Abstract
A series of cyclohumulanoids, i.e., tricocerapicanols A–C (1a–1c), tricoprotoilludenes A (2a) and B (3), tricosterpurol (4), and tricoilludins A–C (5–7) were isolated along with known violascensol (2b) and omphadiol (8) from the culture broth of Daedaleopsis tricolor, an inedible but not toxic mushroom. The structures were fully elucidated on the basis of NMR spectroscopic analysis, and the suggested relative structures were confirmed via density functional theory (DFT)-based chemical shift calculations involving a DP4 probability analysis. In the present study, the 1H chemical shifts were more informative than the 13C chemical shifts to distinguish the diastereomers at C-11. The absolute configurations of 1–5 were determined by comparing the experimental and calculated electronic circular dichroism (ECD) spectra. For 6 and 7, the same chirality was assigned according to their biosynthetic similarities with the other compounds. The successful assignment of some Cotton effects was achieved by utilizing DFT calculations using simple model compounds. The plausible biosynthesis of 1–7 was also discussed on the basis of the structural commonality and general cyclohumulanoid biosynthesis. Compounds 2a and 5 were found to simultaneously induce hyphal swelling and branching at 5.0 μg/mL against a test fungus Cochliobolus miyabeanus.
1. Introduction
Cyclohumulanoids belong to a family of polycyclic sesquiterpenes including illudanes, protoilludanes, suterpuranes, and africanes [1,2]. These compounds are biosynthetically related to humulene, a monocyclic 11-membered sesquiterpene, and are metabolites of plants, fungi, ferns, and marine sponges. In vitro biomimetic cyclizations of humulene conducted in the 1980s, mainly by Shirahama, yielded a series of cyclohumulanoids [3,4,5,6,7]. Modern biosynthetic studies have revealed the enzymes responsible for these processes and the detailed mechanisms [8,9,10]. To date, more than 150 members of this family have been reported [1,7], some of which exhibit notable biological activities. For example, ptaquiloside, a glycosylated illudoid of bracken, acts as an inducer of cancer [11], whereas irofulven, a derivative of illudin [12,13], is under clinical examination as an anticancer drug [14]. Moreover, 5-O-aryl esters of protoilludens exhibit antifungal activity and inhibit plant growth in lettuce [15]. The unique structures of this family of compounds have attracted the interest of synthetic chemists, resulting in numerous reports on their synthesis [16,17,18,19,20,21].
Daedaleopsis tricolor (D. tricolor) is one of the common basidiomycetes, wood-rotting fungi found in Europe [22,23,24,25] and Asia [26,27,28]. This fungus is not toxic but generally inedible. Interestingly, extensive investigations of its constituents have revealed various metabolites such as terpenoids and saccharides [25,26,27,28]. In the present study, we disclosed nine cyclohumulanoids from the culture broth of this fungus collected in the Shirakami mountainous area (Sirakami-Sanchi) of Japan, which UNESCO designated as a World Heritage Site in 1993 [29].
2. Results
D. tricolor sampled in 2019 at the Shirakami Natural Science Park of Hirosaki University located in the Shirakami Mountains, Japan, was cultured in potato dextrose medium for 2 weeks under shaking conditions. The ethyl acetate extracts from both the culture broth and the fungus body were subjected to a series of chromatographic separations to obtain tricocerapicanols A–C (1a–1c), tricoprotoilludenes A (2a) and B (3), tricosterpurol (4), tricoilludins A–C (5–7), and known violascensol (2b) [30] and omphadiol (8) as shown in Figure 1 [31]. Table 1 summarizes the 1H and 13C NMR data of the new compounds. The spectral data of known 2b [30] and 8 [31] agreed with those reported in the literature. Although the molecular ion of 8 was not observed in the electron spray ionization–time of flight–mass spectrometry (ESI-TOFMS) analysis, its formula was confirmed after conversion into its 10-O-benzoate (8-OBz, Supplementary Materials SI-93). Despite being addressed in various studies [30,32], the absolute configuration of 2b was not fully established yet; this was successfully elucidated in the present study. The absolute configuration of 8 is discussed on the basis of our own investigations, although it was previously established synthetically [33].
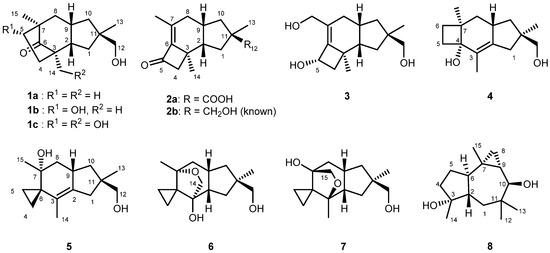
Figure 1.
Structures of cyclohumulanoids isolated from Daedaleopsis tricolor.

Table 1.
Summary of 1H NMR spectral data (δH, type (J in Hz)) in CDCl3 (500 MHz).
The molecular ion of tricocerapicanol A (1a) was observed at m/z 237.1869 in the ESI-TOFMS spectrum (SI-6), suggesting that its molecular formula is C15H24O2 ([M + H]+: 237.1849). This was confirmed by the presence of 15 resonances in the 13C NMR spectrum. A 2D spectral analysis involving heteronuclear single quantum coherence (HSQC, SI-10) and heteronuclear multiple bond coherence (HMBC, SI-11) spectra suggested a cerapicane framework for 1a [34,35]. Its 1H NMR spectral profile (SI-7) resembled that of repraesentin B, which was isolated by Makabe et al. in 2003 from the fungus Lactarius repraesentaneus [36]; however, there were some considerable differences, despite fact that CDCl3 was used as the solvent in both cases. For example, the H-2 of 1a was observed at 2.27 ppm, whereas it appears at 2.37 ppm for repraesentin B. This chemical shift difference suggests that they are different compounds. The irradiation of the H-2 resonance of 1a afforded nuclear Overhauser effects (NOEs) with Hβ-4, Hβ-5, H-9, and H2-12 as shown in Figure 2 (SI-12), revealing that 1a is a C-11 epimer of repraesentin B. Other correlations observed in the 2D nuclear Overhauser effect spectroscopy (NOESY) spectrum not only confirmed the relative structure but also allowed assigning the prochiral methylene 1H signals, except for the conformationally flexible H2-12 (SI-13). Considering other novel compounds isolated in the present study, this compound was named tricocerapicanol A.
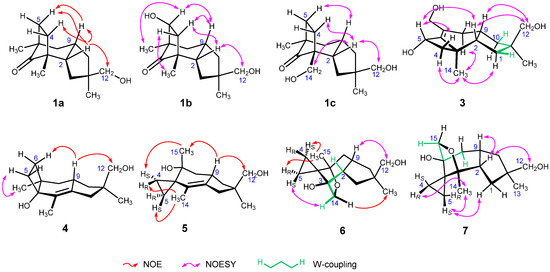
Figure 2.
Representative NOEs, NOESY correlations, and long-range coupling of 1–7.
The ESI-TOFMS spectrum of tricocerapicanol B (1b) exhibited the molecular ion at m/z 253.1802 (SI-15). The mass difference from 1a (Δm/z 15.9933) indicated that 1b contains one more oxygen than 1a. The similar 2D spectral analysis as above allowed establishing that 1b is a 5-hydroxylated derivative of 1a (SI-18, 19, and 20). A NOESY correlation between H-5 and H-9 revealed the α-configuration for 5-OH (SI-21). Although the H-2 and H-9 signals appeared too close (2.28 and 2.35 ppm, respectively) to observe the NOESY correlation between them, these were assigned to take a cis-relationship because Hβ-4 afforded NOESY correlations with both H-2 and H-9. Another NOESY correlation between H-2 and H2-12 defined the relative configuration at C-11.
Tricocerapicanol C (1c) was found in the polar fraction eluted by silica gel column chromatography using 80% EtOAc/hexane. In the 1H NMR spectrum (SI-24), this molecule afforded two sets of broad AB doublets at the medium frequency region (3.14 and 3.20 ppm, J = 10.3 Hz (H2-12); 3.38 and 3.60 ppm, J = 11.0 Hz (H2-14)), suggesting the existence of two hydroxymethyl groups in the molecule. A 2D NMR spectral analysis (SI-26, 27, and 28) allowed us to conclude that 1c is a C-14 hydroxylated congener of 1a, which is in accordance with the observed molecular ion at m/z 253.1802 (C15H25O3+, [M + H]+: 253.1798) in the ESI-TOFMS spectrum (SI-23). The NOESY signals between H-2 and H-12, H-2 and Hβ-4, and Hβ-5 and H-9 were diagnostic of the relative structure depicted in Figure 1. Although the C-5 resonance (29.7 ppm) was overlapped with the solvent signal (acetone-d6, 29.9 ppm), this signal was assigned on the basis of the HSQC and HMBC spectra (SI-27 and 28, respectively).
The 1H NMR spectral data of tricoprotoilludene A (2a, SI-32) resembled those of known protoilludane violascensol 2b (SI-39), except for the absence of the H2-12 signal in 2a [30]. The C-12 resonance was observed at 182.0 ppm, suggesting that 2a is a carboxylic acid derivative of 2b. This was confirmed by ESI-TOFMS spectrum (m/z 249.1482, C15H21O3+, [M + H]+: 249.1485, SI-31). Unfortunately, diagnostic NOEs of 2a were not observed. Since the extracts also afforded 2b, 2a can be assumed to possess the same stereochemical relationship as 2b by considering their biosynthesis. Hence, the prochiral methylene protons were tentatively assigned according to those of 2b. The proposed relative configuration was supported by the theoretical 13C and 1H chemical shifts obtained by density functional theory (DFT) calculations, as will be described later.
The sodium adduct ion of tricoprotoilludene B (3) appeared at m/z 275.1620 in the ESI-TOFMS spectrum (SI-43), suggesting that 3 possesses the molecular formula C15H24O3 ([M + Na]+: 275.1618). The correlation spectroscopy (COSY) spectral data revealed the spin systems H2-1/H-2/C-9(/H2-8)/H2-10 and H2-4/H-5 (SI-46). The C-12 oxymethylene, C-13 methyl, and C-11 quaternary carbon atoms were assigned by the HMBC signals H2-12/C-1, H2-12/C-10, H2-12/C-11, H3-13/C-1, H3-13/C-10, H3-13/C-11, and H3-13/C-12 (SI-48). A W-coupling between Hβ-1 and Hβ-10 (1.4 Hz) not only supported the above assignment but also enabled the configurational discrimination of methylenes H2-1 and H2-10. The H2-15 and H2-8 signals showed HMBC correlations with C-6 and C-7 (146.1 and 134.1 ppm, respectively), and the C-6 resonance further correlated with H2-4, H-5, and H3-14. These results suggest that 3 possesses a protoilludane framework. The NOESY correlations between H-2 and H2-12, H-2 and H-5, Hα-4 and H3-14, Hβ-4 and H-5, and H-9 and H2-12 (SI-49) indicated that 3 has the same relative structure as 2b. Meanwhile, the NOESY correlation between Hα-8 and H3-14 allowed distinguishing the prochiral H2-8 atoms.
Tricosterpurol (4) gave the largest ion at m/z 219.1760 indicating C15H23O+ (calcd. 219.1743) in the ESI-TOFMS spectrum (SI-51). This ion was tentatively assigned as the dehydrated ion because two oxygenated carbon signals were observed at 73.4 and 70.5 ppm (C-4 and C-12, respectively) in the 13C NMR spectrum (SI-53). Thus, C15H24O2 can be assigned as the molecular formula of 4. The methylene protons H2-1 appeared at 2.07 and 2.25 ppm as broad AB doublets (SI-52), which showed HMBC correlations with the C-2 (140.1 ppm) and C-3 (126.6 ppm) sp2 carbons. The COSY spectrum indicated a long-range spin coupling between H2-1 and H3-14 (SI-54), although the signal splittings were not observed in the regular one-dimensional 1H NMR spectrum. These observations suggest a tetrasubstituted C-2/C-3 double bond. In the HMBC spectrum, the quaternary C-4 signal showed correlations with H2-5 and H3-14, and the other quaternary C-7 resonance (43.8 ppm) correlated with H2-6, H2-8, and H3-15, revealing the presence of a C-4/C-5/C-6/C-7 cyclobutane ring. This allowed us to assign a sterpurane framework for 4 [37]. The irradiation of H-9 (2.48 ppm) resulted in NOEs with Hβ-6 and H2-12, which unequivocally defined the relative configuration of 4 (SI-57). An analysis of the NOESY spectrum enabled the full assignment of the 1H and 13C NMR signals, except for the conformationally flexible prochiral H2-12 (SI-58).
Tricoilludin A (5) produced the largest ion at m/z 219.1738 in the ESI-TOFMS spectrum (SI-60), which corresponds to C15H23O+ (calcd. 219.1743). Similarly to 4, the presence of two oxygenated carbon signals at 70.4 and 70.6 ppm in the 13C NMR spectrum indicates that this signal can be assigned to the dehydrated molecular ion (SI-62). Accordingly, the molecular formula of 4 was determined to be C15H24O2. The 1H NMR spectrum showed two characteristic sets of methylene proton signals at a low-frequency region (at 0.69 and 0.75 ppm (H2-4) and at 0.48 and 0.81 ppm (H2-5), SI-61) which showed the HMBC correlations with a quaternary carbon at 30.2 ppm (SI-65). The carbon atoms bonded to these protons resonated also at low frequency (5.7 and 7.7 ppm). These results reveal the presence of a spiro-cyclopropane ring (C-4/C-5/C-6) in the molecule. Since these cyclopropane methylene protons further correlated with quaternary carbons at 70.4 (C-7) and 125.2 ppm (C-3) in the HMBC spectrum, the cyclopropane ring can be assumed to be sandwiched between them. Further analysis of the 2D NMR spectra allowed establishing an illudane framework for 5, [38] as illustrated in Figure 1. The irradiation of H-9 (2.55 ppm) resulted in NOEs with H2-12 and H3-15 (SI-66), which unequivocally defined the relative configuration. The presence of NOE signals between HR-4 and H3-14, HS-4 and H3-15, and HS-5 and H3-14 enabled distinguishing both 1H and 13C signals on the prochiral cyclopropane ring. However, like in other compounds, the prochiral methylene protons at C-12 could not be distinguished.
Tricoilludin B (6) afforded the sodium adduct ion at m/z 275.1623 along with the dehydrated ion at m/z 235.1700 in the ESI-TOFMS spectrum (SI-68), suggesting its molecular formula to be C15H24O3 ([M + Na]+: 275.1618, [M + H−H2O]+: 235.1693). This molecule also showed ethylene protons at smaller frequencies than 1.0 ppm in the 1H NMR spectrum (SI-69), which is consistent with an illudane framework. Notably, H-2 was newly observed at 2.32 ppm, and C-14 (71.2 ppm) was oxygenated. The HMBC correlation between Hα-14 (3.97 ppm) and C-7 (83.1 ppm) is indicative of an ether linkage between C-7 and C-14 (SI-73). This was supported by the acetylation of 6 under conventional conditions (Ac2O, pyridine) affording only a monoacetate (SI-76-80). Irradiation of Hα-14 resulted in an NOE with H3-13, which unequivocally defined the relative configuration of 6 (SI-74). A long-range spin coupling between H-2 and Hβ-14 (2.0 Hz, W-coupling) supported this configuration. The full assignment of the 1H and 13C NMR signals, except for the conformationally flexible H2-12 (SI-75), was achieved on the basis of a NOESY spectrum (SI-75).
The ESI-TOFMS spectrum of tricoilludin C (7) showed the molecular ion at m/z 253.1793 and the dehydrated ion at m/z 235.1689 (SI-82), suggesting the same molecular formula as that of 6 (C15H24O3, [M + H]+: 253.1798, [M + H−H2O]+: 235.1693). Furthermore, the 1H NMR spectra of both compounds were similar (SI-83). Nevertheless, a notable difference was found; one of the oxymethylene signals at 3.57 ppm (Hα-15) of 7 showed a long-range coupling with Hα-8 (1.70 ppm, 4JHα-8/Hα-15 = 1.8 Hz), whereas a similar long-range coupling but between H-2 and Hβ-14 was observed for 6. Accordingly, 7 contains a cyclic ether between C-3 and C-15 instead of the ether linkage between C-7 and C-14 in 6. The HMBC spectrum supported the proposed relative structure (SI-87). The NOESY correlations observed between H-2 and H-9, H-2 and H2-12, and H-9 and Hβ-15 allowed establishing the relative configuration of 7 shown in Figure 2 (SI-88). Moreover, the NOESY signals between HR-4 and H3-14 and between HS-5 and Hα-1 enabled us to differentiate the prochiral H2-1, H2-4, and H2-5 methylene protons.
The relative structures of 1a–7 and known 2b and 8 were further investigated by DFT-based NMR chemical shift calculations because the variety of the framework of 1a–8 constituted interesting examples to evaluate the reliability of this methodology [39,40,41,42] and to clearly establish the relative structure of 2a. Calculations were performed with the NMR chemical shift calculation protocol equipped on Spartan’18 without changing the default settings (Hehre’s protocol) [43]. The protocol features the chemical shift calculations using ωB97X-D/6-31G*, the evaluation of the energies of individual conformers with ωB97X-V/6-311+G(2df,2p)[6-311G*]//ωB97X-D/6-31G*, and the empirical correction based on the type of carbons, attached atoms, and bond lengths. Some molecules (1a, 1b, 4, and 5) were calculated using the ent-forms ((11S)-enantiomers) of the natural products because the chirality investigation was performed later. However, in this article, their NMR and electronic circular dichroism (ECD) properties are discussed after interpretation that the natural products are all (11R)-enantiomers for convenience. In the statistical analysis of 1H NMR signals, the methylene protons of the proposed diastereomers were arranged according to the observed NOEs except for unassigned protons such as H2-12 of 1a–7 and H2-15 of 3, which were set to reduce the chemical shift difference with the calculated values. Similarly, all prochiral nuclei in other diastereomers were arranged so that the chemical shift difference with the calculated values was smaller. Note that the signals were arranged so that the incorrect structures became rather advantageous in the analysis. The calculated 1H and 13C chemical shifts were directly subjected to statistical and DP4 analysis without empirical correction [44], since Hehre’s protocol involved more sophisticated corrections [43]. Although this protocol has been proved to be more accurate than Goodman’s method [39], Goodman’s standard deviation (σ: 2.306 and 0.185 ppm for δ13C and δ1H, respectively) and freedom (ν: 11.38 and 14.18 for δ13C and δ1H, respectively) were applied in the DP4 analysis [44] because these parameters have not been published for Hehre’s protocol. Accordingly, the DP4 analysis in the present study tended to be slightly less sensitive toward structural differences than that using the appropriate parameters (SI-98-122).
Table 2 summarizes the results. The proposed structures of 1a–8 afforded satisfying small root-mean-square of the deviation (RMSD) values for δ13C and δ1H from the experimental data. The δ13C DP4 probability for 6 was not the highest (47.9%), and the 11-epimer gave the best score (52.1%). However, the δ13C RMSD values for these isomers were small enough (1.1 and 1.0 ppm, respectively) when considering the average error of this protocol (2.0 ppm) [43]. In other words, the small difference of the calculated δ13C values between 6 and its 11-epimer hindered their differentiation. In contrast, the δ1H DP4 probability for 6 was quite high (98.5%), which compensated for the unsatisfactory results of the δ13C DP4 analysis. Although the relative configuration of 2a based on the spectroscopic analysis remained unclear, both δ1H and δ13C DP4 scores strongly supported the proposed structure (δ13C: 87.3%, δ1H: 99.9%, δ13C + δ1H: 100%). The 13C + 1H DP4 values were above 99% probability in all cases. It is worth noting that the δ13C DP4 probability of 8 was the highest and exclusive (99.9%) among the 32 possible diastereomers. As described above, these chemical shift calculations not only supported the relative structures of 1a–8 obtained by the spectral analysis but also demonstrated the efficiency of this methodology.
Table 2.
DFT-based NMR chemical shift analysis.
The calculation results must be discussed more holistically. The δ1H DP4 values generally gave higher scores than the δ13C DP4 values for the correct diastereomers of 1a–7. The 13C chemical shift is known to depend on parameters such as the electronegativity of neighboring functional groups, bond angles, and bond lengths. Since the substituents methyl and hydroxymethyl groups at C-11 are located at the sterically less hindered end of the molecules, the effect of its geometrical difference on the conformations of the other moieties in 1a–7 and on the δ13C values can be considered insignificant. In contrast, the anisotropic effect of 12-OH significantly affects the magnetic shielding of the nearby hydrogen atoms. This effect is small for carbon nuclei probably because carbons are located inside the molecules and are usually shielded by hydrogen atoms. This would explain why only the 1H chemical shifts are sensitive to the geometry of 12-OH. Despite the high δ13C DP4 score obtained for 8, its δ1H DP4 is less conclusive (30.7%). This result is usual in calculations of this type and demonstrates the difficulty of the stereostructural elucidation only based on δ1H DP4 values.
Then, the absolute configurations of the natural products were investigated. Tricocerapicanol A (1a) provided a characteristic negative Cotton effect (Δε −5.7) at 297 nm attributed to the R-band (n→π∗ transition) of the C-5 carbonyl group in the ECD spectrum (Figure 3, spectra A). This was reproduced by a B3LYP/def2-TZVP model [45,46] when the (11R)-enantiomer of 1 was applied (Figure 3, spectra C, SI-124). However, the calculated and experimental spectra were considerably different in the 200–230 nm region. The Cotton effect in this region depended on the individual stable conformers (Figure 3, spectra D), whereas the negative Cotton effects at approximately 300 nm were constant regardless of the conformers, suggesting that the Cotton effect at approximately 300 nm is more reliable for the chirality assessment than that at approximately 200 nm. It is known that the octant rule can be applied for Cotton effects at the R-band of the ketone group [47]. The bicyclo[1–3]-8-octanone framework in 1a is highly symmetric; thus, this moiety is not responsible for the Cotton effect. Similarly, the Cotton effects of the 14- and 15-methyl groups should cancel each other’s the Cotton effects. Meanwhile, Hα-1 (highlighted with a green sphere in the 3D and 2D models in Figure 3), which is located at the front right lower octant, could be expected to contribute to the negative Cotton effect at approximately 300 nm. This assumption was verified with the simplified virtual models I (the model without 12-CH2OH and 13-CH3), II (the model in which the Hα-1 of model I was replaced with a chlorine atom), and III (the Δ1,12 model); the calculations of model I afforded a negative Cotton effect at approximately 300 nm similar to that of the experimental spectrum of 1a, and the intensity of the negative Cotton effect increased in model II because of the presence of a polarized C–Cl bond there, whereas that of model III was inversed because of the absence of Hα-1 (see SI-145), according to the calculations (SI-145, 146, and 147).
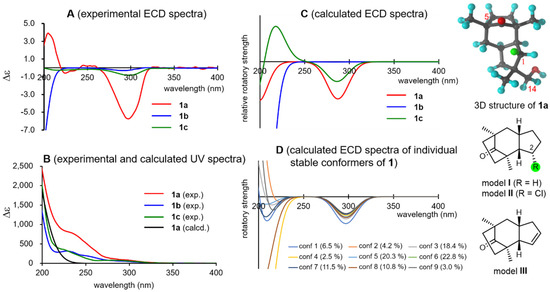
Figure 3.
Experimental UV/ECD spectra of 1a, 1b, and 1c (spectra A and B), calculated ECD spectra (spectra C), calculated ECD spectra of the individual conformers of 1a (spectra D), three-dimensional structure of 1a, and structures of models I–III.
In contrast, 1b and 1c showed only faint, negative Cotton effects at approximately 300 nm (Figure 3, spectra A), which were moderately well reproduced by the calculations (Figure 3, spectra C, SI-126–131). Consequently, the 5-OH group of 1b, located at the rear left upper octant, contributes to the positive Cotton effect and cancels the negative Cotton effect observed in 1a. Although the reason for this observation remains unclear, the 14-OH group of 1c likely weakens the negative Cotton effect observed in 1a. Note that the DFT calculations can describe these effects more quantitatively. On the basis of these results, it can be concluded that 1a, 1b, and 1c possess the (11R)-configuration.
Violascensol (2b) was first isolated by Vidari’s group in 1998 and was reported to show a negative Cotton effect at 332 nm [30]. However, its absolute configuration could not be determined on the basis of this observation and was tentatively assigned as 11R for 2b according to its structural and biosynthetic resemblance to related natural products. In 2002, Ferlek mentioned that the general helicity rule for cisoidal enones is not applicable to 2b in their review discussing the chirotopical properties of cisoidal enones [32]. Both 2a and 2b exhibited negative Cotton effects at approximately 335 nm, which were in accordance with Vidari’s report (Figure 4, spectra A and B), along with positive and negative Cotton effects at around 260 and 220 nm, respectively. All these Cotton effects were nicely reproduced for the (11R)-enantiomers of both 2a and 2b using a B3LYP/def2-TZVP model (SI-134–137). Interestingly, the simplified virtual model IV consisting of the substructure highlighted in red in 2a and 2b (Figure 4) also well reproduced the experimental ECD spectrum when the dihedral angle O/C-5/C-6/C-7 was set to 15° (SI-148 and 149), which was nearly identical to that of the stable conformer of 2a (the numbering followed that used for 2a). Consequently, the three Cotton effects observed in 2a and 2b are most likely caused by the chiral torsion of their cisoidal α,β-unsaturated ketone moiety.
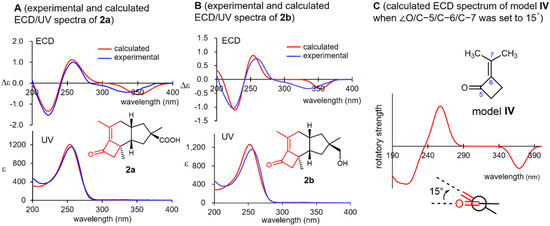
Figure 4.
Experimental and calculated ultraviolet/electronic circular dichroism (ECD) spectra of 2a (spectra A) and 2b (spectra B) and virtual ECD spectrum of model IV by setting the torsion O/C-5/C-6/C-7 to 15° (spectrum C).
Compounds 3, 4, and 5 weakly absorbed UV light at approximately 200 nm, which can be attributed to the K-band (π→π* transition) of their isolated double bonds (Figure 5, spectra A–C). Coincidentally, all these compounds showed positive Cotton effects at this wavelength region in their ECD spectra, which were well reproduced when the (11R)-enantiomers of 3–5 were subjected to ECD calculations (SI-136–142). Although these compounds involved more than 30 stable conformers within 10 kJ/mol from the global minimum, the 10 most stable conformers showed positive Cotton effects at around 210 nm in all cases (spectra D–F), which confirms the reliability of the above argument. In the UV/ECD spectral reproductions for 3, and the BHLYP functional provided a better match than B3LYP (SI-152), whereas the latter functional afforded satisfying UV/ECD spectra for other compounds. This highlights the necessity of using various functionals for a reliable elucidation of the chirality. The suggested chirality of 3 was consistent with that of 2a and 2b, which was expected from the viewpoint of their biosynthesis. For 3–5, attempts at applying Scott’s empirical octant rule for olefins [48] were not conclusive.
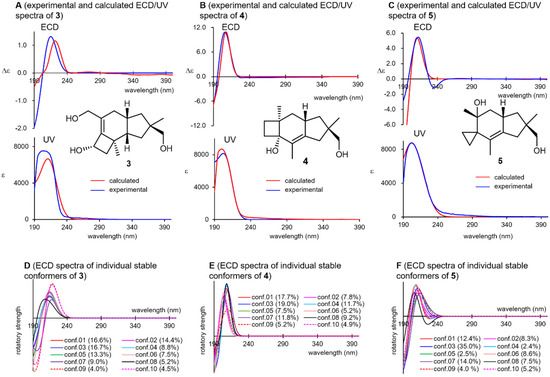
Figure 5.
Experimental and calculated ultraviolet/electronic circular dichroism (ECD) spectrum of 3–5 (spectra A–C) and their constituent ECD spectra of the individual stable conformers (spectra D–F).
The absolute configuration of omphadiol (8) was established to be the (10R)-enantiomer in the enantioselective total synthesis reported by Romo [33]. Nevertheless, its 10-O-benzoate 8-OBz was independently investigated in the present study. Natural 8 showed no remarkable Cotton effects at the UV/vis region, whereas 8-OBz showed a characteristic ECD spectrum, as shown in Figure 6. Such complexed Cotton effects most likely stem from the interaction of the π character of the cyclopropane ring [49] with the benzoyl chromophore. The experimental ECD spectrum was successfully reproduced by the DFT B3LYP/def2-TZVP functional (SI-143 and 144), confirming the (10R)-configuration. Interestingly, the experimental ECD spectrum of 8-OBz could be roughly reproduced with the simplified virtual model V, which only contains the substructure highlighted in red in 8-OBz (Figure 6), when the dihedral angle C-7/C-9/C-10/O was set to 170°, which is the angle of the corresponding atoms in the stable conformation of 8 (SI-150 and 151). These demonstrated that the DFT-based modeling calculation is an efficient tool not only for chirality determinations but also for revealing the major factors causing the Cotton effects.
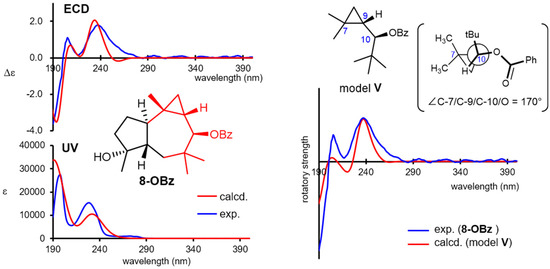
Figure 6.
Experimental and calculated ultraviolet/electronic circular dichroism (ECD) spectra of 8-OBz and calculated ECD spectrum of model V by setting the dihedral angle C-7/C-9/C-10/O to 170°.
Although tricoilludins B (6) and C (7) do not possess either appropriate functionals or chromophores for chirality elucidation, the (11R)-configuration could be assigned by considering that they were isolated from the same culture broth as 5.
These molecules belong to the cyclohumulanoid family, and except for 8, contain a C-1/C-2/C-9/C-10/C-11 cyclopentane ring sharing methyl (C-13) and hydroxymethyl (C-12) groups at C-11 with (R)-configuration, although the absolute configurations of 6 and 7 were not unequivocally established. A protoilludane derivative 10 can be envisaged as the key biosynthetic intermediate for the compounds obtained in the present study. Protoilludane 10 is likely derived from farnesyl pyrophosphate via humulenyl cation 9 as shown in Scheme 1. According to the established configurations, the oxidation of C-12 occurs stereoselectively right after cyclization because all compounds except for 8 contain an (11R)-hydroxymethyl group. Humulenyl cation 9 also produces 8 in another tandem cyclization at C-7/C-9 and C-2/C-6; however, a different type of enzyme may be responsible for this process [8,10]. Protoilludanes 2a, 2b, and 3 are obtained by oxidation(s) of 10. Epoxide 11 can be proposed as a versatile intermediate yielding cerapicanes 1a–c, sterpurane 4, and illudanes 5–7. An epoxide ring opening reaction at C-6 induces a ring contraction of cyclobutene into a cyclopropane, affording illudanes 5–7. Another epoxide ring opening at C-7 promotes a ring expansion into cyclopentane, generating cerapicanium cation 12. Deprotonation at 5-OH affords ceapicanes 1a–1c, whereas another deprotonation at H-2 produces a concerted olefin formation and ring contraction into cyclobutane to give tricosterpurol (4).
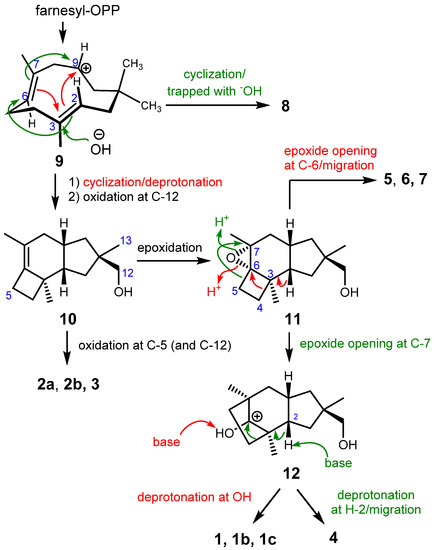
Scheme 1.
Plausible biosynthesis.
Finally, an antifungal assay was performed using Cochliobolus miyabeanus as the test fungus to reveal that 2a and 5 simultaneously produced swelling and branching at a concentration of 5 μg/mL (SI-153), whereas other compounds did not show obvious inhibitory effect even at higher concentrations.
3. Experimental
General Experimental Procedures. UV spectra were obtained on a HITACHI U-2010 spectrometer (Hitachi High-Tech, Tokyo, Japan) using a 10 mm length cell. ECD spectra were recorded on a JASCO J-1100 spectropolarimeter (JASCO Cooperation, Tokyo, Japan) with a 10 mm length cell. Fourier transform infrared spectroscopy was conducted using a HORIBA FT-720 spectrometer (Horiba Ltd, Kyoto, Japan) and a KBr cell. 1H and 13C NMR spectra were recorded on a JEOL JNM-ECX500 spectrometer (1H: 500 MHz, 13C: 125 MHz, (JEOL Ltd., Tokyo, Japan). Tetramethylsilane (0 ppm) was used as the internal standard for both types of spectra when CDCl3 was used as the solvent. When acetone-d6 was used, the signals of CHD2COCD3 (2.05 ppm) and 13CD3COCD3 (29.92 ppm) were used as the internal standard for the 1H and 13C NMR spectra, respectively. ESI-TOFMS spectra were obtained using a HITACHI NanoFrontier LD spectrometer equipped with a HITACHI 2100 high-performance liquid chromatography (HPLC) pump, a HITACHI L-2420 UV detector (Hitachi High-Tech, Tokyo, Japan), and a HITACHI L-2300 column oven. Calibration was performed with a mixture of tetrabutylammonium ion (m/z 242.2848), reserpine (m/z 609.2807), and Ultramark 1621. Silica gel thin-layer chromatography (TLC) analyses were conducted using Merck TLC silica gel 60 F254 plates (No. 5715) (Merck & Co., Kenilworth, NJ, USA). Silica gel column chromatography was performed using silica gel Merck 707734. Chemicals and solvents were purchased from FUJIFILM Wako Pure Chemical Cooperation and Sigma-Aldrich Co. LLC (St. Louis, MO, USA) and used without further purification. Conformation searches and chemical shift calculations were performed with Spartan’18 (Wavefunction, Irvine, CA, USA) using a PC (operating system: Windows 7 Professional; CPU: Intel Xeon E5-2697 v2 processor, 2.70 GHz, 12 cores; RAM: 128 GB). ECD spectra were calculated using TmoleX 2021 (Dassault Systèms, Vélizy-Villacoublay, France) on a PC workstation (operating system: CentOS 7.1.1; CPU: Intel Xeon E5-2687W V4, 3.0 GHz, 12 cores × 2; RAM: 256 GB). The calculated ECD spectra were constructed using Microsoft Excel for Microsoft Office365 on a commercial PC (Windows 10, Microsoft, Redomond, WA, USA).
Fungus.D. tricolor was sampled in 2019 at the Shirakami Natural Science Park of Hirosaki University located in the Shirakami Mountains, Japan.
Isolation.D. tricolor was cultured in a potato dextrose medium (200 mL in a 500 mL baffled Erlenmeyer flask × 45) on a rotary shaker (110 rpm) at 26 °C for 14 days. MeOH (150 mL) was then added to each flask to precipitate the fungus body. After filtration through cotton gauze, the MeOH of the combined filtrate was removed with a rotary evaporator. The resulting aqueous suspension was extracted with ethyl acetate (3 L × 3), and the organic layer was combined and concentrated under vacuum conditions to obtain the crude extracts (1.7 g). These operations were repeated to obtain the second crude extracts (1.6 g). The first crude extracts were diluted with EtOAc (100 mL), and then, silica gel (~6.0 g) was added. The resulting suspension was carefully concentrated under reduced pressure. The obtained residual powder was loaded on a silica gel column (300 g) and eluted with 0→100% EtOAc/hexane to give fraction A to fraction G. Fraction C eluted with 20–30% EtOAc/hexane in the first column chromatography (113 mg) was further subjected to ODS medium-pressure column chromatography (YAMAZENE ODS universal column type S, 50–100% MeOH/H2O for 20 min, flow rate 5 mL/min) to give 1a (11.6 mg). Fraction D eluted with 50–60% EtOAc/hexane in the first column chromatography (205 mg) was similarly subjected to the ODS medium-pressure column chromatography (YAMAZENE ODS universal column type S, 50–100% MeOH/H2O for 30 min, flow rate 5 mL/min) to give fractions D-1 to D-4. Fraction D-2 (35.0 mg) was subjected to a preparative silica gel TLC (30% EtOAc/hexane, two developments) to give 8 (1.5 mg, Rf = 0.3), 2b (1.8 mg, Rf = 0.45), and 2a (1.0 mg, Rf = 0.2). Fraction E eluted with 70–80% EtOAc/hexane in the first column chromatography (186 mg) was further subjected to ODS medium-pressure column chromatography (YAMAZENE ODS universal column type S, 50–100% MeOH/H2O for 20 min, flow rate 5 mL/min) to give crude 1b (40 mg). Analytical 1b (10 mg) was obtained by preparative silica gel chromatography (70% EtOAc/hexane). The second crude extracts were also dispersed in silica gel (6.0 g) and subjected to silica gel column chromatography as described above to obtain fraction A′ to fraction J′. Similar separations afforded 1a (13.4 mg) and 2a (1.4 mg). Fraction A′ eluted with 10% EtOAc/hexane (8.0 mg) was subjected to preparative silica gel chromatography (30% EtOAc/hexane) to obtain 5 (5.5 mg, Rf = 0.3). Fraction J′ eluted with 80% EtOAc/hexane (434 mg) was dispersed on diatomaceous earth powder (1.0 g) in MeOH (20 mL), and the solvent was then carefully evaporated under reduced pressure. The obtained dried powder was placed in a small column and connected to a YAMAZENE ODS universal column type L, which was developed using gradient conditions (5→100% MeOH/H2O (containing 1% AcOH) for 250 min, flow rate 3 mL/min). The eluents were fractionated into 85 test tubes. Each fraction was checked with silica gel TLC and integrated into eight fractions (J′-1 to J′-8). Fraction J′-2 eluted with 10–15% MeOH/H2O (22.7 mg) was further subjected to silica gel column chromatography (~5 g, 50% EtOAc/hexane) to yield 6 (1.2 mg).
Fraction J′-6 eluted with 40–50% MeOH/H2O (91.8 mg) was subjected to ODS medium-pressure chromatography (YAMAZENE ODS universal column type M, 20→60% MeOH/H2O for 60 min, flow rate 8 mL/min). The fractions eluted with 30% and 35% MeOH/H2O were recovered to obtain 7 (3.6 mg) and crude 3 (24.3 mg), respectively. The latter fraction was subjected to ODS HPLC (Wakopak® Ultra C18-5; φ 20 mm × 250 mm, 20% CH3CN/H2O (containing 0.1% TFA), flow rate 10 mL/min, detected at 214 nm) to yield 3 (10.9 mg, tR = 28.0 min). Fraction J′-7 eluted with 50–60% MeOH/H2O (115 mg) was subjected to silica gel column chromatography (~5 g, 30% EtOAc/hexane) to yield 4 (3.9 mg). Fraction J′-8 eluted with 70–80% MeOH/H2O (25.1 mg) was subjected to ODS HPLC (Wakopak® Ultra C18-5; φ 20 mm × 250 mm, 20→40% CH3CN/H2O (containing 0.1% TFA) for 30 min, flow rate 10 mL/min, detected at 220 nm) to give 1c (1.4 mg, tR = 22.7 min).
Physical data of 1a: ECD (2.85 × 10−4 mol/L in CH3CN): 297 nm (Δε −5.7); UV (2.85 × 10−4 mol/L in CH3CN): 236 nm (sh, ε 760), 196 nm (ε 2,800); IR (film) 3400, 2954, 2927, 2868, 1733, 1455, 1029 cm−1; 1H NMR and 13C NMR data in CDCl3 are shown in Table 1. ESI-TOFMS (rel. int (%), assignment) m/z 254.2139 (35, [M + NH4]+: 254.2115), 237.1869 (100, [M + H]+: 237.1849).
Physical data of 1b: ECD (7.0 × 10−4 mol/L in CH3CN) 295 nm (Δε −0.3); UV (7.0 × 10−4 mol/L in CH3CN) 230 (sh, ε 310), 196 nm (ε 1600); IR (film) 3450, 2930, 2867, 1732, 1450, 1045 cm−1; 1H NMR and 13C NMR data in CDCl3 are shown in Table 1; ESI-TOFMS (rel. int (%), assignment) m/z 527.3369 (45, [2 M + Na]+: 527.3343), 270.2065 (50, [M + Na]+: 270.1618), 253.1802 (100, [M + H-H2O]+: 253.1798).
Physical data of 1c: ECD (2.78 × 10−4 mol/L in CH3CN) 300 nm (Δε −0.8); UV (2.78 × 10−4 mol/L in CH3CN) 230 (sh, ε 310), 195 nm (ε 2500); IR (film) 3365, 2927, 2870, 1732, 1043 cm−1; 1H NMR and 13C NMR data in CDCl3 are shown in Table 1; ESI-TOFMS (rel. int (%), assignment) m/z 527.3374 (12, [2M + Na]+: 527.3343), 253.1802 (100, [M + H-H2O]+: 253.1798).
Physical data of 2a: ECD (2.82 × 10 −4 mol/L in CH3CN) 335 nm (Δε −0.4); 261 nm (Δε +1.0), 224 nm (Δε −1.5); UV (2.82 × 10 −4 mol/L in CH3CN) 258 nm (ε 1720); IR (film) 2956, 2923, 1737, 1675 cm−1; 1H NMR and 13C NMR data in CDCl3 are shown in Table 1; ESI-TOFMS (rel. int (%), assignment) m/z 519.2747 (30, [2M + Na]+: 519.2717), 271.1297 (20, [M + Na]+: 271.1305), 249.1482 (100, [M + H]+: 249.1485).
Physical data of 2b: ECD (6.35 × 10−4 mol/L in CH3CN) 333 nm (Δε −0.4), 258 nm (Δε +0.8), and 225 nm (Δε −1.0); UV (6.35 × 10−4 mol/L in CH3CN) 258 nm (ε 1140); IR (film) 3425, 2925, 2867, 1728, 1668, 1043 cm−1; 1H NMR (500 MHz, CDCl3) δ 1.06 (1H, brd, J = 12.2 Hz, H2-10), 1.13 (3H, s, H3-13), 1.18 (3H, s, H3-14), 1.43 (1H, dd, J = 9.3, 13.5 Hz, H2-1), 1.43 (1H, dd, J = 9.8, 13.6 Hz, H2-8), 1.73 (1H, ddd, J = 1.5, 8.8, 13.5 Hz, H2-1), 1.92 (1H, ddd, J = 1.0, 7.3, 12.2 Hz, H2-10), 2.01 (3H, s, H3-15), 2.18 (1H, ddd, J = 8.8, 9.3, 13.5 Hz, H-2), 2.25 (1H, dq, J = 6.5, 14.9 Hz, H2-8), 2.41 (1H, m, H-9), 2.66 (1H, d, J = 16.8 Hz, H2-4), 2.69 (1H, d, J = 16.8 Hz, H2-4), 3.40 (1H, d, J = 10.7 Hz, H2-12), 3.43 (1H, d, J = 10.7 Hz, H2-12). 13C NMR (CDCl3) δ 20.4 (C-14), 20.4 (C-15), 24.5 (C-13), 35.8 (C-8), 36.2 (C-1), 36.8 (C-3), 41.8 (C-9), 43.6 (C-10), 45.5 (C-11), 47.4 (C-2), 60.9 (C-4), 69.4 (C-12), 143.3 (C-7), 150.7 (C-6), 197.2 (C-5). The spectral data are consistent with those in the literature [30]. ESI-TOFMS (rel. int (%), assignment) m/z 491.3080 (30, [2M + Na]+: 491.3132), 469.3252 (35, [2M + H]+: 469.3312), 235.1666 (100, [M + H]+: 235.1693).
Physical data of 3: ECD (6.35 × 10−4 mol/L in CH3CN) 214 nm (ε +1.3); UV (6.35 × 10−4 mol/L in CH3CN); 205 nm (ε 7500); IR (film) 3350, 2925, 2870, 1463, 1040 cm−1; 1H NMR and 13C NMR data in CDCl3 are shown in Table 1; ESI-TOFMS (rel. int (%), assignment) m/z 527.3391 (15, [2M + Na]+: 527.3343), 275.1620 (100, [M + Na]+: 275.1618).
Physical data of 4: ECD (1.06 × 10−4 mol/L in CH3CN) 206 nm (Δε +10.9); UV (1.06 × 10−4 mol/L in CH3CN) 202 nm (ε 8100); IR (film) 3310, 2920, 2863, 1455, 1040 cm−1; 1H NMR and 13C NMR data in CDCl3 are shown in Table 1; ESI-TOFMS (rel. int (%), assignment) m/z 219.1760 (100, [M + H-H2O]+: 219.1743), 201.1653 (55, [M + H-2H2O]+: 201.1638).
Physical data of 5: ECD (1.31 × 10−4 mol/L in CH3CN) 212 nm (ε +5.3); UV (1.31 × 10−4 mol/L in CH3CN) 203 nm (ε 8600). IR (film) 3394, 2954, 2850 cm−1. 1H NMR and 13C NMR data in CDCl3 are shown in Table 1. ESI-TOFMS (rel. int (%), assignment) m/z 219.1738 (100, [M + H-H2O]+: 219.1743), 201.1633 (45, [M + H-2H2O]+: 201.1638).
Physical data of 6: This compound showed no considerable absorption in the UV region. IR (film) 3400, 2923, 2869, 1025 cm−1; the 1H and 13C NMR data in CDCl3 are shown in Table 1; ESI-TOFMS (rel. int (%), assignment) m/z 275.1623 (45, [M + Na]+: 275.1623), 235.1700 (50, [M + H-H2O]+: 235.1693), 217.1592 (65, [M + H-2H2O]+: 217.1587), 167.0133 (100, not assigned).
Acetylation of 6: A solution of 6 (~0.5 mg) in pyridine (0.3 mL) was stirred with acetic anhydride (0.1 mL) at room temperature for 5 h. The reaction mixture was concentrated under reduced pressure, and the resulting residue was subjected to preparative TLC (Rf = 0.5, 80% EtOAc/hexane) to obtain the 12-O-acetate of 6. NMR spectra were measured using a SHIGEMI symmetrical MICRO NMR tube (SHIGEMI CO., LTD, Tokyo, Japan). IR (film) 3446, 2923, 2850, 1733 cm−1, 1H NMR (500 MHz, CDCl3) δ 0.30 (1H, dt, J = 10.0, 5.7 Hz), 0.52 (1H, dt, J = 5.7, 10.0 Hz), 0.66 (2H, overlapped), 0.94 and 1.16 (each 3H, s), 1.50–1.72 (5H, overlapped), 1.81 (1H, dd, J = 9.5, 14.7 Hz), 2.08 (3H, s), 2.33 (1H, m), 2.46 (1H, m), 3.47 (1H, dd, J = 1.9, 7.9 Hz), 3.78 and 3.90 (each 1H, AB doublet, J = 10.7 Hz), 3.95 (1H, d, J = 7.9 Hz), 13C NMR (125 MHz, CDCl3) d 2.3, 3.2, 21.0, 21.7, 24.8, 34.2, 35.3, 37.6, 39.1, 41.5, 43.9, 46.4, 70.8, 71.2, 78.0, 83.0, 171.5; ESI-TOFMS (rel. int (%), assignment) m/z 317.1737 (100, [M + Na]+: 317.1723).
Physical data of 7: This compound afforded no considerable absorption in the UV region. IR (film) 3370, 2930, 2870, 1455, 1478, 1022 cm−1; the 1H and 13C NMR data in CDCl3 are shown in Table 1; ESI-TOFMS (rel. int (%), assignment) m/z 253.1793 (25, [M + H]+: 253.1798), 235.1689 (100, [M+H-H2O]+: 235.1693), 217.1580 (65, [M + H-2H2O]+: 217.1587).
Physical data of 8: 1H NMR (500 MHz, CDCl3) δ 0.45 (1H, t, J = 4.5 Hz, H-8), 0.55 (1H, dt, J = 5.8, 8.4 Hz, H-9), 0.74 (1H, dt, J = 8.0, 4.5 Hz, H-8), 0.97 (3H, s, H3-12), 0.99 (3H, s, H3-15), 1.02 (3H, s, H3-12), 1.19 (t, J = 12.6 Hz, H-1), 1.27 (3H, s, H3-14), 1.42 (1H, overlapped, H-6), 1.45 (1H, dd, J = 2.5, 12.6 Hz, H-1), 1.60 (1H, overlapped, H-4), 1.61 (1H, overlapped, H-2), 1.70 (2H, overlapped, H-4, H-5), 1.80 (overlapped, H-5), 3.14 (1H, d, J = 8.0 Hz, H-10), 13C NMR (125 MHz, CDCl3) δ 19.1 (C-12), 19.4 (C-15), 19.5 (C-7), 22.6 (C-8), 23.2 (C-5), 25.7 (C-14), 28.7 (C-13), 29.7 (C-9), 38.0 (C-11), 41.4 (C-4), 42.1 (C-1), 48.2 (C-2), 49.6 (C-6), 81.0 (C-10), 81.1 (C-3). The spectral data are consistent with those in the literature [31]. ESI-TOFMS did not provide considerable ions.
Benzoylation of 8: A solution of 8 (~1.0 mg) in pyridine (0.1 mL) was stirred with benzoyl chloride (2.0 μL) and 4-(dimethylamino)pyridine (1.0 mg) at room temperature for 30 min. MeOH (1.0 mL) was then added, and the mixture was stirred at room temperature. After 1 min, diethylether (1.0 mL) was added, the resulting suspension was filtered through cotton, and the filtrate was concentrated under reduced pressure. Preparative silica gel TLC of the residue (30% EtOAc/hexane) afforded the 10-O-benzoate of 8 (~1.1 mg). The yield of this compound was estimated according to the UV absorption at 230 nm by assuming an ε value of 15,300. ECD 4.66 × 10−5 mol/L (CH3CN) 239 nm (Δε +1.7) and 206 nm (Δε +1.0), UV 229 nm (ε 15300). IR (film) 3506, 3433, 2958, 2925, 2852, 1714, 1277, 1115 cm−1. 1H NMR (CDCl3) δ 0.71 (1H, H-9), 0.73 (2H, H2-8), 0.97 (3H, s, H3-13), 1.02 (3H, s, H3-15), 1.16 (3H, s, H3-12), 1.29 (3H, s, H3-14), 1.36 (1H, t, J = 7.8 Hz, H-4), 1.51 (1H, H2-1), 1.61 (1H, H-2), 1.62 (1H, H-6), 1.71 (1H, H-4), 1.73 (1H, H-5), 1.88 (1H, m, H-5), 4.77 (1H, d, J = 8.1 Hz, H-10), 7.44 (2H, aromatic protons), 7.55 (2H, aromatic protons), 8.07 (1H, aromatic protons). 13C NMR (CDCl3) δ 19.4 (C-15), 19.6 (C-7), 20.5 (C-12), 23.2 (C-5), 23.3 (C-8), 25.7 (C-14), 27.1 (C-9), 28.7 (C-13), 37.9 (C-11), 41.5 (C-4), 41.9 (C-1), 48.1 (C-6), 49.3 (C-2), 81.0 (C-3), 82.7 (C-10), 128.3, 129.6, 131.0, 132.7 (aromatic carbons), 165.7 (C=O). ESI-TOFMS (rel. int (%), assignment) m/z 360.2539 (25, [M + NH4]+: 360.2533), 203.1787 (100, [M + H–OBz–H2O]+: 203.1794).
Chemical shift calculations. (11S)-1a, (11S)-1b, (11R)-1c, (11R)-2a, (11R)-2b, (11R)-3, (11S)-4, (11S)-5, (11R)-6, (11R)-7, (10R)-8 and their possible diastereomers were built on Spartan’18 and were directly subjected to the chemical shift calculation protocol with a default setting of the program [39], which automatically performed the conformational search with MMFF, followed by the collection of the candidate conformers by setting the threshold at 40 kJ/mol from the global minimum conformer; structure re-optimization employing the HF/3-21G model, followed by conformer narrowing by setting the threshold at 40 kJ/mol from the global minimum conformer; energy estimation using the ωB97X-D/6-31G* model, followed by conformer narrowing by setting the threshold at 15 kJ/mol from the global minimum conformer; structural optimization with the ωB97X-D/6-31G* model, followed by conformer narrowing by setting the threshold at 10 kJ/mol from the global minimum conformer; energy estimation applying the ωB97X-V/6-311+G(2df,2p)[6-311G*] model, followed by conformer narrowing by setting the threshold at 10 kJ/mol from the global minimum conformer; chemical shift calculations using ωB97X-D/6-31G*, followed by the empirical correction [39]. The obtained chemical shifts were directly compared with the experimental data, and the result data (RMSD, maximum deviation, and DP4) were calculated using Microsoft Excel for Microsoft 365. The methylene protons of the proposed diastereomers except for H-12 were arranged according to the assignments on the basis of the NOE analysis, and H2-12 and all the methylene signals in other isomers were set to reduce the chemical shift difference with the calculated values. The obtained 1H and 13C chemical shifts were directly subjected to statistical analysis without corrections on the basis of the slopes and intercepts. In the DP4 analysis, the original parameters provided by Goodman’s method (σ: 2.306 ppm for 13C and 0.185 ppm for 1H; ν: 11.38 for 13C and 14.18 for 1H) were used [44].
ECD spectral calculations. The stable conformer sets obtained in the chemical shift calculations were further optimized with B3LYP/def2-TZVP on TmoleX 2021. Only the most 10 stable conformers were subjected to the ECD calculations when more than 10 stable conformers within 10 kJ/mol from the global minimum conformers were obtained with ωB97X-V/6-311+G(2df,2p)[6-311G*//ωB97X-D/6-31G* (1b, 1c, 3, 4, 5, 6, and 7). After vibrational analysis, UV/ECD calculations were performed with B3LYP/def2-TZVP. The UV and ECD spectra of the individual conformers were constructed according to the frequencies, oscillator strength, and rotatory strengths using the NORM.DIST function in Microsoft Excel for Microsoft 365. The widths of the UV and ECD bands were set to reproduce the spectra appropriately. The UV/ECD spectra of the individual conformers were constructed by Boltzmann averaging on the basis of the free energy. The wavelengths of the UV spectra were corrected according to the experimental spectra, and those of the ECD spectra were corrected using the identical number. The ECD spectra of 1a, 1b, 4, and 5 were expressed as the values obtained after multiplying by −1.
Model I was constructed on Spartan’18 by removing nonessential atoms from the most stable conformer of 1a. For model II, Hα-1 of model I was replaced with chlorine. Model III was prepared from model I by introducing a double bond between C-1 and C-11. The resulting structures were then optimized with B3LYP/def2-TZVP and subjected to ECD calculations as described above. The most stable conformation of 8-OBz was obtained by performing a conformational search with MMFF, and the 6 stable conformers were subjected to the ECD calculations in a similar manner to that above. Model IV and model V were prepared by removing nonessential atoms from the most stable conformations of 2a and 8-OBz, respectively. The structures obtained by setting the dihedral angles O/C-5/C-6/C-7 in model IV and C-7/C-9/C-10/O in model V to 15° and 170°, respectively, were optimized with B3LYP/def2-TZVP by fixing the set torsions and then subjected to UV/ECD calculations. The calculated UV/ECD spectra were constructed similarly to those above.
Antifungal assay. A series of suspensions of spores of Cochliobolus miyabeanus (1.0 mL) containing 1, 5, 25, and 100 μg/mL of samples in Petri dishes were prepared and incubated at 25 °C for 24 h. Then, microscopic observation was performed using an Olympus CKX-41 binocular inverted microscope equipped with a ×10 objective lens and ×10 eyepieces.
4. Conclusions
In summary, nine cyclohumulanoids were successfully identified from D. tricolor. The present study proves the usefulness of DFT-based modeling calculations for the determination of the relative and absolute configurations of polycyclic terpenoids and the suitability of calculations using simplified models for elucidating the reasons behind the Cotton effects.
Supplementary Materials
The following are available online; (1) copies of IR, ESI-TOFMS, 1H, 13C, COSY, HSQC, HMBC, NOE (NOESY) NMR spectra for 1a, 1b, 1c, 2a, 2b, 3, 4, 5, 6, monoacetate of 6, 7, 8, and 8-OBz, calculated Boltzmann distributions, wavelengths, oscillator strengths, and rotatory strengths for ent-1a, ent-1b, 1c, 2a, 2b, 3, ent-4, ent-5, and 8-OBz, and the parameters used for UV/ECD spectral compositions, and pictures for hyphal growth inhibitions of C. miyabeanus by 2a and 5 at 5.0 μg/mL (in a PDF file, 153 pages), and (2) NMR statistical analysis details for ent-1a, ent-b, 1c, 2a, 2b, 3, ent-4, ent-5, 6, 7, and 8, and their geometries for the stable conformers, and those for stable conformer for their all diastereomers in a structure-data file format (Excel files).
Author Contributions
Conceptualization, M.H. and K.K.; experiments. R.K.; calculations, M.H. and R.K., source of fungus, A.T.; writing, M.H. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Most of the data in this article are disclosed in the Supplementary Material. Further data presented in this study are available on request from the corresponding author.
Acknowledgments
We thank Kota Inose and Mami Nishiyama for their experimental support. We are also grateful to Professor Hideaki Oikawa of Hokkaido University for his valuable suggestions about terpene biosynthesis.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are not available from the authors.
References
- da Costa, R.G.M.; Lopes, C.; Oliveira, P.A.; Bastos, M.M.S.M. Illudane-Type Sesquiterpenes: Challenginges and Opportunities for Toxicology and Chemotherapy. In New Development in Terpene Research; Hu, J., Ed.; Nova publishers, Inc.: New York, NY, USA, 2014; pp. 135–184. ISBN 978-1-62948-760-1. [Google Scholar]
- Cadelis, M.M.; Copp, B.R.; Wiles, S. A Review of Fungal Protoilludane Sesquiterpenoid Natural Products. Antibiotics 2020, 9, 928. [Google Scholar] [CrossRef]
- Mlotkiewicz, J.A.; Murray-Rust, J.; Murray-Rust, P.; Parker, W.; Riddell, F.G.; Roberts, J.S.; Sattar, A. A biogenetically significant cyclization of humulene-4,5-epoxide. Tetrahedron Lett. 1979, 20, 3887–3890. [Google Scholar] [CrossRef]
- Shirahama, H.; Hayano, K.; Kanemoto, Y.; Misumi, S.; Ohtsuka, T.; Hashiba, N.; Furusaki, A.; Murata, S.; Noyori, R.; Matsumoto, T. Conformationally Selective Transannular Cyclizations of Humulene 9,10-Epoxide. Synthesis of the Two Skeletally Different Cyclohumulanoids: Dl-Bicyclohumulenone and Dl-Africanol. Tetrahedron Lett. 1980, 21, 4835–4838. [Google Scholar] [CrossRef]
- Shirahama, H.; Osawa, E.; Matsumoto, T. Conformational studies on humulene by means of empirical force field calculations. Role of stable conformers of humulene in biosynthetic and chemical reactions. J. Am. Chem. Soc. 1980, 102, 3208–3213. [Google Scholar] [CrossRef]
- Murata, Y.; Ohtsuka, T.; Shirahama, H.; Matsumoto, T. Chemical Conversion of humulene to Cyclohumulanoids along the Biosynthetic Pathway Sterpurene, a Hydrocarbon from Silver-Leaf Disease Fungus. Tetrahedron Lett. 1981, 22, 4313–4314. [Google Scholar] [CrossRef]
- Morisaki, N.; Furukawa, J.; Kobayashi, H.; Iwasaki, S.; Nozoe, S.; Okuda, S. Cyclobutyl Cation Rearrangements of 6-Protoilluden-8a-ol, 7-Protoilluden-6-ol and Related Compounds. Chem. Pharm. Bull. 1987, 35, 2678–2685. [Google Scholar] [CrossRef][Green Version]
- Tantillo, D.J. Biosynthesis via carbocations: Theoretical Studies on Terpene Formation. Nat. Prod. Rep. 2011, 28, 1035–1053. [Google Scholar] [CrossRef] [PubMed]
- Quin, M.B.; Flynn, C.M.; Wawrzyn, G.T.; Choudhary, S.; Schmidt-Dannert, C. Mushroom Hunting by Using Bioinformatics: Application of a Predictive Framework Facilitates the Selective Identification of Sesquiterpene Synthases in Basidiomycota. ChemBioChem 2013, 14, 2480–2491. [Google Scholar] [CrossRef]
- Quin, M.; Flynn, C.M.; Schmidt-Dannert, C. Traversing the fungal terpenome. Nat. Prod. Rep. 2014, 31, 1449–1473. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Ojika, M.; Kigoshi, H. Ptaquiloside, the major toxin of bracken, and related terpene glycosides: Chemistry, biology and ecology. Nat. Prod. Rep. 2007, 24, 798–813. [Google Scholar] [CrossRef]
- MacDonald, J.R.; Muscoplat, C.C.; Dexter, D.L.; Mangold, G.L.; Chen, S.-F.; Kelner, M.J.; McMorris, T.C.; Von Hoff, D.D.; Macdonald, J.R.; Muscoplat, C.C.; et al. Preclinical Antitumor Activity of 6-Hydroxymethylacylfulvene, a Semisynthetic Derivative of the Mushroom Toxin Illudin S. Cancer Res. 1997, 57, 279–283. [Google Scholar] [PubMed]
- Niwa, H.; Ojika, M.; Wakamatsu, K.; Yamada, K.; Hirono, I.; Matsushita, K. Ptaquiloside, a novel norsesquiterpene glucoside from bracken, Pteridium aquilinum var. latiusculum. Tetrahedron Lett. 1983, 24, 4117–4120. [Google Scholar] [CrossRef]
- Jiang, H.; Greenberg, R.A. Morning for Irofulven, What Could be fiNER? Clin. Cancer Res. 2021, 27, 1833–1835. [Google Scholar] [CrossRef] [PubMed]
- Kobori, H.; Sekiya, A.; Suzuki, T.; Choi, J.-H.; Hirai, H.; Kawagishi, H. Bioactive Sesquiterpene Aryl Esters from the Culture Broth of Armillaria sp. J. Nat. Prod. 2015, 78, 163–167. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, G.; Eggert, U.; Kalesse, M. Synthesis of (+)-Omphadiol and (+)-Pyxidatol C. Org. Lett. 2016, 18, 2320–2322. [Google Scholar] [CrossRef]
- Furusaki, A.; Shirahama, H.; Matsumoto, T. Determination of The Chirality of Illudin S by the Bijvoet X-Ray Method. Chem. Lett. 1973, 2, 1293–1294. [Google Scholar] [CrossRef]
- Padwa, A.; Sandanayaka, V.P.; Curtis, E.A. Synthetic studies toward Illudins and Ptaquilosin. A highly convergent approach via the dipolar cycloaddition of carbonyl ylides. J. Am. Chem. Soc. 1994, 116, 2667–2668. [Google Scholar] [CrossRef]
- Zorn, C.; Anichini, B.; Goti, A.; Brandi, A.; Kozhushkov, S.I.; de Meijere, A.; Citti, L. Studies on the Synthesis of Aza Analogues of Illudins by Cycloadditions to Highly Strained Methylenecyclopropanes. J. Org. Chem. 1999, 64, 7846–7855. [Google Scholar] [CrossRef]
- Padwa, A.; Curtis, E.A.; Sandanayaka, V.P. An Approach toward the Illudin Family of Sesquiterpenes Using the Tandem Cyclization−Cycloaddition Reaction of Rhodium Carbenoids. J. Org. Chem. 1997, 62, 1317–1325. [Google Scholar] [CrossRef]
- Aungst, J.R.A.; Chan, A.C.; Funk, R.L. Total Synthesis of the Sesquiterpene (±)-Illudin C via an Intramolecular Nitrile Oxide Cycloaddition. Org. Lett. 2001, 3, 2611–2613. [Google Scholar] [CrossRef]
- Menttrida, S.; Krisai-Greilhuber, I.; Voglmayr, H. Molecular evaluation of species delimitation and barcoding of Daedaleopsis confragosa specimens in Austria. Österr. Z. Pilzk. 2015, 24, 173–179. [Google Scholar]
- Koukol, O.; Kotlaba, F.; Pouzar, Z. Taxonomic evaluation of the polypore Daedaleopsis tricolor based on morphology and molecular data. Czech Mycol. 2014, 66, 107–119. [Google Scholar] [CrossRef]
- Bernicchia, A.; Fugazzola, M.A.; Gemelli, V.; Mantovani, B.; Lucchetti, A.; Cesari, M.; Speroni, E. DNA recovered and sequenced from an almost 7000 y-old Neolithic polypore, Daedaleopsis tricolor. Mycol. Res. 2006, 110, 14–17. [Google Scholar] [CrossRef] [PubMed]
- Rösecke, J.; König, W.A. Constituents of the fungi Daedalea quercina and Daedaleopsis confragosa var. tricolor. Phytochemistry 2000, 54, 757–762. [Google Scholar] [CrossRef]
- Zhao, J.-Y.; Ding, J.-H.; Li, Z.-H.; Feng, T.; Zhang, H.-B.; Liu, J.-K. Two new compounds from cultures of the basidiomycete Daedaleopsis tricolor. J. Asian Nat. Prod. Res. 2019, 22, 941–946. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.-M.; Hae-Ryong, J.; Min, T.-J. Purification, Structure Determination and Biological Activities of 20(29)-lupen-3-one from Daedaleopsis tricolor (Bull. ex Fr.) Bond. et Sing. Bull. Korean Chem. Soc. 2001, 22, 59–62. [Google Scholar]
- Zhao, J.-Y.; Feng, T.; Li, Z.-H.; Dong, Z.-J.; Zhang, H.-B.; Liu, J.-K. Sesquiterpenoids and an ergosterol from cultures of the fungus Daedaleopsis tricolor. Nat. Prod. Bioprospect. 2013, 3, 271–276. [Google Scholar] [CrossRef]
- Sirakami-Sanchi. Available online: https://whc.unesco.org/en/list/663/ (accessed on 16 July 2021).
- Vidari, G.; Garlaschelli, L.; Rossi, A.; Vita-Finzi, P. New protoilludane sesquiterpenes from Lactarius violascens. Tetrahedron Lett. 1998, 39, 1957–1960. [Google Scholar] [CrossRef]
- McMorris, T.C.; Lira, R.; Gantzel, P.K.; Kelner, M.J.; Dawe, R. Sesquiterpenes from the Basidiomycete Omphalotus illudens. J. Nat. Prod. 2000, 63, 1557–1559. [Google Scholar] [CrossRef]
- Frelek, J.; Szczepek, W.J.; Neubrech, S.; Schultheis, B.; Brechtel, J.; Kuball, H.-G. Chiroptical Properties of cisoid Enones from Circular Dichroism (CD) and Anisotropic Circular Dichroism (ACD) Spectroscopy. Chem. Eur. J. 2002, 8, 1899–1907. [Google Scholar] [CrossRef]
- Liu, G.; Romo, D. Total Synthesis of (+)-Omphadiol. Angew. Chem. Int. Ed. 2011, 50, 7537–7540. [Google Scholar] [CrossRef] [PubMed]
- Hanssen, H.-P.; Abraham, W.-R. Sesquiterpene alcohols with novel skeletons from the fungus Ceratocystis piceae (Ascomycotina). Tetrahedron 1988, 44, 2175–2180. [Google Scholar] [CrossRef]
- Liu, H.; Tan, H.; Chen, K.; Chen, Y.; Li, S.; Li, H.; Zhang, W. Cerrenins A-C, cerapicane and isohirsutane sesquiterpenoids from the endophytic fungus Cerrena sp. Fitoterapia 2018, 129, 173–178. [Google Scholar] [CrossRef]
- Hirota, M.; Shimizu, Y.; Kamo, T.; Makabe, H.; Shibata, H. New Plant Growth Promoters, Repraesentins A, B and C, from Lactarius repraesentaneus. Biosci. Biotechnol. Biochem. 2003, 67, 1597–1600. [Google Scholar] [CrossRef] [PubMed]
- Ayer, W.A.; Saeedi-Ghomi, M.H. l-Sterpurene-3,12,14-triol and 1-sterpurene, metabolites of silver-leaf disease fungus Stereum purpureum. Can. J. Chem. 1981, 59, 2536–2538. [Google Scholar] [CrossRef]
- McMorris, T.C.; Anchel, M. The Structures of the Basidiomycete Metabolites Illudin S and Illudin M. J. Am. Chem. Soc. 1963, 85, 831–832. [Google Scholar] [CrossRef]
- Hehre, W.J.; Kuunzinger, P.; Deppmeier, B.; Driessen, A.; Uchida, N.; Hashimoto, M.; Fukushi, E.; Takata, Y. Efficient Protocol for Accurately Calculating 13C Chemical Shifts of Conformationally Flexible Natural Products: Scope, Assessment and Limitations. J. Nat. Prod. 2019, 82, 2299–2306. [Google Scholar] [CrossRef]
- Wang, B.; Dossey, A.T.; Walse, S.S.; Edison, A.; Merz, J.K.M. Relative Configuration of Natural Products Using NMR Chemical Shifts. J. Nat. Prod. 2009, 72, 709–713. [Google Scholar] [CrossRef]
- Bifulco, G.; Dambruoso, P.; Gomez-Paloma, L.; Riccio, R. Determination of Relative Configuration in Organic Compounds by NMR Spectroscopy and Computational Methods. Chem. Rev. 2007, 107, 3744–3779. [Google Scholar] [CrossRef]
- Barone, G.; Gomez-Paloma, L.; Duca, D.; Silvestri, A.; Riccio, R.; Bifulco, G. Structure Validation of Natural Products by Quantum-Mechanical GIAO Calculations of 13C NMR Chemical Shifts. Chem. A Eur. J. 2002, 8, 3233–3239. [Google Scholar] [CrossRef]
- Wavefunction-Inc. Tutorial and User’s Guide, Spartan’18 for Windows, Macintosh and Linux; Wavefunction Inc.: Irvine, CA, USA, 2016. [Google Scholar]
- Smith, S.G.; Goodman, J.M. Assigning Stereochemistry to Single Diastereoisomers by GIAO NMR Calculation: The DP4 Probability. J. Am. Chem. Soc. 2010, 132, 12946–12959. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Murphy, W.S. The octant rule: Its place in organic stereochemistry. J. Chem. Educ. 1975, 52, 774. [Google Scholar] [CrossRef]
- Scott, A.I.; Wrixon, A.D. A symmetry rule for chiral olefins. Tetrahedron 1970, 26, 3695–3715. [Google Scholar] [CrossRef]
- de Meijere, A. Bonding Properties of Cyclopropane and Their Chemical Consequences. Angew. Chem. Int. Ed. 1979, 18, 809–826. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).