
Simplified and Rapid Determination of Primaquine and 5,6-Orthoquinone Primaquine by UHPLC-MS/MS: Its Application to a Pharmacokinetic Study

 , and
, and
Abstract
1. Introduction
2. Material and Methods
2.1. Chemicals
2.2. Instrumentation and Chromatographic Conditions
2.3. Standard Stock Solutions Preparation
2.4. Method Validation
2.5. Application of Method
2.5.1. Subjects and Sample Collection
2.5.2. Sample Preparation
2.5.3. Pharmacokinetic Parameters Analysis
3. Results
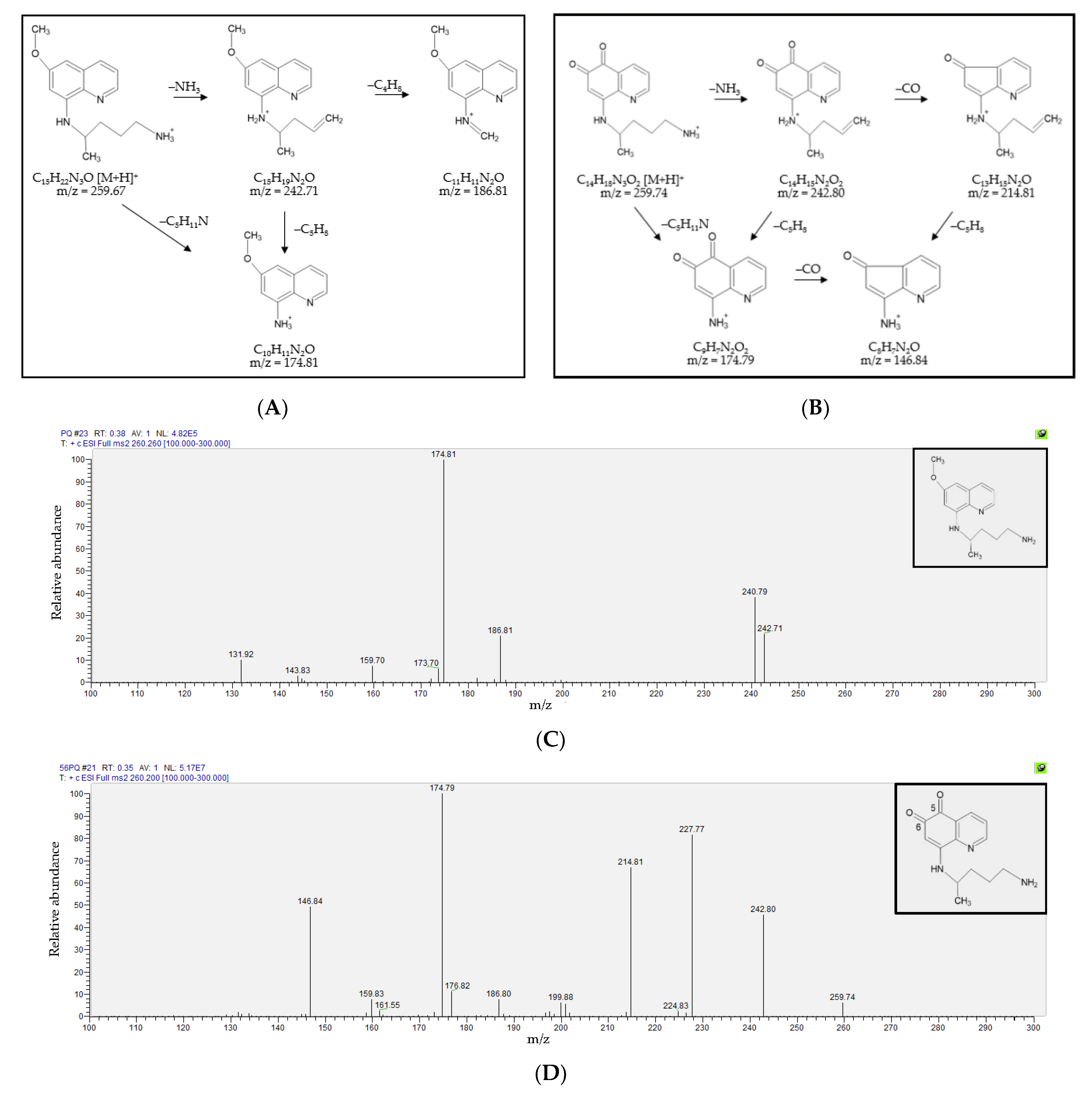
3.1. Fragmentation Patterns and MS/MS Spectra of PQ and 5,6-PQ
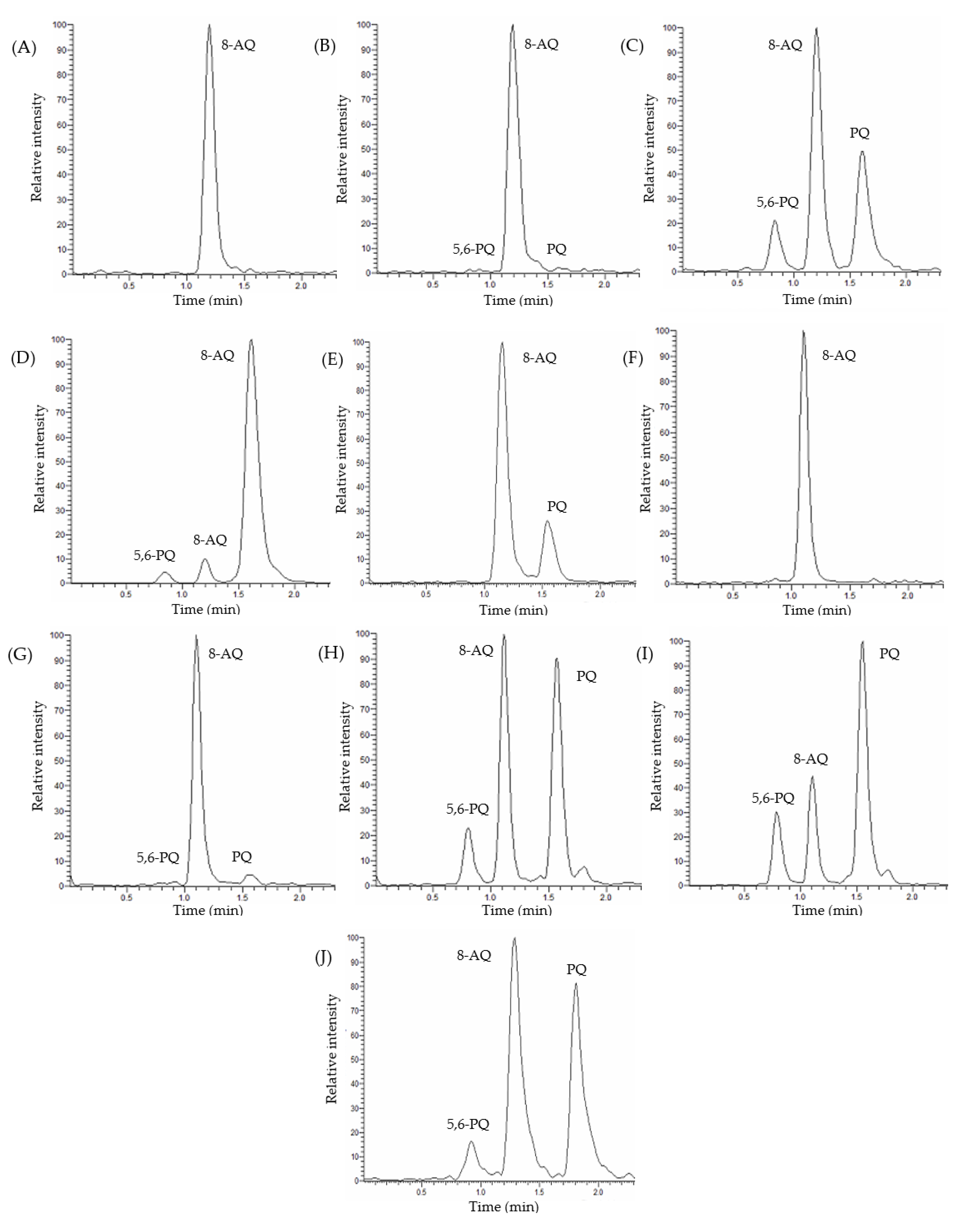
3.2. Detection and Quantification of Primaquine, 5,6-Orthoquinone Primaquine and 8-Aminoquinoline
3.3. Method Validation
3.3.1. Selectivity and Specificity
3.3.2. Linearity and Sensitivity
3.3.3. Matrix Factor and Recovery
3.3.4. Accuracy and Precision
3.3.5. Stability
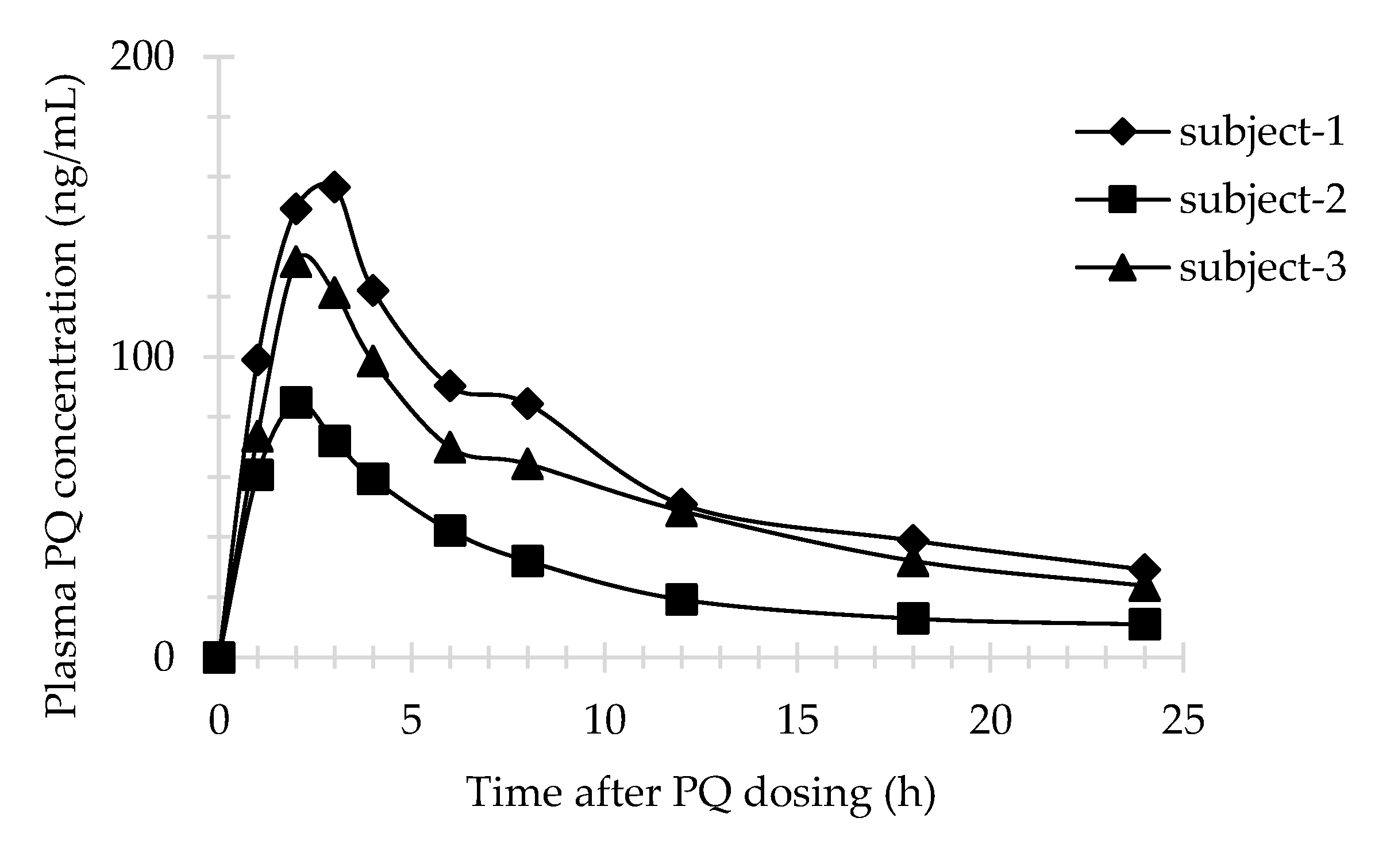
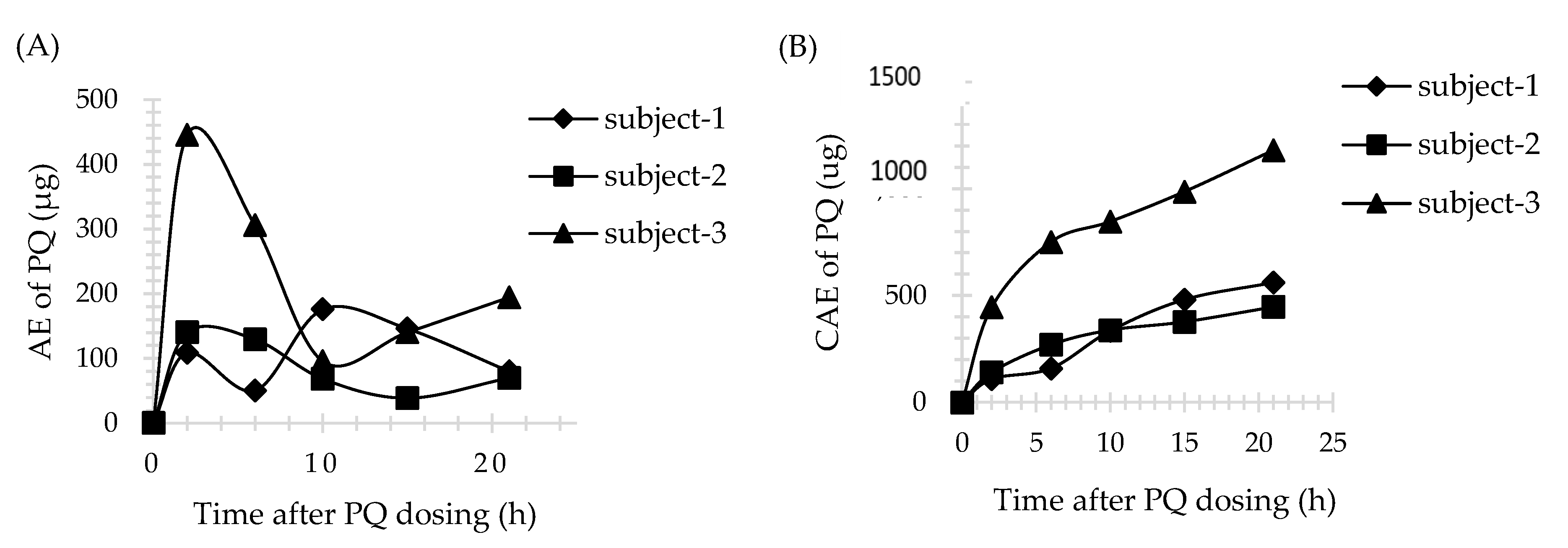
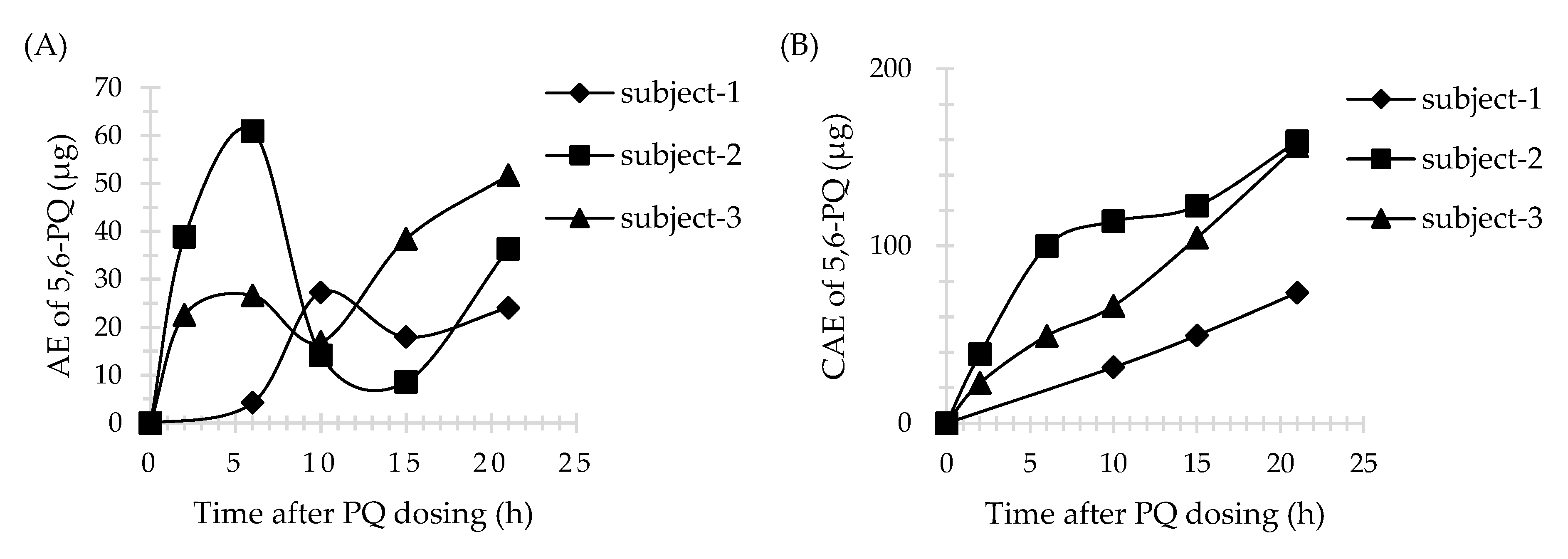
3.4. Application of Method
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- World Health Organization. WHO Guidelines for Malaria; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- Vinetz, J.M.; Clain, J.; Bounkeua, V.; Eastman, R.T.; Fidock, D. Chemotherapy of Malaria. In Goodman & Gilman’s the Pharmacological Basis of Therapeutics, 12th ed.; Brunton, L.L., Chabner, B.A., Knollmann, B.C., Eds.; McGraw-Hill: New York, NY, USA, 2011; pp. 1259–1282. [Google Scholar]
- Potter, B.M.; Xie, L.H.; Vuong, C.; Zhang, J.; Zhang, P.; Duan, D.; Luong, T.T.; Bandara Herath, H.M.; Dhammika Nanayakkara, N.P.; Tekwani, B.L.; et al. Differential CYP 2D6 metabolism alters primaquine pharmacokinetics. Antimicrob. Agents Chemother. 2015, 59, 2380–2387. [Google Scholar] [CrossRef] [PubMed]
- Constantino, L.; Paixão, P.; Moreira, R.; Portela, M.; Rosario, V.D.; Iley, J. Metabolism of primaquine by liver homogenate fractions: Evidence for monoamine oxidase and cytochrome P450 involvement in the oxidative deamination of primaquine to carboxyprimaquine. Exp. Toxicol. Pathol. 1999, 51, 299–303. [Google Scholar] [CrossRef]
- Pybus, B.S.; Marcsisin, S.R.; Jin, X.; Deye, G.; Sousa, J.C.; Li, Q.; Caridha, D.; Zeng, Q.A.; Reichard, G.; Ockenhouse, C.; et al. The metabolism of primaquine to its active metabolite is dependent on CYP 2D6. Malar. J. 2013, 12, 212. [Google Scholar] [CrossRef] [PubMed]
- Marcsisin, S.R.; Reichard, G.; Pybus, B.S. Primaquine pharmacology in the context of CYP 2D6 pharmacogenomics: Current state of the art. Pharmacol. Ther. 2016, 161, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Pybus, B.S.; Sousa, J.C.; Jin, X.A.; Ferguson, J.E.; Christian, R.; Barnhart, R.; Vuong, C.; Sciotti, R.J.A.; Reichard, G.; Kozar, M.P.; et al. CYP450 phenotyping and accurate mass identification of metabolites of the 8-aminoquinoline, anti-malarial drug primaquine. Malar. J. 2012, 11, 259. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Potter, B.; Luong, T.-L.; Nelson, J.; Vuong, C.; Potter, C.; Xie, L.; Zhang, J.; Zhang, P.; Sousa, J.; et al. Pre-clinical evaluation of CYP 2D6 dependent drug–drug interactions between primaquine and SSRI/SNRI antidepressants. Malar. J. 2016, 15, 280. [Google Scholar] [CrossRef]
- Ingram, R.J.H.; Crenna-Darusallam, C.; Soebianto, S.; Noviyanti, R.; Baird, J.K. The clinical and public health problem of relapse despite primaquine therapy: Case review of repeated relapses of Plasmodium vivax acquired in Papua New Guinea. Malar. J. 2014, 13, 488. [Google Scholar] [CrossRef]
- Silvino, A.C.R.; Costa, G.L.; De Araújo, F.C.F.; Ascher, D.; Pires, D.; Fontes, C.; Carvalho, L.H.; De Brito, C.F.A.; Sousa, T.N. Variation in Human Cytochrome P-450 Drug-Metabolism Genes: A Gateway to the Understanding of Plasmodium vivax Relapses. PLoS ONE 2016, 11, e0160172. [Google Scholar] [CrossRef] [PubMed]
- Brasil, L.W.; Rodrigues-Soares, F.; Santoro, A.B.; Almeida, A.C.G.; Kühn, A.; Ramasawmy, R.; Lacerda, M.V.G.; Monteiro, W.M.; Suarez-Kurtz, G. CYP2D6 activity and the risk of recurrence of Plasmodium vivax malaria in the Brazilian Amazon: A prospective cohort study. Malar. J. 2018, 17, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, B.P.; Pett, H.; Tiono, A.B.; Murry, D.; Sirima, S.B.; Niemi, M.; Bousema, T.; Drakeley, C.; Ter Heine, R. Age, Weight, and CYP2D6 Genotype Are Major Determinants of Primaquine Pharmacokinetics in African Children. Antimicrob. Agents Chemother. 2017, 61, e02590-16. [Google Scholar] [CrossRef] [PubMed]
- Tekwani, B.L.; Avula, B.; Sahu, R.; Chaurasiya, N.D.; Khan, S.I.; Jain, S.; Fasinu, P.S.; Herath, H.B.; Stanford, D.; Nanayakkara, N.D.; et al. Enantioselective Pharmacokinetics of Primaquine in Healthy Human Volunteers. Drug Metab. Dispos. 2015, 43, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Avula, B.; Tekwani, B.L.; Chaurasiya, N.D.; Fasinu, P.; Nanayakkara, N.P.D.; Herath, H.M.T.B.; Wang, Y.-H.; Bae, J.-Y.; Khan, S.I.; ElSohly, M.A.; et al. Metabolism of primaquine in normal human volunteers: Investigation of phase I and phase II metabolites from plasma and urine using ultra-high performance liquid chromatography-quadrupole time-of-flight mass spectrometry. Malar. J. 2018, 17, 294. [Google Scholar] [CrossRef] [PubMed]
- Spring, M.D.; Sousa, J.C.; Li, Q.; A Darko, C.; Morrison, M.N.; Marcsisin, S.R.; Mills, K.T.; Potter, B.M.; Paolino, K.M.; Twomey, P.S.; et al. Determination of Cytochrome P450 Isoenzyme 2D6 (CYP2D6) Genotypes and Pharmacogenomic Impact on Primaquine Metabolism in an Active-Duty US Military Population. J. Infect. Dis. 2019, 220, 1761–1770. [Google Scholar] [CrossRef] [PubMed]
- Avula, B.; Khan, S.I.; Tekwani, B.L.; Nanayakkara, N.D.; McChesney, J.D.; Walker, L.A.; Khan, I.A. Analysis of primaquine and its metabolite carboxyprimaquine in biological samples: Enantiomeric separation, method validation and quantification. Biomed. Chromatogr. 2010, 25, 1010–1017. [Google Scholar] [CrossRef] [PubMed]
- Page-Sharp, M.; Ilett, K.F.; Betuela, I.; Davis, T.M.; Batty, K.T. Simultaneous determination of primaquine and carboxyprimaquine in plasma using solid phase extraction and LC–MS assay. J. Chromatogr. B 2012, 902, 142–146. [Google Scholar] [CrossRef] [PubMed]
- Khan, W.; Wang, Y.-H.; Nanayakkara, N.D.; Herath, H.B.; Catchings, Z.; Khan, S.; Fasinu, P.S.; ElSohly, M.A.; McChesney, J.D.; Khan, I.A.; et al. Quantitative determination of primaquine-5,6-ortho-quinone and carboxyprimaquine-5,6-ortho-quinone in human erythrocytes by UHPLC-MS/MS. J. Chromatogr. B 2021, 1163, 122510. [Google Scholar] [CrossRef] [PubMed]
- U.S. Department of Health and Human Services Food and Drug Administration, Center for Drug Evaluation and Research, Center for Veterinary Medicine. Bioanalytical Method Validation: Guidance for Industry. [Internet]. 2018. Available online: https://www.fda.gov/files/drugs/published/Bioanalytical-Method-Validation-Guidance-for-Industry.pdf (accessed on 25 March 2021).
- European Medicines Agency. Guideline on Bioanalytical Method Validation. [internet]. 2012. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-bioanalytical-method-validation_en.pdf (accessed on 26 June 2021).
- Mihaly, G.W.; Ward, S.; Edwards, G.; Orme, M.L.; Breckenridge, A.M. Pharmacokinetics of primaquine in man: Identification of the carboxylic acid derivative as a major plasma metabolite. Br. J. Clin. Pharmacol. 1984, 17, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Singhasivanonl, V.; Sabcharoenl, A.; Attanathl, P.; Chongsuphajaisiddhil, T.; Diquee, B.; Turk, P. Pharmacokinetics of prima-quine in healthy volunteers. S. Asian J. Trop. Med. Public Health 1991, 22, 527–533. [Google Scholar]
- Mihaly, G.W.; Ward, S.; Edwards, G.; Nicholl, D.D.; Orme, M.L.; Breckenridge, A.M. Pharmacokinetics of primaquine in man. I. Studies of the absolute bioavailability and effects of dose size. Br. J. Clin. Pharmacol. 1985, 19, 745–750. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analytes | Human Plasma | Human Urine | ||
|---|---|---|---|---|
| Matrix Effect (%) (mean ± SD/CV) | Recovery (%) (mean ± SD/CV) | Matrix Effect (%) (mean ± SD/CV) | Recovery (%) (mean ± SD/CV) | |
| PQ | ||||
| LQC | 100 ± 16/6.38 | 77.8 ± 8.91/11.45 | 87 ± 5/1.94 | 108.39 ± 9.92/9.15 |
| MQC | - | 95.30 ± 13.95/14.64 | - | 111.91 ± 3.45/3.08 |
| HQC | 116 ± 5/2.04 | 91.59 ± 3.60/3.93 | 89 ± 3/1.65 | 102.24 ± 9.51/9.30 |
| 5,6-PQ | ||||
| LQC | 104 ± 12/7.11 | 97.93 ± 12.62/12.88 | 86 ± 5/2.33 | 97.49 ± 8.17/8.38 |
| MQC | - | 84.58 ± 12.45/14.72 | - | 108.00 ± 3.24/3.00 |
| HQC | 87 ± 2/4.88 | 80.47 ± 5.72/7.11 | 87 ± 3/2.38 | 108.51 ± 2.70/2.49 |
| Analytes | Human Plasma | Human Urine | ||
|---|---|---|---|---|
| Intra-Day (n = 5) Accuracy/Precision (%, mean ± SD/%, CV) | Inter-Day (n = 15) Accuracy/Precision (%, mean ± SD/%, CV) | Intra-Day (n = 5) Accuracy/Precision (%, mean ± SD/%, CV) | Inter-Day (n = 15) Accuracy/Precision (%, mean ± SD/%, CV) | |
| PQ | ||||
| LLOQ | 98.32 ± 2.18/2.22 | 99.17 ± 7.65/3.04 | 108.66 ± 1.12/1.03 | 102.62 ± 7.4/3.22 |
| LQC | 112.08 ± 1.54/1.37 | 103.98 ± 5.95/3.76 | 97.19 ± 6.31/6.49 | 99.46 ± 3.27/4.70 |
| MQC | 92.05 ± 4.63/5.03 | 94.45 ± 0.72/5.90 | 105.02 ± 7.8/7.43 | 98.14 ± 6.06/5.85 |
| HQC | 100.46 ± 1.84/1.83 | 100.52 ± 2.11/3.10 | 107.58 ± 2.93/2.72 | 95.46 ± 10.89/3.44 |
| 5,6-PQ | ||||
| LLOQ | 94.35 ± 1.72/1.82 | 100.47 ± 11.82/2.27 | 114.18 ± 1.68/1.47 | 109.52 ± 7.74/4.13 |
| LQC | 99.56 ± 3.98/4.00 | 97.98 ± 6.90/6.55 | 112.82 ± 4.24/3.76 | 111.42 ± 1.7/7.76 |
| MQC | 90.10 ± 10.98/12.19 | 89.19 ± 1.01/7.83 | 94.82 ± 4.78/5.04 | 92.41 ± 2.37/9.13 |
| HQC | 93.66 ± 6.34/6.77 | 93.24 ± 4.29/6.95 | 94.53 ± 5.3/5.61 | 94.18 ± 2.64/4.51 |
| Subject ID | BW (kg) | BMI (kg/m2) | Cmax (ng/mL) | Tmax (h) | AUC (ng/mL*h) | AUC0-inf (ng/mL*h) |
|---|---|---|---|---|---|---|
| 1 | 86 | 28.08 | 156.62 | 3 | 1597.57 | 1613.65 |
| 2 | 67.2 | 26.25 | 84.84 | 2 | 693.67 | 709.54 |
| 3 | 65 | 25.71 | 131.73 | 2 | 1315.37 | 1326.87 |
| Subject ID | PQ | 5,6-PQ | ||||
|---|---|---|---|---|---|---|
| AEmax (μg) | Tmax (h) a | CAE (μg) | AEmax (μg) | Tmax (h) a | CAE (μg) | |
| 1 | 86 | 28.08 | 156.62 | 3 | 1597.57 | 1613.65 |
| 2 | 67.2 | 26.25 | 84.84 | 2 | 693.67 | 709.54 |
| 3 | 65 | 25.71 | 131.73 | 2 | 1315.37 | 1326.87 |
| Parameters | Analytes | Avula et al., 2011 [16] | Page-Sharp et al., 2012 [17] | Khan et al., 2021 [18] | This Study | |
|---|---|---|---|---|---|---|
| LOD | PQ 5,6-PQ | 2 ng/mL N/D | 1 μg/mL N/D | N/D 1.62 ± 0.23 ng/mL | 10.74 ± 4.32 ng/mL 2.99 ± 1.82 ng/mL | 8.77 ± 3.74 ng/mL 1.05 ± 0.59 ng/mL |
| LOQ | PQ 5,6-PQ | 5 ng/mL N/D | 2 μg/mL N/D | N/D 4.95 ± 0.61 ng/mL | 25 ng/mL | |
| Recovery | PQ 5,6-PQ | 87–88% N/D | 79–117% N/D | N/D 84–98% | 78–95% 80–98% | 102–112% 97–109% |
| Matrix effect | PQ 5,6-PQ | N/D | 97–118% N/D | N/D | 100–116% 87–104% | 87–89% 86–87% |
| Accuracy | PQ 5,6-PQ | 101–112% N/D | 94–124% N/D | N/D 96–103.2% | 92–112% 90–100% | 95–109% 92–114% |
| %RSD | PQ 5,6-PQ | 0.2–9.1% N/D | 4.8–9.7% N/D | N/D 5.49% | 1.37–3.9% 1.82–12.19% | 1.03–7.43% 1.47–7.76% |
| Stability | PQ 5,6-PQ | N/D | N/D | N/D 13 days | 7 days | |
| Matrix | Mice plasma | Human plasma | Human erythrocyte | Human plasma | Human urine | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pookmanee, W.; Thongthip, S.; Tankanitlert, J.; Mungthin, M.; Sukasem, C.; Wittayalertpanya, S. Simplified and Rapid Determination of Primaquine and 5,6-Orthoquinone Primaquine by UHPLC-MS/MS: Its Application to a Pharmacokinetic Study. Molecules 2021, 26, 4357. https://doi.org/10.3390/molecules26144357
Pookmanee W, Thongthip S, Tankanitlert J, Mungthin M, Sukasem C, Wittayalertpanya S. Simplified and Rapid Determination of Primaquine and 5,6-Orthoquinone Primaquine by UHPLC-MS/MS: Its Application to a Pharmacokinetic Study. Molecules. 2021; 26(14):4357. https://doi.org/10.3390/molecules26144357
Chicago/Turabian StylePookmanee, Waritda, Siriwan Thongthip, Jeeranut Tankanitlert, Mathirut Mungthin, Chonlaphat Sukasem, and Supeecha Wittayalertpanya. 2021. "Simplified and Rapid Determination of Primaquine and 5,6-Orthoquinone Primaquine by UHPLC-MS/MS: Its Application to a Pharmacokinetic Study" Molecules 26, no. 14: 4357. https://doi.org/10.3390/molecules26144357
APA StylePookmanee, W., Thongthip, S., Tankanitlert, J., Mungthin, M., Sukasem, C., & Wittayalertpanya, S. (2021). Simplified and Rapid Determination of Primaquine and 5,6-Orthoquinone Primaquine by UHPLC-MS/MS: Its Application to a Pharmacokinetic Study. Molecules, 26(14), 4357. https://doi.org/10.3390/molecules26144357