides a Novel Therapy for Treating Hematologic Cancers. Clin. Cancer Res. 2014, 20, 1834–1845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano-Saenz, S.; Palacios, C.; Delgado-Bellido, D.; López-Jiménez, L.; Garcia-Diaz, A.; Soto-Serrano, Y.; Casal, J.I.; Bartolome, R.A.; Fernández-Luna, J.L.; López-Rivas, A.; et al. PIM kinases mediate resistance of glioblastoma cells to TRAIL by a p62/SQSTM1-dependent mechanism. Cell Death Dis. 2019, 10, 51. [Google Scholar] [CrossRef] [Green Version]
- Magnuson, N.S.; Wang, Z.; Ding, G.; Reeves, R. Why target PIM1 for cancer diagnosis and treatment? Future Oncol. 2010, 6, 1461–1478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roskoski, R. Properties of FDA-approved small molecule protein kinase inhibitors: A 2021 update. Pharmacol. Res. 2021, 165, 105463. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Murray, J.M.; Bussiere, D.E. Targeting the Purinome. Methods Mol. Biol. 2009, 575, 47–92. [Google Scholar] [CrossRef]
- Lavogina, D.; Enkvist, E.; Uri, A. Bisubstrate Inhibitors of Protein Kinases: From Principle to Practical Applications. Chem. Med. Chem. 2010, 5, 23–34. [Google Scholar] [CrossRef]
- Lamba, V.; Ghosh, I. New Directions in Targeting Protein Kinases: Focusing Upon True Allosteric and Bivalent Inhibitors. Curr. Pharm. Des. 2012, 18, 2936–2945. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kim, J.; Jo, J.; Chang, J.W.; Sim, J.; Yun, H. Recent advances in development of hetero-bivalent kinase inhibitors. Eur. J. Med. Chem. 2021, 216, 113318. [Google Scholar] [CrossRef] [PubMed]
- Ivan, T.; Enkvist, E.; Viira, B.; Manoharan, G.B.; Raidaru, G.; Pflug, A.; Alam, K.A.; Zaccolo, M.; Engh, R.A.; Uri, A. Bifunctional Ligands for Inhibition of Tight-Binding Protein–Protein Interactions. Bioconjugate Chem. 2016, 27, 1900–1910. [Google Scholar] [CrossRef]
- Ekambaram, R.; Enkvist, E.; Vaasa, A.; Kasari, M.; Raidaru, G.; Knapp, S.; Uri, A. Selective Bisubstrate Inhibitors with Sub-nanomolar Affinity for Protein Kinase Pim-1. Chem. Med. Chem. 2013, 8, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Kestav, K.; Lavogina, D.; Raidaru, G.; Chaikuad, A.; Knapp, S.; Uri, A. Bisubstrate Inhibitor Approach for Targeting Mitotic Kinase Haspin. Bioconjugate Chem. 2015, 26, 225–234. [Google Scholar] [CrossRef]
- Lavogina, D.; Kestav, K.; Chaikuad, A.; Heroven, C.; Knapp, S.; Uri, A. Co-crystal structures of the protein kinase haspin with bisubstrate inhibitors. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2016, 72, 339–345. [Google Scholar] [CrossRef] [Green Version]
- Vaasa, A.; Lust, M.; Terrin, A.; Uri, A.; Zaccolo, M. Small-molecule FRET probes for protein kinase activity monitoring in living cells. Biochem. Biophys. Res. Commun. 2010, 397, 750–755. [Google Scholar] [CrossRef]
- Enkvist, E.; Lavogina, D.; Raidaru, G.; Vaasa, A.; Viil, I.; Lust, M.; Viht, K.; Uri, A. Conjugation of Adenosine and Hexa-(d-arginine) Leads to a Nanomolar Bisubstrate-Analog Inhibitor of Basophilic Protein Kinases. J. Med. Chem. 2006, 49, 7150–7159. [Google Scholar] [CrossRef]
- Ramos-Molina, B.; Lick, A.N.; Shirazi, A.N.; Oh, D.; Tiwari, R.; El-Sayed, N.S.; Parang, K.; Lindberg, I. Cationic Cell-Penetrating Peptides Are Potent Furin Inhibitors. PLoS ONE 2015, 10, e0130417. [Google Scholar] [CrossRef] [Green Version]
- DeRouchey, J.; Hoover, B.; Rau, D.C. A Comparison of DNA Compaction by Arginine and Lysine Peptides: A Physical Basis for Arginine Rich Protamines. Biochemistry 2013, 52, 3000–3009. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.-H.; Zhang, P.; Kim, H.J.; Mitrea, D.M.; Sarkar, M.; Freibaum, B.D.; Cika, J.; Coughlin, M.; Messing, J.; Molliex, A.; et al. C9orf72 Dipeptide Repeats Impair the Assembly, Dynamics, and Function of Membrane-Less Organelles. Cell 2016, 167, 774–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chico, D.E.; Given, R.L.; Miller, B.T. Binding of cationic cell-permeable peptides to plastic and glass. Peptides 2003, 24, 3–9. [Google Scholar] [CrossRef]
- Enkvist, E.; Kriisa, M.; Roben, M.; Kadak, G.; Raidaru, G.; Uri, A. Effect of the structure of adenosine mimic of bisubstrate-analog inhibitors on their activity towards basophilic protein kinases. Bioorganic Med. Chem. Lett. 2009, 19, 6098–6101. [Google Scholar] [CrossRef]
- De Antoni, A.; Maffini, S.; Knapp, S.; Musacchio, A.; Santaguida, S. A small-molecule inhibitor of Haspin alters the kinetochore functions of Aurora B. J. Cell Biol. 2012, 199, 269–284. [Google Scholar] [CrossRef] [Green Version]
- Pogacic, V.; Bullock, A.N.; Fedorov, O.; Filippakopoulos, P.; Gasser, C.; Biondi, A.; Meyer-Monard, S.; Knapp, S.; Schwaller, J. Structural Analysis Identifies Imidazo[1,2-b]Pyridazines as PIM Kinase Inhibitors with In vitro Antileukemic Activity. Cancer Res. 2007, 67, 6916–6924. [Google Scholar] [CrossRef] [Green Version]
- Pietsch, M.; Viht, K.; Schnitzler, A.; Ekambaram, R.; Steinkrüger, M.; Enkvist, E.; Nienberg, C.; Nickelsen, A.; Lauwers, M.; Jose, J.; et al. Unexpected CK2β-antagonistic functionality of bisubstrate inhibitors targeting protein kinase CK2. Bioorganic Chem. 2020, 96, 103608. [Google Scholar] [CrossRef]
- Bullock, A.N.; Debreczeni, J.; Amos, A.L.; Knapp, S.; Turk, B.E. Structure and Substrate Specificity of the Pim-1 Kinase. J. Biol. Chem. 2005, 280, 41675–41682. [Google Scholar] [CrossRef] [Green Version]
- Bullock, A.N.; Debreczeni, J.É.; Fedorov, O.Y.; Nelson, A.; Marsden, B.D.; Knapp, S. Structural Basis of Inhibitor Specificity of the Human Protooncogene Proviral Insertion Site in Moloney Murine Leukemia Virus (PIM-1) Kinase. J. Med. Chem. 2005, 48, 7604–7614. [Google Scholar] [CrossRef]
- Schröder, M.; Bullock, A.N.; Fedorov, O.; Bracher, F.; Chaikuad, A.; Knapp, S. DFG-1 Residue Controls Inhibitor Binding Mode and Affinity, Providing a Basis for Rational Design of Kinase Inhibitor Selectivity. J. Med. Chem. 2020, 63, 10224–10234. [Google Scholar] [CrossRef]
- Tao, Z.-F.; Hasvold, L.A.; Leverson, J.D.; Han, E.K.; Guan, R.; Johnson, E.F.; Stoll, V.S.; Stewart, K.D.; Stamper, G.; Soni, N.; et al. Discovery of 3H-Benzo[4,5]thieno[3,2-d]pyrimidin-4-ones as Potent, Highly Selective, and Orally Bioavailable Inhibitors of the Human Protooncogene Proviral Insertion Site in Moloney Murine Leukemia Virus (PIM) Kinases. J. Med. Chem. 2009, 52, 6621–6636. [Google Scholar] [CrossRef] [PubMed]
- Tsuganezawa, K.; Watanabe, H.; Parker, L.; Yuki, H.; Taruya, S.; Nakagawa, Y.; Kamei, D.; Mori, M.; Ogawa, N.; Tomabechi, Y.; et al. A Novel Pim-1 Kinase Inhibitor Targeting Residues That Bind the Substrate Peptide. J. Mol. Biol. 2012, 417, 240–252. [Google Scholar] [CrossRef]
- Vaasa, A.; Viil, I.; Enkvist, E.; Viht, K.; Raidaru, G.; Lavogina, D.; Uri, A. High-affinity bisubstrate probe for fluorescence anisotropy binding/displacement assays with protein kinases PKA and ROCK. Anal. Biochem. 2009, 385, 85–93. [Google Scholar] [CrossRef]
- Kasari, M.; Ligi, K.; Williams, J.G.; Vaasa, A.; Enkvist, E.; Viht, K.; Pålsson, L.-O.; Uri, A. Responsive microsecond-lifetime photoluminescent probes for analysis of protein kinases and their inhibitors. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2013, 1834, 1330–1335. [Google Scholar] [CrossRef]
- Introduction to Fluorescence. In Principles of Fluorescence Spectroscopy; Lakowicz, J.R. (Ed.) Springer US: Boston, MA, USA, 2006; pp. 1–26. ISBN 978-0-387-31278-1. [Google Scholar]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The Protein Kinase Complement of the Human Genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [Green Version]
- Rahnel, H.; Viht, K.; Lavogina, D.; Mazina, O.; Haljasorg, T.; Enkvist, E.; Uri, A. A Selective Biligand Inhibitor of CK2 Increases Caspase-3 Activity in Cancer Cells and Inhibits Platelet Aggregation. Chem. Med. Chem. 2017, 12, 1723–1736. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, R.S.K.; He, P.; Modi, V.; Duong-Ly, K.C.; Ma, H.; Peterson, J.R.; Dunbrack, R.L.; Levy, R.M. Conformational Analysis of the DFG-Out Kinase Motif and Biochemical Profiling of Structurally Validated Type II Inhibitors. J. Med. Chem. 2015, 58, 466–479. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Xiu, J.; Ren, C.; Yu, Z. Protein kinase PIM2: A simple PIM family kinase with complex functions in cancer metabolism and therapeutics. J. Cancer 2021, 12, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Manoharan, G.B.; Enkvist, E.; Uri, A. Combining chemical and genetic approaches for development of responsive FRET-based sensor systems for protein kinases. Biophys. Chem. 2016, 211, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Douglass, E.F.; Miller, C.J.; Sparer, G.; Shapiro, H.; Spiegel, D.A. A Comprehensive Mathematical Model for Three-Body Binding Equilibria. J. Am. Chem. Soc. 2013, 135, 6092–6099. [Google Scholar] [CrossRef] [Green Version]
- Navrátil, V.; Schimer, J.; Tykvart, J.; Knedlík, T.; Vik, V.; Majer, P.; Konvalinka, J.; Šácha, P. DNA-linked Inhibitor Antibody Assay (DIANA) for sensitive and selective enzyme detection and inhibitor screening. Nucleic Acids Res. 2016, 45, e10. [Google Scholar] [CrossRef] [Green Version]
- Seo, H.B.; Gu, M.B. Aptamer-based sandwich-type biosensors. J. Biol. Eng. 2017, 11, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Xu, G.; Zhang, L.; Zhao, J.; Ji, P.; Li, Y.; Liu, B.; Zhang, J.; Zhao, Q.; Sun, Y.; et al. Development of a double monoclonal antibody–based sandwich enzyme-linked immunosorbent assay for detecting canine distemper virus. Appl. Microbiol. Biotechnol. 2020, 104, 10725–10735. [Google Scholar] [CrossRef]
- Santio, N.M.; Landor, S.K.-J.; Vahtera, L.; Ylä-Pelto, J.; Paloniemi, E.; Imanishi, S.Y.; Corthals, G.; Varjosalo, M.; Manoharan, G.B.; Uri, A.; et al. Phosphorylation of Notch1 by Pim kinases promotes oncogenic signaling in breast and prostate cancer cells. Oncotarget 2016, 7, 43220–43238. [Google Scholar] [CrossRef] [Green Version]
- Lemke, E.A.; Schultz, C. Principles for designing fluorescent sensors and reporters. Nat. Chem. Biol. 2011, 7, 480–483. [Google Scholar] [CrossRef]
- Letribot, B.; Akué-Gédu, R.; Santio, N.M.; El-Ghozzi, M.; Avignant, D.; Cisnetti, F.; Koskinen, F.J.; Gautier, A.; Anizon, F.; Moreau, P. Use of copper(I) catalyzed azide alkyne cycloaddition (CuAAC) for the preparation of conjugated pyrrolo[2,3-a]carbazole Pim kinase inhibitors. Eur. J. Med. Chem. 2012, 50, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Fan, J.; Wang, B.; Xiao, M.; Li, Y.; Du, J.; Peng, X. Highly Selective Red-Emitting Fluorescent Probe for Imaging Cancer Cells in Situ by Targeting Pim-1 Kinase. ACS Appl. Mater. Interfaces 2018, 10, 1499–1507. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Cao, Z.; Yuan, Y.; Li, W.; Cui, X.; Chen, Z.; Hu, X.; Yu, A. Design and synthesis of a novel mitochondria-targeted osteosarcoma theranostic agent based on a PIM1 kinase inhibitor. J. Control. Release 2021, 332, 434–447. [Google Scholar] [CrossRef] [PubMed]
- Uri, A.; Raidaru, G.; Subbi, J.; Padari, K.; Pooga, M. Identification of the ability of highly charged nanomolar inhibitors of protein kinases to cross plasma membranes and carry a protein into cells. Bioorganic Med. Chem. Lett. 2002, 12, 2117–2120. [Google Scholar] [CrossRef]
- Heitz, F.; Morris, M.C.; Divita, G. Twenty years of cell-penetrating peptides: From molecular mechanisms to therapeutics. Br. J. Pharmacol. 2009, 157, 195–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamoto, K.; Morishita, T.; Aburai, K.; Sakai, K.; Abe, M.; Nakase, I.; Futaki, S.; Sakai, H. Key Process and Factors Controlling the Direct Translocation of Cell-Penetrating Peptide through Bio-Membrane. Int. J. Mol. Sci. 2020, 21, 5466. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, T.; Futaki, S. Current Understanding of Direct Translocation of Arginine-Rich Cell-Penetrating Peptides and Its Internalization Mechanisms. Chem. Pharm. Bull. 2016, 64, 1431–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lecher, J.C.; Nowak, S.J.; McMurry, J.L. Breaking in and busting out: Cell-penetrating peptides and the endosomal escape problem. Biomol. Concepts 2017, 8, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Palušová, V.; Renzová, T.; Verlande, A.; Vaclová, T.; Medková, M.; Cetlová, L.; Sedláčková, M.; Hříbková, H.; Slaninová, I.; Krutá, M.; et al. Dual Targeting of BRAF and mTOR Signaling in Melanoma Cells with Pyridinyl Imidazole Compounds. Cancers 2020, 12, 1516. [Google Scholar] [CrossRef]
- Eerola, S.K.; Santio, N.M.; Rinne, S.; Kouvonen, P.; Corthals, G.L.; Scaravilli, M.; Scala, G.; Serra, A.; Greco, D.; Ruusuvuori, P.; et al. Phosphorylation of NFATC1 at PIM1 target sites is essential for its ability to promote prostate cancer cell migration and invasion. Cell Commun. Signal. 2019, 17, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Santio, N.M.; Vainio, V.; Hoikkala, T.; Mung, K.L.; Lång, M.; Vahakoski, R.; Zdrojewska, J.; Coffey, E.T.; Kremneva, E.; Rainio, E.; et al. PIM1 accelerates prostate cancer cell motility by phosphorylating actin capping proteins. Cell Commun. Signal. 2020, 18, 1–18. [Google Scholar] [CrossRef]
- Enkvist, E.; Vaasa, A.; Kasari, M.; Kriisa, M.; Ivan, T.; Ligi, K.; Raidaru, G.; Uri, A. Protein-Induced Long Lifetime Luminescence of Nonmetal Probes. ACS Chem. Biol. 2011, 6, 1052–1062. [Google Scholar] [CrossRef]
- Harrington, L.; Cheley, S.; Alexander, L.T.; Knapp, S.; Bayley, H. Stochastic detection of Pim protein kinases reveals electrostatically enhanced association of a peptide substrate. Proc. Natl. Acad. Sci. USA 2013, 110, E4417–E4426. [Google Scholar] [CrossRef] [Green Version]
- Ekambaram, R.; babu Manoharan, G.; Enkvist, E.; Ligi, K.; Knapp, S.; Uri, A. PIM kinase-responsive microsecond-lifetime photoluminescent probes based on selenium-containing heteroaromatic tricycle. RSC Adv. 2015, 5, 96750–96757. [Google Scholar] [CrossRef]
- Kuipers, B.J.H.; Gruppen, H. Prediction of Molar Extinction Coefficients of Proteins and Peptides Using UV Absorption of the Constituent Amino Acids at 214 nm To Enable Quantitative Reverse Phase High-Performance Liquid Chromatography−Mass Spectrometry Analysis. J. Agric. Food Chem. 2007, 55, 5445–5451. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, O.; Niesen, F.H.; Knapp, S. Kinase Inhibitor Selectivity Profiling Using Differential Scanning Fluorimetry. Methods Mol. Biol. 2012, 795, 109–118. [Google Scholar] [CrossRef]
- Kabsch, W. XDS. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Evans, P.R.; Murshudov, G.N. How good are my data and what is the resolution? Acta Crystallogr. Sect. D Biol. Crystallogr. 2013, 69, 1204–1214. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phasercrystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [Green Version]
- Fedorov, O.; Marsden, B.; Pogacic, V.; Rellos, P.; Müller, S.; Bullock, A.N.; Schwaller, J.; Sundström, M.; Knapp, S. A systematic interaction map of validated kinase inhibitors with Ser/Thr kinases. Proc. Natl. Acad. Sci. USA 2007, 104, 20523–20528. [Google Scholar] [CrossRef] [Green Version]
- Debreczeni, J.E.; Bullock, A.N.; von Delft, F.; Sundstrom, M.; Arrowsmith, C.; Edwards, A.; Weigelt, J.; Knapp, S. RCSB PDB-2J2I: Crystal Structure of the Humab PIM1 in Complex with LY333531. Available online: https://www.rcsb.org/structure/2J2I (accessed on 30 May 2021).
- Emsley, P.; Crispin, M. Structural analysis of glycoproteins: Building N-linked glycans withCoot. Acta Crystallogr. Sect. D Struct. Biol. 2018, 74, 256–263. [Google Scholar] [CrossRef] [Green Version]
- Kovalevskiy, O.; Nicholls, R.A.; Long, F.; Carlon, A.; Murshudov, G.N. Overview of refinement procedures withinREFMAC5: Utilizing data from different sources. Acta Crystallogr. Sect. D Struct. Biol. 2018, 74, 215–227. [Google Scholar] [CrossRef] [Green Version]
- International Centre for Kinase Profiling. Available online: http://www.kinase-screen.mrc.ac.uk/services/premier-screen (accessed on 17 July 2021).
- Ursu, A.; Childs-Disney, J.L.; AngelBello, A.J.; Costales, M.G.; Meyer, S.M.; Disney, M.D. Gini Coefficients as a Single Value Metric to Define Chemical Probe Selectivity. ACS Chem. Biol. 2020, 15, 2031–2040. [Google Scholar] [CrossRef]
- Belgrader, P.; Hansford, D.; Kovacs, G.T.; Venkateswaran, K.; Mariella, R.; Milanovich, F.; Nasarabadi, S.; Okuzumi, M.; Pourahmadi, F.; Northrup, M.A. A Minisonicator To Rapidly Disrupt Bacterial Spores for DNA Analysis. Anal. Chem. 1999, 71, 4232–4236. [Google Scholar] [CrossRef] [PubMed]
- Laasfeld, T.; Ehrminger, R.; Tahk, M.-J.; Veiksina, S.; Kõlvart, K.R.; Min, M.; Kopanchuk, S.; Rinken, A. Budded baculoviruses as a receptor display system to quantify ligand binding with TIRF microscopy. Nanoscale 2021, 13, 2436–2447. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, M.; Imamoto, N.; Sakata-Sogawa, K. Highly inclined thin illumination enables clear single-molecule imaging in cells. Nat. Methods 2008, 5, 159–161. [Google Scholar] [CrossRef] [PubMed]
- Soulez, F.; Denis, L.; Tourneur, Y.; Thiebaut, E. Blind deconvolution of 3D data in wide field fluorescence microscopy. In Proceedings of the 2012 9th IEEE International Symposium on Biomedical Imaging (ISBI), Barcelona, Spain, 30 April–5 May 2012; pp. 1735–1738. [Google Scholar]
- De Chaumont, F.; Dallongeville, S.; Chenouard, N.; Hervé, N.; Pop, S.; Provoost, T.; Meas-Yedid, V.; Pankajakshan, P.; LeComte, T.; Le Montagner, Y.; et al. Icy: An open bioimage informatics platform for extended reproducible research. Nat. Methods 2012, 9, 690–696. [Google Scholar] [CrossRef] [PubMed]
- Lagache, T.; Sauvonnet, N.; Danglot, L.; Olivo-Marin, J.-C. Statistical analysis of molecule colocalization in bioimaging. Cytom. Part A 2015, 87, 568–579. [Google Scholar] [CrossRef] [PubMed]
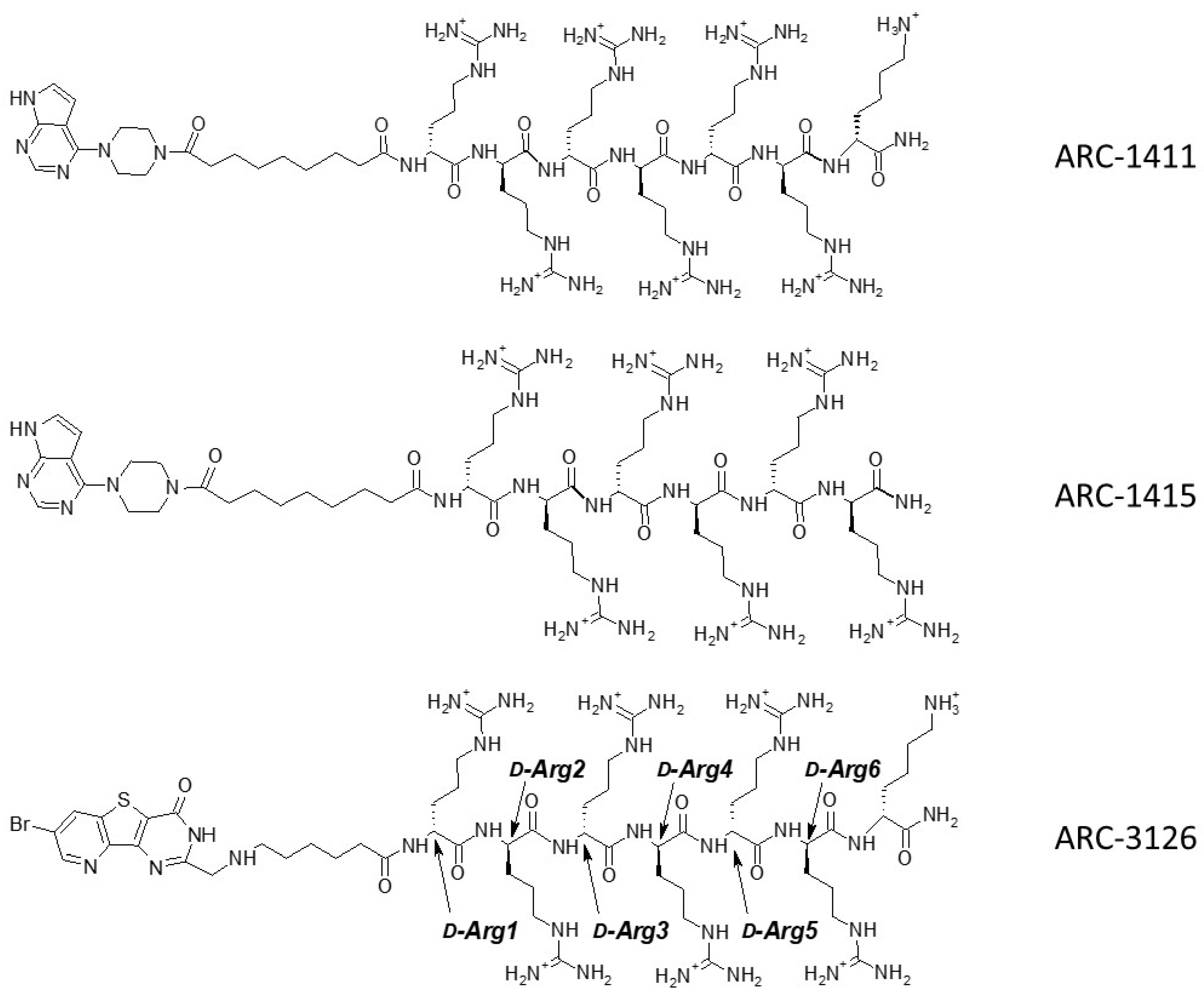
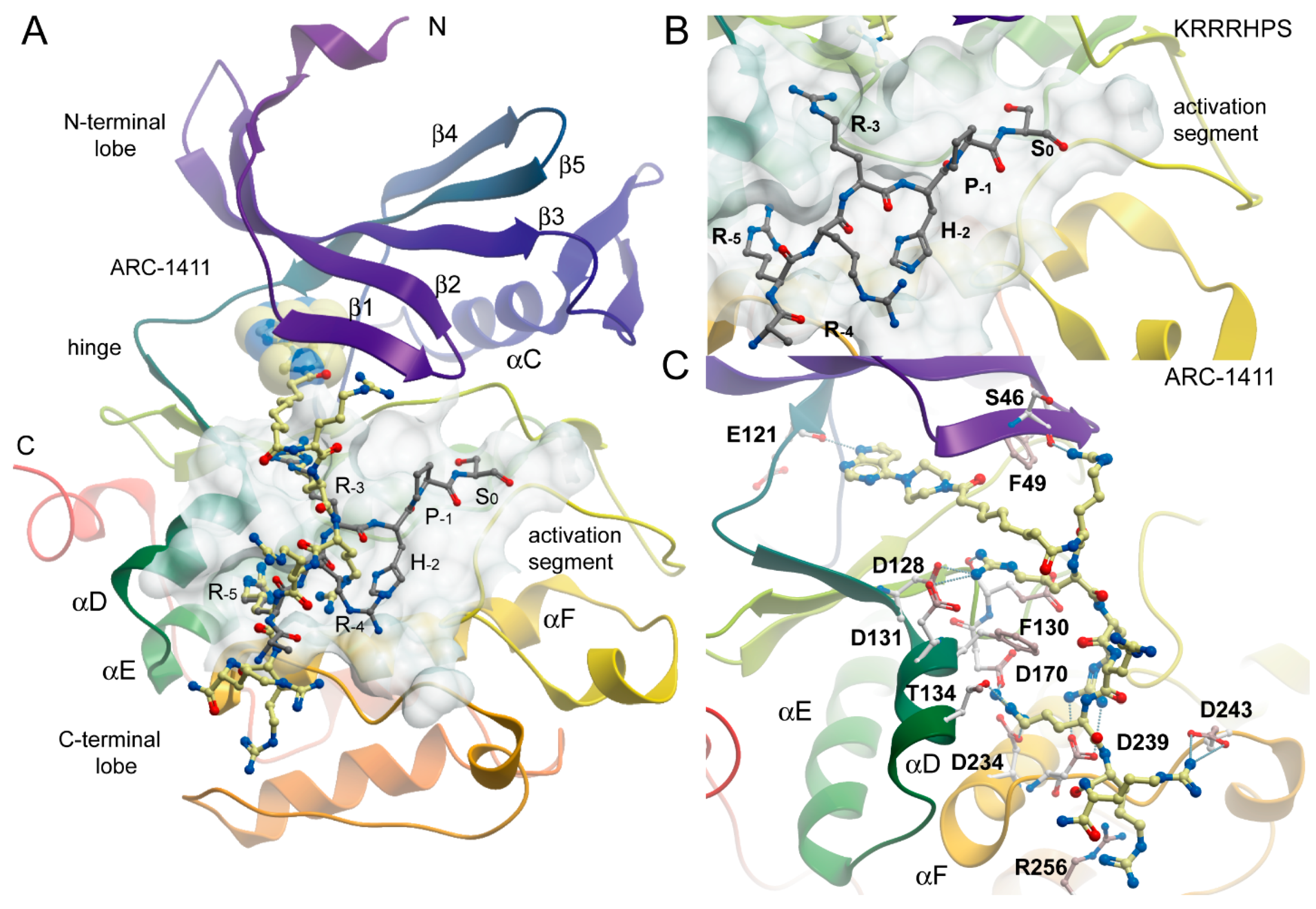
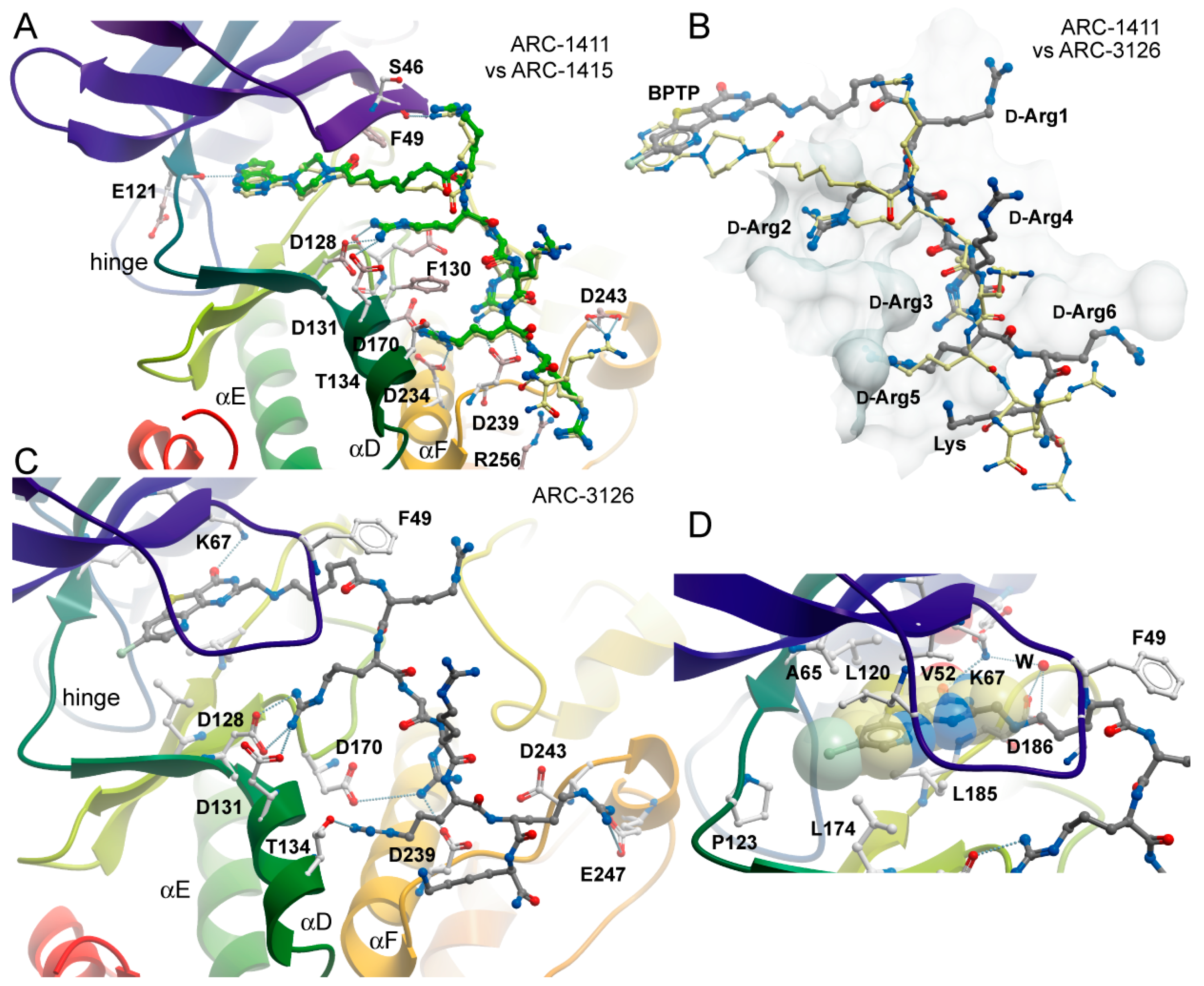
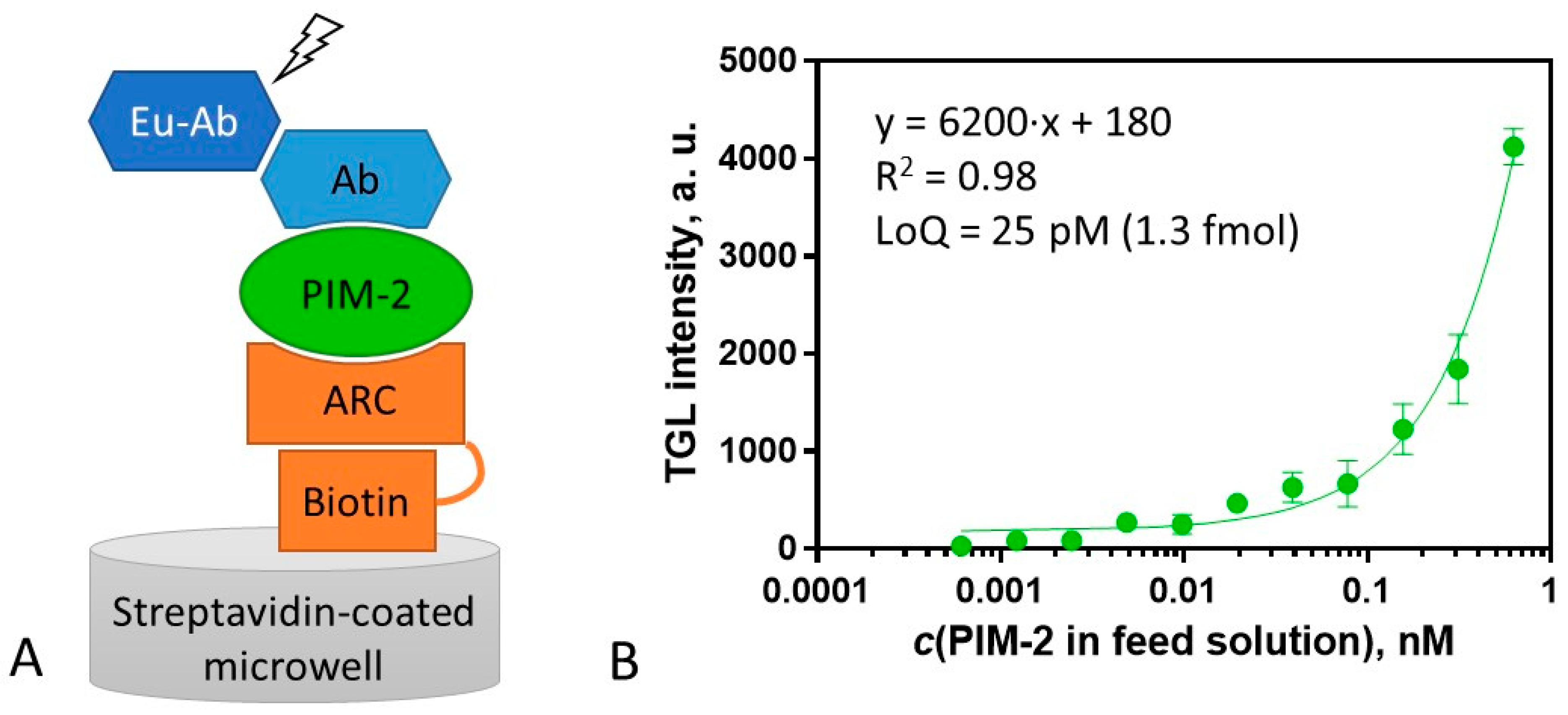
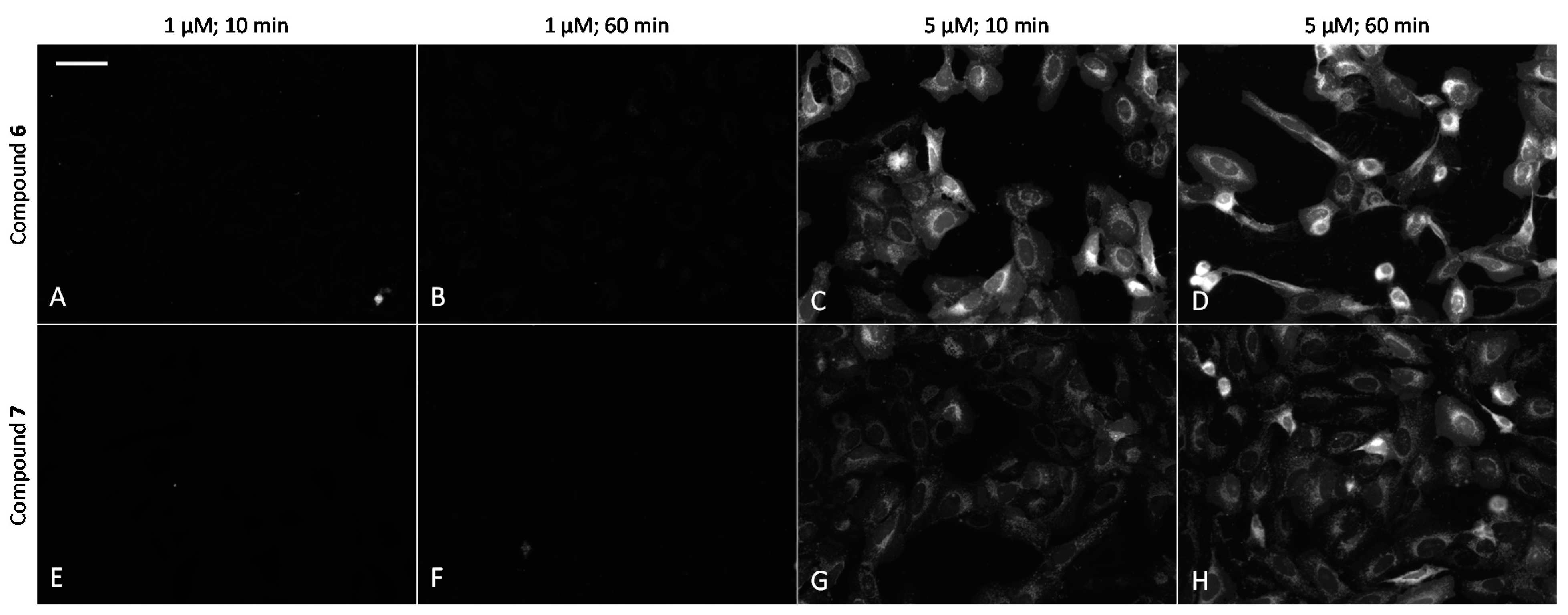
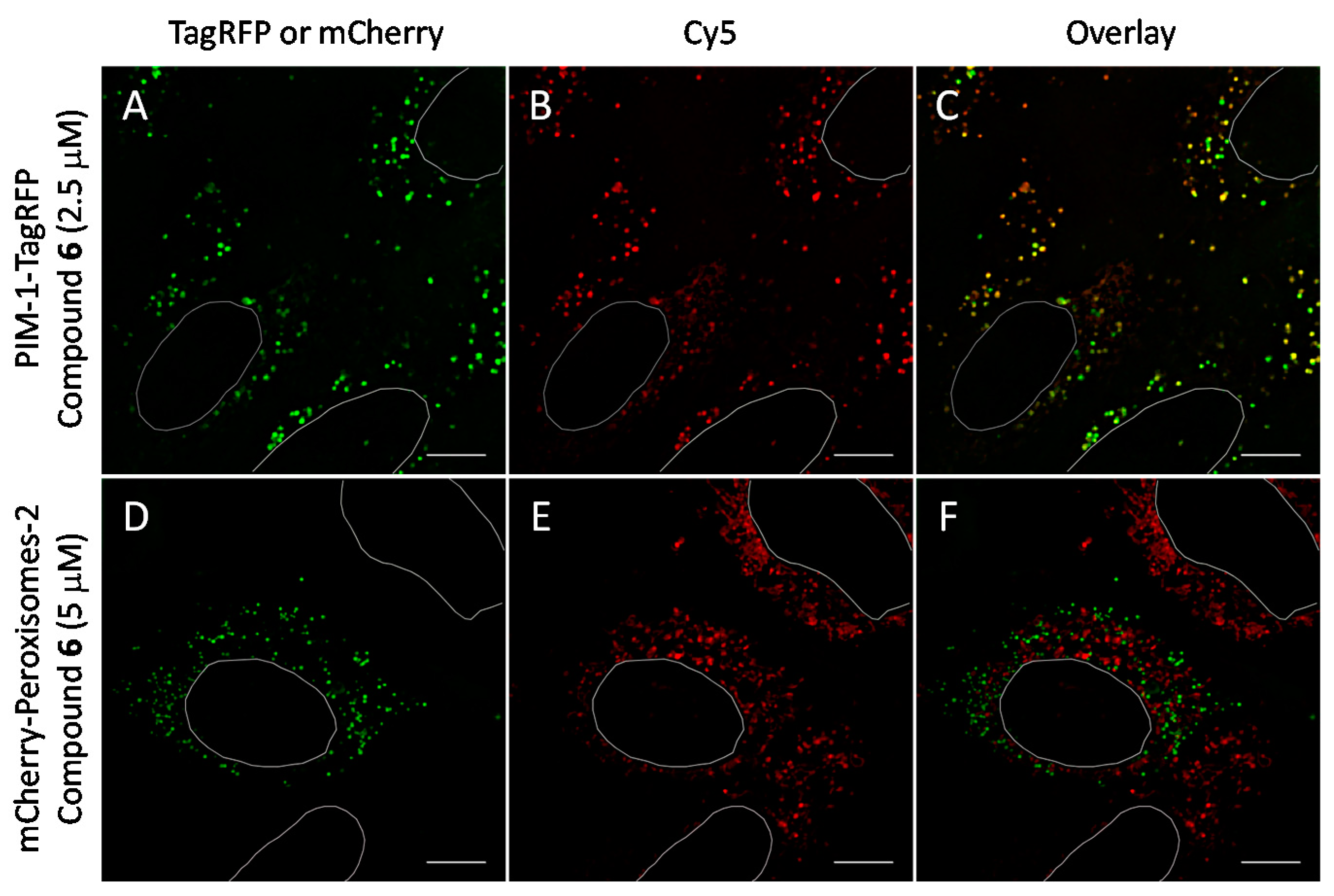
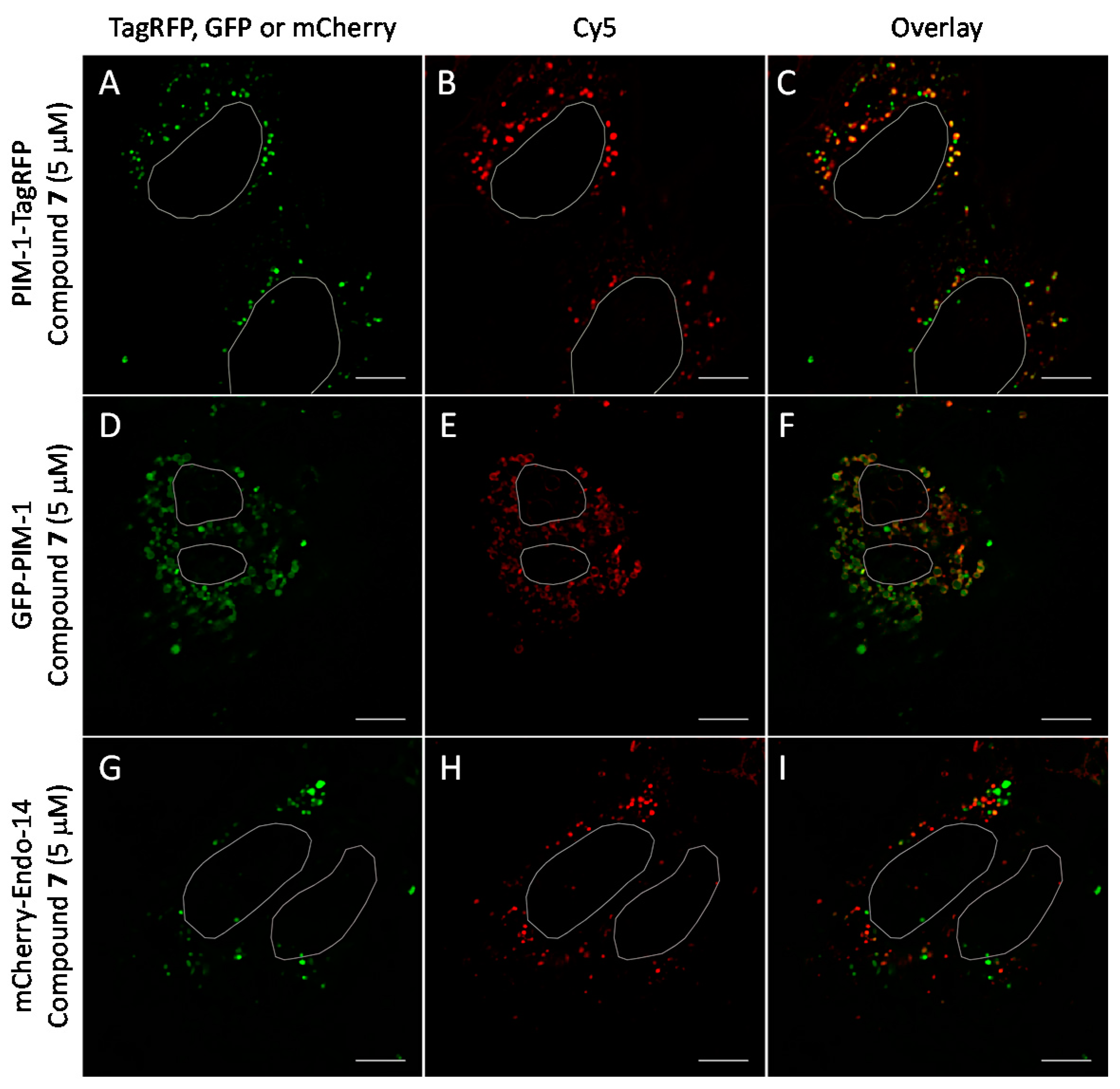
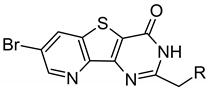
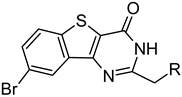
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).