
Disproportionation of H2O2 Mediated by Diiron-Peroxo Complexes as Catalase Mimics

 ,
,
Abstract
:1. Introduction
2. Results and Discussions
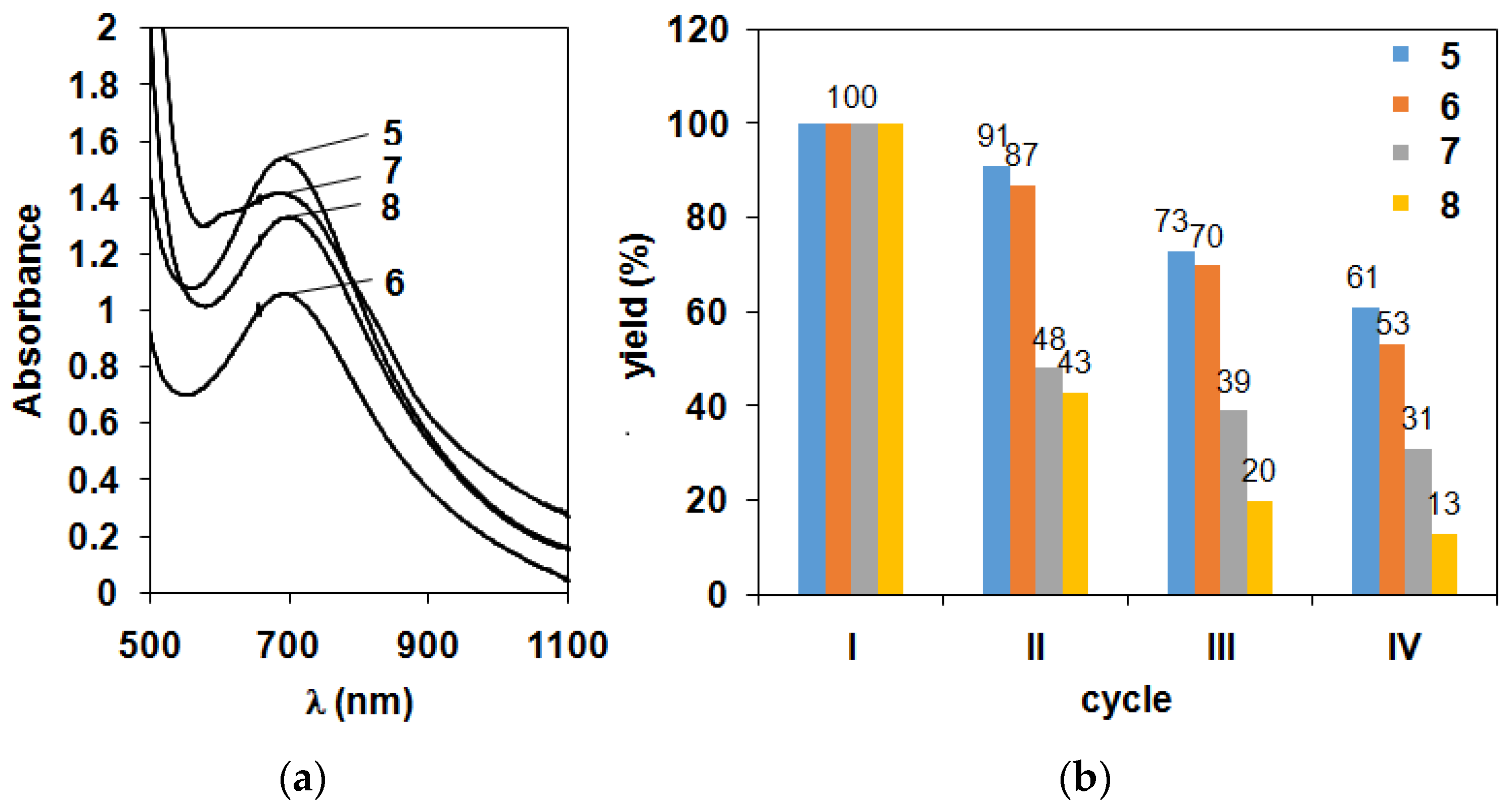
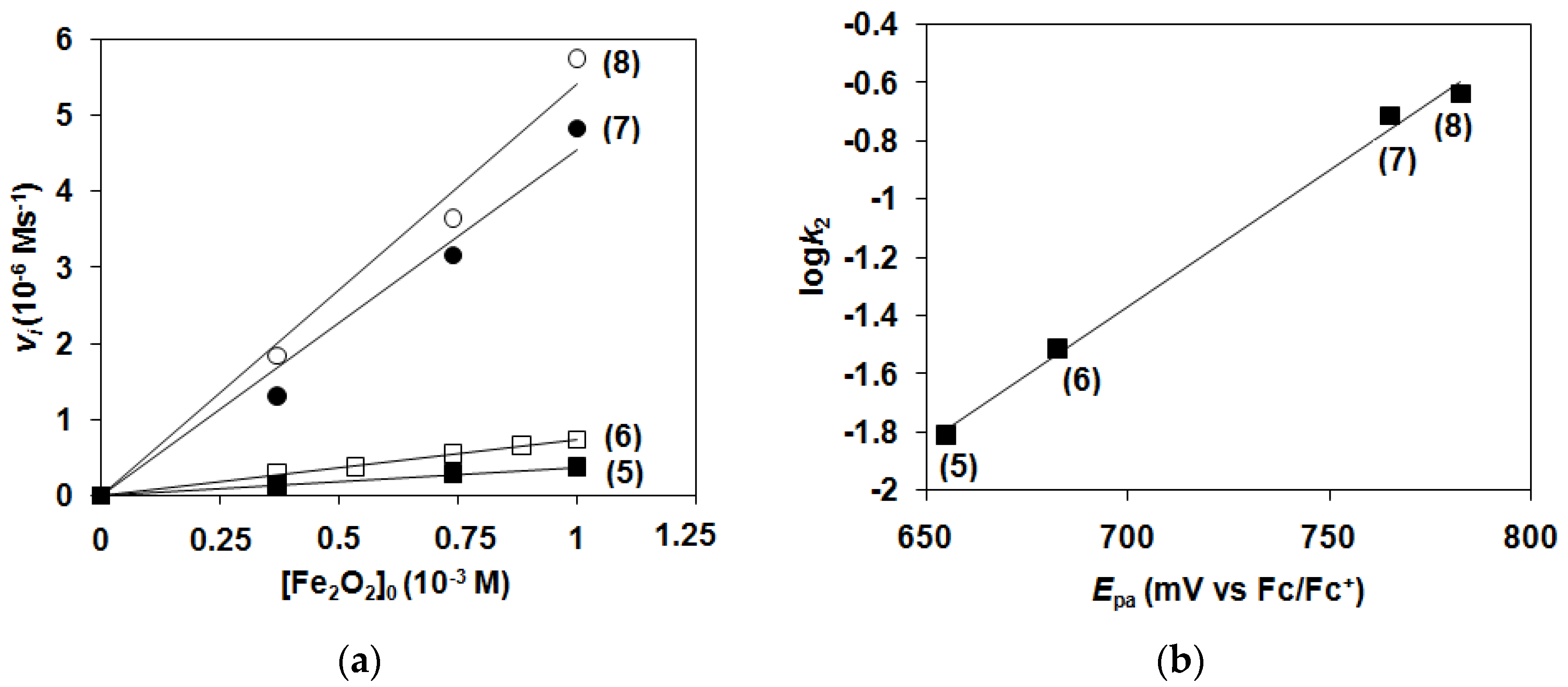
2.1. Kinetic Studies on the Formation of Diiron-Peroxo Complexes (5–8)
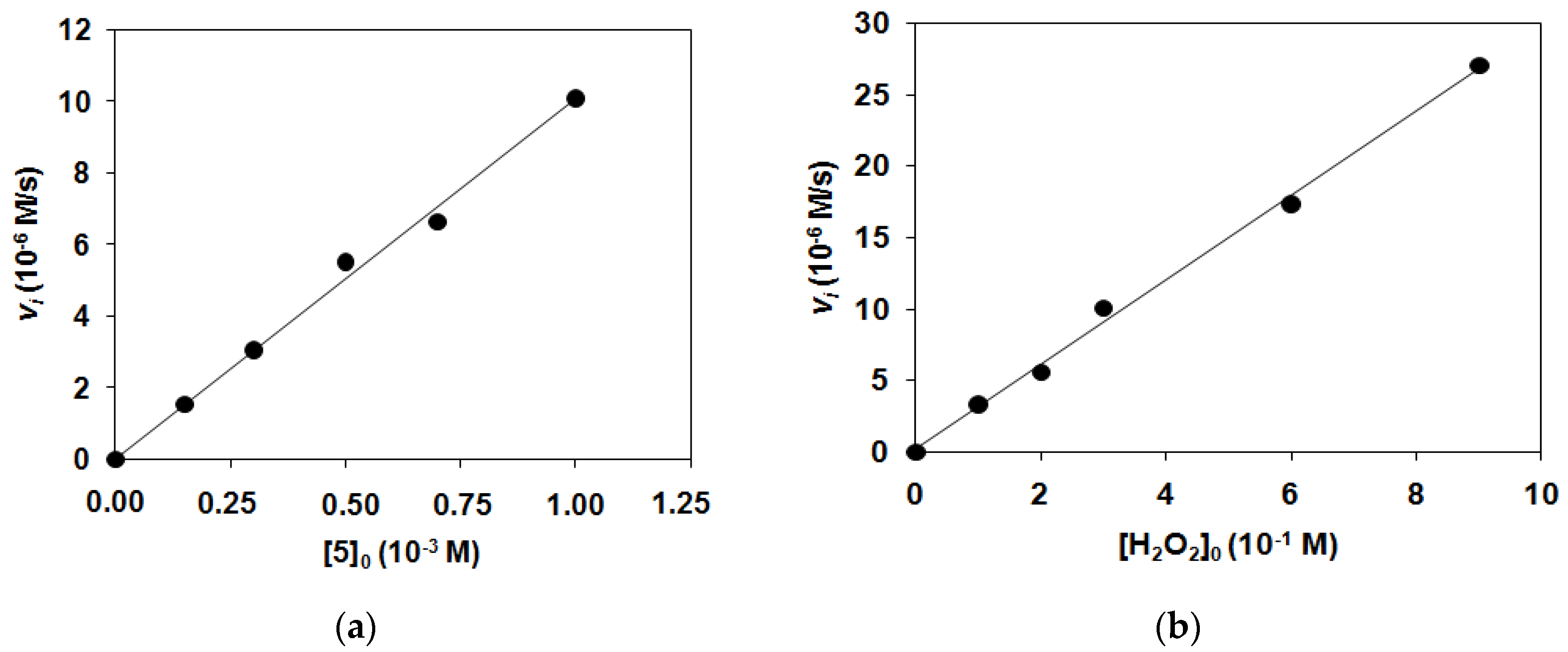
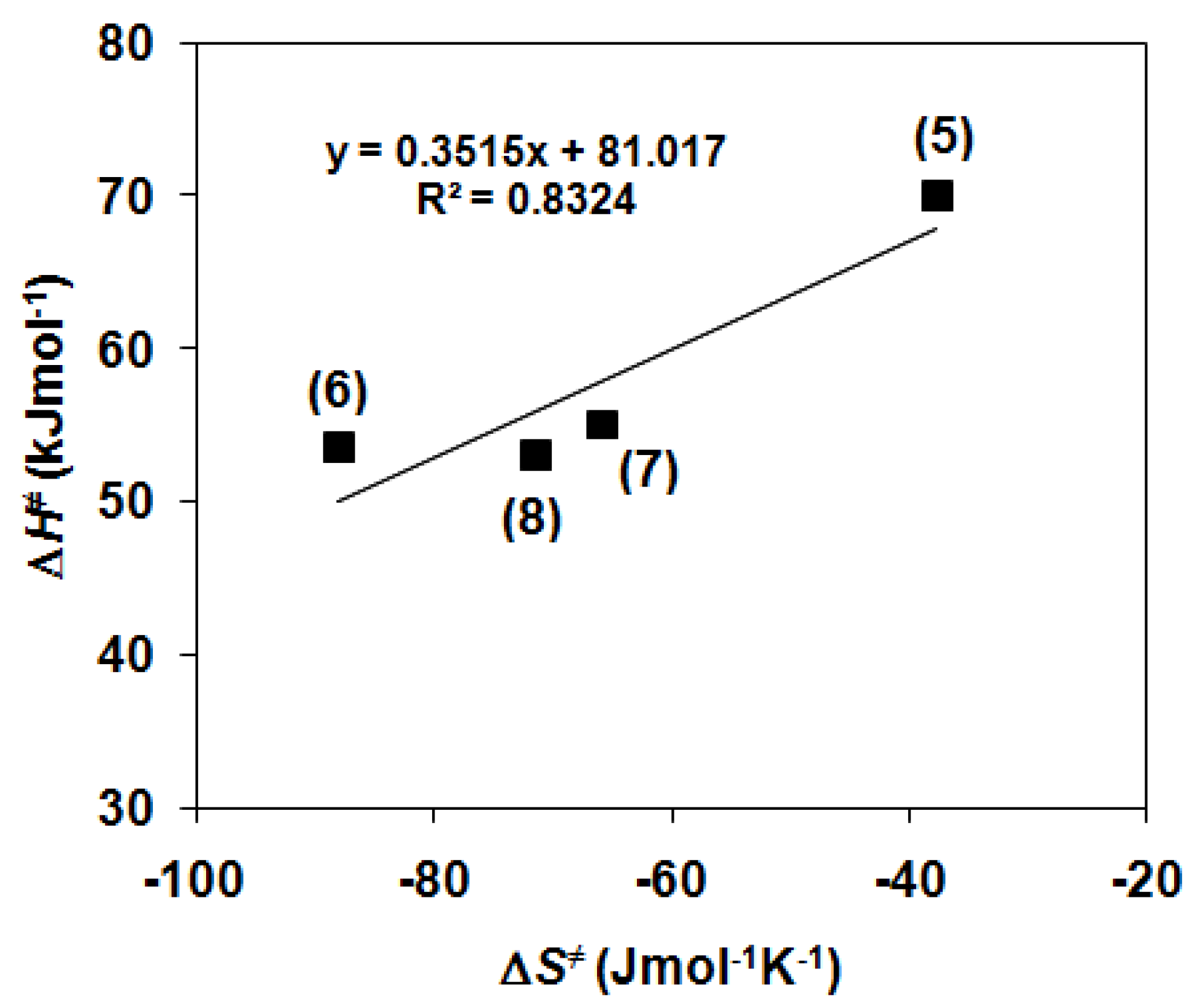
2.2. Catalase-Like Activity of Peroxo-Diiron Complexes
3. Experimental
3.1. Materials and Methods
3.2. Characterization of Ligands and Their Complexes
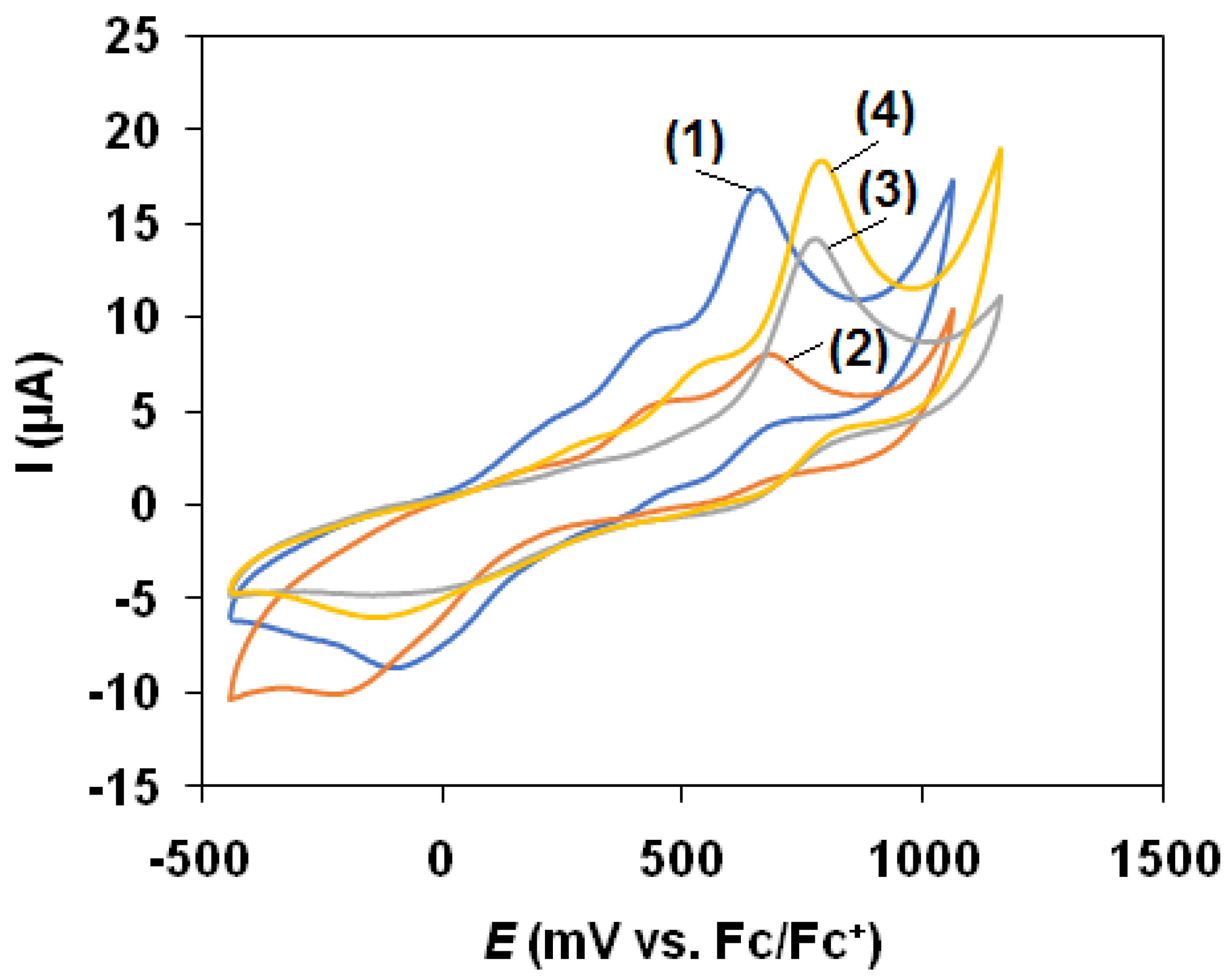
3.3. Cyclic Voltammetry
3.4. Generation of Diiron-Peroxo Complexes (5–8)
3.5. Diiron-Peroxo-Mediated Disproportionaton of H2O2
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Beyer, W.F.; Fridovich, I. Catalases-with and without heme. Basic Life Sci. 1988, 49, 651–661. [Google Scholar] [PubMed]
- Nicholls, P.; Fita, I.; Loewen, P.C. Enzymology and structure of catalases. Adv. Inorg. Chem. 2001, 51, 51–106. [Google Scholar] [CrossRef]
- Kono, Y.; Fridovich, I. Isolation and characterization of the pseudocatalase of Lactobacillus plantarum. J. Biol. Chem. 1983, 258, 6015–6019. [Google Scholar] [CrossRef]
- Barynin, V.V.; Whittaker, M.M.; Antonyuk, S.V.; Lamzin, V.S.; Harrison, P.M.; Artymiuk, P.J.; Whittaker, J.W. Crystal Structure of Manganese Catalase from Lactobacillus plantarum. Structure 2001, 9, 725–738. [Google Scholar] [CrossRef]
- Antonyuk, S.V.; Melik-Adman, V.R.; Popov, A.N.; Lamzin, V.S.; Hempstead, P.D.; Harrison, P.M.; Artymyuk, P.J.; Barynin, V.V. Three-dimensional structure of the enzyme dimanganese catalase from Thermus thermophilus at 1 Å resolution. Crystallogr. Rep. 2000, 45, 105–113. [Google Scholar] [CrossRef]
- Barynin, V.V.; Grebenko, A.I. T-catalase is nonheme catalase of the extremely thermophilic bacterium Thermus thermophilus HB8. Dokl. Akad. Nauk SSSR 1986, 286, 461–464. [Google Scholar]
- Allgood, G.S.; Perry, J.J. Characterization of a manganese-containing catalase from the obligate thermophile Thermoleophilum album. J. Bacteriol. 1986, 168, 563–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amo, T.; Atomi, H.; Imanaka, T. Unique Presence of a Manganese Catalase in a Hyperthermophilic Archaeon, Pyrobaculum calidifontis VA1. J. Bacteriol. 2002, 184, 3305–3312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whittaker, J.W. Non-heme manganese catalase-the “other” catalase. Arch. Biochem. Biophys. 2012, 525, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Cardenas, J.P.; Quatrini, R.; Holmes, D.S. Aerobic Lineage of the Oxidative Stress Response Protein Rubrerythrin Emerged in an Ancient Microaerobic, (Hyper)Thermophilic Environment. Front. Microbiol. 2016, 7, 1822–1831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bihani, S.C.; Chakravarty, D.; Ballal, A. KatB, a cyanobacterial Mn-catalase with unique active site configuration: Implications for enzyme function. Free Radic. Biol. Med. 2016, 93, 118–129. [Google Scholar] [CrossRef]
- Signorella, S.; Palopoli, C.; Ledesma, G. Rationally designed mimics of antioxidant manganoenzymes: Role of structural features in the quest for catalysts with catalaseand superoxide dismutase activity. Coord. Chem. Rev. 2018, 365, 75–102. [Google Scholar] [CrossRef]
- Wu, A.J.; Penner-Hahn, J.E.; Pecoraro, V.L. Structural, Spectroscopic, and Reactivity Models for the Manganese Catalases. Chem. Rev. 2004, 104, 903–938. [Google Scholar] [CrossRef] [PubMed]
- Tovmasyan, A.; Maia, C.G.C.; Weitner, T.; Carballal, S.; Sampaio, R.S.; Lieb, D.; Ghazaryan, R.; Ivanovic-Burmazovic, I.; Radi, R.; Reboucas, J.S.; et al. A comprehensive evaluation of catalase-like activity of different classes of redox-active therapeutics. Free Radic. Biol. Med. 2015, 86, 308–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batinic-Haberle, I.; Tovmasyan, A.; Spasojevic, I. An educational overview of the chemistry, biochemistry and therapeutic aspects of Mn porphyrins—From superoxide dismutation to HO-driven pathways. Redox Biol. 2015, 5, 43–65. [Google Scholar] [CrossRef] [Green Version]
- Kaizer, J.; Baráth, G.; Speier, G.; Réglier, M.; Giorgi, M. Synthesis, structure and catalase mimics of novel homoleptic manganese(II) complexes of 1,3-bis(2′-pyridylimino)isoindoline, Mn(4R-ind)2 (R = H, Me). Inorg. Chem. Commun. 2007, 10, 292–294. [Google Scholar] [CrossRef]
- Kaizer, J.; Kripli, B.; Speier, G.; Párkányi, L. Synthesis, structure, and catalase-like activity of a novel manganese(II) complex: Dichloro[1,3-bis(2’-benzimidazolylimino)isoindoline]manganese(II). Polyhedron 2009, 28, 933–936. [Google Scholar] [CrossRef]
- Kaizer, J.; Csay, T.; Kővári, P.; Speier, G.; Párkányi, L. Catalase mimics of a manganese(II) complex: The effect of axial ligands and pH. J. Mol. Catal. A Chem. 2008, 280, 203–209. [Google Scholar] [CrossRef]
- Kripli, B.; Garda, Z.; Sólyom, B.; Tircsó, G.; Kaizer, J. Formation, stability and catalase-like activity of mononuclear manganese(II) and oxomanganese(IV) complexes in protic and aprotic solvents. NJC 2020. [Google Scholar] [CrossRef]
- Stadtman, E.R.; Berlett, B.S.; Chock, P.B. Manganese-dependent disproportionation of hydrogen peroxide in bicarbonate buffer. Proc. Natl. Acad. Sci. USA 1990, 87, 384–388. [Google Scholar] [CrossRef] [Green Version]
- Kripli, B.; Sólyom, B.; Speier, G.; Kaizer, J. Stability and Catalase-Like Activity of a Mononuclear Non-Heme Oxoiron(IV) Complex in Aqueous Solution. Molecules 2019, 24, 3236. [Google Scholar] [CrossRef] [Green Version]
- Szávuly, M.I.; Surducan, M.; Nagy, E.; Surányi, M.; Speier, G.; Silaghi-Dumitrescu, R.; Kaizer, J. Functional models of nonheme enzymes: Kinetic and computational evidence for the formation of oxoiron(IV) species from peroxo-diiron(III) complexes, and their reactivity towards phenols and H2O2. Dalton Trans. 2016, 45, 14709–14718. [Google Scholar] [CrossRef] [PubMed]
- Pap, J.S.; Draksharapu, A.; Giorgi, M.; Browne, W.R.; Kaizer, J.; Speier, G. Stabilisation of mu-peroxido-bridged Fe(III) intermediates with non-symmetric bidentate N-donor ligands. Chem. Commun. 2014, 50, 1326–1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pap, J.S.; Cranswick, M.A.; Balogh-Hergovich, É.; Baráth, G.; Giorgi, M.; Rohde, G.T.; Kaizer, J.; Speier, G.; Que, L., Jr. An Iron(II)[1,3-bis(2’-pyridylimino)isoindoline] Complex as a Catalyst for Substrate Oxidation with H2O2—Evidence for a Transient Peroxidodiiron(III) Species. Eur. J. Inorg. Chem. 2013, 3858–3866. [Google Scholar] [CrossRef] [Green Version]
- Kripli, B.; Csendes, F.V.; Török, P.; Speier, G.; Kaizer, J. Stoichiometric Aldehyde Deformylation Mediated by Nucleophilic Peroxo-diiron(III) Complex as a Functional Model of Aldehyde Deformylating Oxygenase. Chem. Eur. J. 2019, 25, 14290–14294. [Google Scholar] [CrossRef]
- Török, P.; Unjaroen, D.; Csendes, F.V.; Giorgi, M.; Browne, W.R.; Kaizer, J. A nonheme peroxo-diiron(III) complex exhibiting both nucleophilic and electrophilic oxidation of organic substrates. Dalton Trans. 2021, 50, 7181–7185. [Google Scholar] [CrossRef] [PubMed]
- Kripli, B.; Szávuly, M.; Csendes, F.V.; Kaizer, J. Functional models of nonheme diiron enzymes: Reactivity of the mu-oxo-mu-1,2-peroxo-diiron(III) intermediate in electrophilic and nucleophilic reactions. Dalton Trans. 2020, 49, 1742–1746. [Google Scholar] [CrossRef] [PubMed]
- Oloo, W.N.; Fielding, A.J.; Que, L., Jr. Rate determining water assisted O-O bond cleavage of a FeIII-OOH intermediate in a bioinspired nonheme iron-catalyzed oxidation. J. Am. Chem. Soc. 2013, 135, 6438–6441. [Google Scholar] [CrossRef]
- Meena, B.I.; Kaizer, J. Design and Fine-Tuning Redox Potentials of Manganese(II) Complexes with Ioindoline-Based Ligands: H2O2 Oxidation and Oxidative Bleaching Performance in Aqueous Solution. Catalysts 2020, 10, 404. [Google Scholar] [CrossRef] [Green Version]
- Jensen, M.P.; Payeras, A.M.I.; Fiedler, A.T.; Costas, M.; Kaizer, J.; Stubna, A.; Münck, E.; Que, L., Jr. Kinetic Analysis of the Conversion of Nonheme (Alkylperoxo)iron(III) Species to Iron(IV) Complexes. Inorg. Chem. 2007, 46, 2398–2408. [Google Scholar] [CrossRef] [Green Version]
- Csonka, R.; Speier, G.; Kaizer, J. Isoindoline-derived ligands and applications. RSC Adv. 2015, 5, 18401–18419. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cycles | Yield (5) and (O2)/% | Yield (6)/% | Yield (7)/% | Yield (8)/% |
|---|---|---|---|---|
| I. | 100 (95) | 100 | 100 | 100 |
| II. | 91 (92) | 87 | 48 | 43 |
| III. | 73 (78) | 70 | 39 | 20 |
| IV. | 61 (64) | 53 | 31 | 13 |
| V. | 41 (27) | 36 | 26 | 12 |
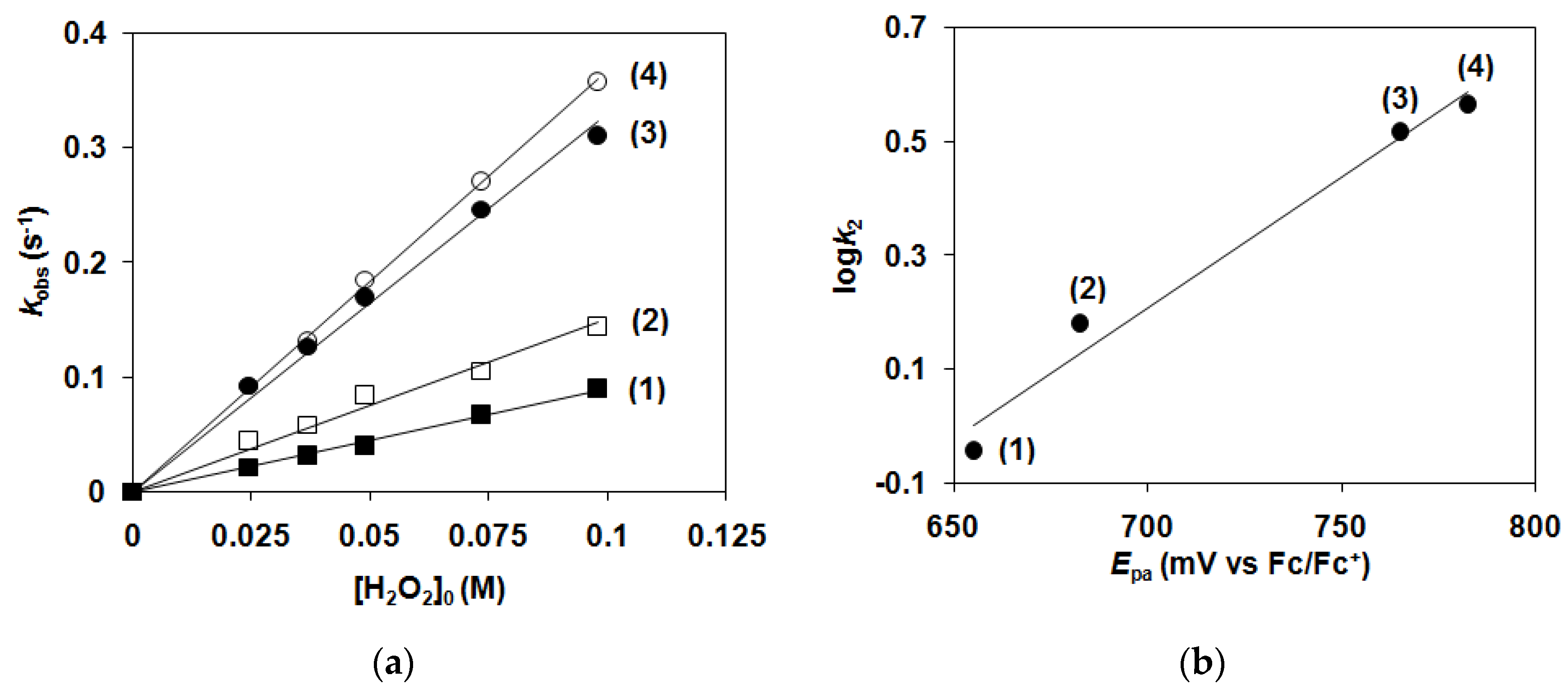
| Nr. | Complex | T/°C | [1–4]0/mM | [H2O2]0/M | kobs10−2 s−1 | k2/M−1 s−1 |
|---|---|---|---|---|---|---|
| 1 | 1 | 15 | 2 | 0.0243 | 2.12 | 0.87 ± 0.03 |
| 2 | 1 | 15 | 2 | 0.0367 | 3.15 | 0.86 ± 0.04 |
| 3 | 1 | 15 | 2 | 0.0489 | 4.07 | 0.83 ± 0.03 |
| 4 | 1 | 15 | 2 | 0.0734 | 6.76 | 0.92 ± 0.04 |
| 5 | 1 | 15 | 2 | 0.0979 | 9.04 | 0.92 ± 0.03 |
| 6 | 2 | 15 | 2 | 0.0243 | 4.45 | 1.83 ± 0.09 |
| 7 | 2 | 15 | 2 | 0.0367 | 5.83 | 1.58 ± 0.05 |
| 8 | 2 | 15 | 2 | 0.0489 | 8.44 | 1.72 ± 0.06 |
| 9 | 2 | 15 | 2 | 0.0734 | 10.5 | 1.43 ± 0.04 |
| 10 | 2 | 15 | 2 | 0.0979 | 14.45 | 1.48 ± 0.04 |
| 11 | 3 | 15 | 2 | 0.0243 | 9.22 | 3.79 ± 0.15 |
| 12 | 3 | 15 | 2 | 0.0367 | 12.64 | 3.44 ± 0.13 |
| 13 | 3 | 15 | 2 | 0.0489 | 16.95 | 3.46 ± 0.14 |
| 14 | 3 | 15 | 2 | 0.0734 | 24.61 | 3.35 ± 0.14 |
| 15 | 3 | 15 | 2 | 0.0979 | 31.07 | 3.17 ± 0.11 |
| 16 | 4 | 15 | 2 | 0.0243 | 9.25 | 3.81 ± 0.15 |
| 17 | 4 | 15 | 2 | 0.0367 | 13.17 | 3.59 ± 0.14 |
| 18 | 4 | 15 | 2 | 0.0489 | 18.48 | 3.78 ± 0.16 |
| 19 | 4 | 15 | 2 | 0.0734 | 27.07 | 3.69 ± 0.15 |
| 20 | 4 | 15 | 2 | 0.0979 | 35.83 | 3.66 ± 0.15 |
| Nr. | Complex | T/°C | [1–4]0/mM | [H2O2]0/M | kobs/10−3 s−1 | k2/10−2 M−1 s−1 |
|---|---|---|---|---|---|---|
| 1 | 1 | 15 | 0.738 | 0.0243 | 0.358 | 1.47 ± 0.06 |
| 2 | 1 | 15 | 1.48 | 0.0243 | 0.387 | 1.59 ± 0.07 |
| 3 | 1 | 15 | 2 | 0.0243 | 0.370 | 1.52 ± 0.06 |
| 4 | 1 | 5 | 2 | 0.0243 | 0.116 | 0.477 ± 0.02 |
| 5 | 1 | 10 | 2 | 0.0243 | 0.160 | 0.658 ± 0.03 |
| 6 | 1 | 20 | 2 | 0.0243 | 0.523 | 2.15 ± 0.11 |
| 7 | 1 | 25 | 2 | 0.1 | 3.33 | 3.33 ± 0.11 |
| 8 | 1 | 25 | 2 | 0.2 | 5.57 | 2.79 ± 0.08 |
| 9 | 1 | 25 | 2 | 0.3 | 10.1 | 3.36 ± 0.09 |
| 10 | 1 | 25 | 2 | 0.6 | 17.3 | 2.89 ± 0.07 |
| 11 | 1 | 25 | 2 | 0.9 | 27.1 | 3.01 ± 0.06 |
| 12 | 1 | 25 | 0.3 | 0.3 | 10.3 | 3.42 ± 0.12 |
| 13 | 1 | 25 | 0.6 | 0.3 | 10.1 | 3.38 ± 0.11 |
| 14 | 1 | 25 | 1 | 0.3 | 11.1 | 3.68 ± 0.14 |
| 15 | 1 | 25 | 1.4 | 0.3 | 9.47 | 3.16 ± 0.13 |
| 16 | 1 | 10 | 2 | 0.3 | 1.99 | 0.663 ± 0.02 |
| 17 | 1 | 15 | 2 | 0.3 | 3.63 | 1.21 ± 0.05 |
| 18 | 1 | 20 | 2 | 0.3 | 6.12 | 2.04 ± 0.08 |
| 19 | 2 | 15 | 0.738 | 0.0243 | 0.823 | 3.39 ± 0.11 |
| 20 | 2 | 15 | 1.07 | 0.0243 | 0.692 | 2.85 ± 0.09 |
| 21 | 2 | 15 | 1.48 | 0.0243 | 0.772 | 3.18 ± 0.10 |
| 22 | 2 | 15 | 1.77 | 0.0243 | 0.744 | 3.06 ± 0.09 |
| 23 | 2 | 15 | 2 | 0.0243 | 0.730 | 3.00 ± 0.11 |
| 24 | 2 | 5 | 2 | 0.0243 | 0.332 | 1.37 ± 0.04 |
| 25 | 2 | 10 | 2 | 0.0243 | 0.413 | 1.70 ± 0.05 |
| 26 | 2 | 20 | 2 | 0.0243 | 1.09 | 4.49 ± 0.27 |
| 27 | 3 | 15 | 0.738 | 0.0243 | 3.92 | 16.1 ± 0.72 |
| 28 | 3 | 15 | 1.48 | 0.0243 | 4.71 | 19.4 ± 0.62 |
| 29 | 3 | 15 | 2 | 0.0243 | 4.83 | 19.9 ± 0.68 |
| 30 | 3 | 5 | 2 | 0.0243 | 2.42 | 9.96 ± 0.35 |
| 31 | 3 | 10 | 2 | 0.0243 | 3.41 | 14.01 ± 0.63 |
| 32 | 3 | 20 | 2 | 0.0243 | 8.90 | 36.6 ± 2.01 |
| 33 | 4 | 15 | 0.738 | 0.0243 | 5.49 | 22.6 ± 0.79 |
| 34 | 4 | 15 | 1.48 | 0.0243 | 5.44 | 22.4 ± 0.83 |
| 35 | 4 | 15 | 2 | 0.0243 | 5.75 | 23.8 ± 0.85 |
| 36 | 4 | 5 | 2 | 0.0243 | 2.89 | 11.9 ± 0.48 |
| 37 | 4 | 10 | 2 | 0.0243 | 4.20 | 17.3 ± 0.67 |
| 38 | 4 | 20 | 2 | 0.0243 | 10.2 | 42.01 ± 1.77 |
| 1 (5) | 2 (6) | 3 (7) | 4 (8) | |
|---|---|---|---|---|
| Epa(FeII/FeIII)/mV vs. Fc/Fc+ | 655 | 683 | 765 | 783 |
| Epc(FeII/FeIII)/(mV vs. Fc/Fc+ | −90 | −223 | −155 | −143 |
| ΔE/mV | 745 | 905 | 920 | 925 |
| yield/% | 85 | 100 | 92 | 85 |
| TON ([S]/[Fe]) | 10.4 | 12.2 | 11.3 | 10.4 |
| TOF ([S]/[Fe]/h) | 288 | 418 | 354 | 234 |
| k2(cat)/10−2 M−1 s−1 (15 °C) | 1.52 | 3.05 | 19.9 | 23.8 |
| EA/kJ mol−1 | 72.3 | 55.9 | 57.5 | 55.5 |
| ΔS≠/J mol−1 K−1 | −37.7 | −88.1 | −65.9 | −71.4 |
| ΔH≠/kJ mol−1 | 70 | 53.5 | 55 | 53.1 |
| ΔG≠/kJ mol−1 | 81 | 78.9 | 74 | 73.7 |
| kdecay/10−2s−1 (15 °C) | 0.13 | 0.15 | 1.22 | 1.41 |
| kform/M−1 s−1 (15 °C) | 0.93 | 1.43 | 3.16 | 3.66 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lakk-Bogáth, D.; Török, P.; Csendes, F.V.; Keszei, S.; Gantner, B.; Kaizer, J. Disproportionation of H2O2 Mediated by Diiron-Peroxo Complexes as Catalase Mimics. Molecules 2021, 26, 4501. https://doi.org/10.3390/molecules26154501
Lakk-Bogáth D, Török P, Csendes FV, Keszei S, Gantner B, Kaizer J. Disproportionation of H2O2 Mediated by Diiron-Peroxo Complexes as Catalase Mimics. Molecules. 2021; 26(15):4501. https://doi.org/10.3390/molecules26154501
Chicago/Turabian StyleLakk-Bogáth, Dóra, Patrik Török, Flóra Viktória Csendes, Soma Keszei, Beatrix Gantner, and József Kaizer. 2021. "Disproportionation of H2O2 Mediated by Diiron-Peroxo Complexes as Catalase Mimics" Molecules 26, no. 15: 4501. https://doi.org/10.3390/molecules26154501