
Trans-(−)-Kusunokinin: A Potential Anticancer Lignan Compound against HER2 in Breast Cancer Cell Lines?

 and
and
Abstract
:
1. Introduction
2. Results
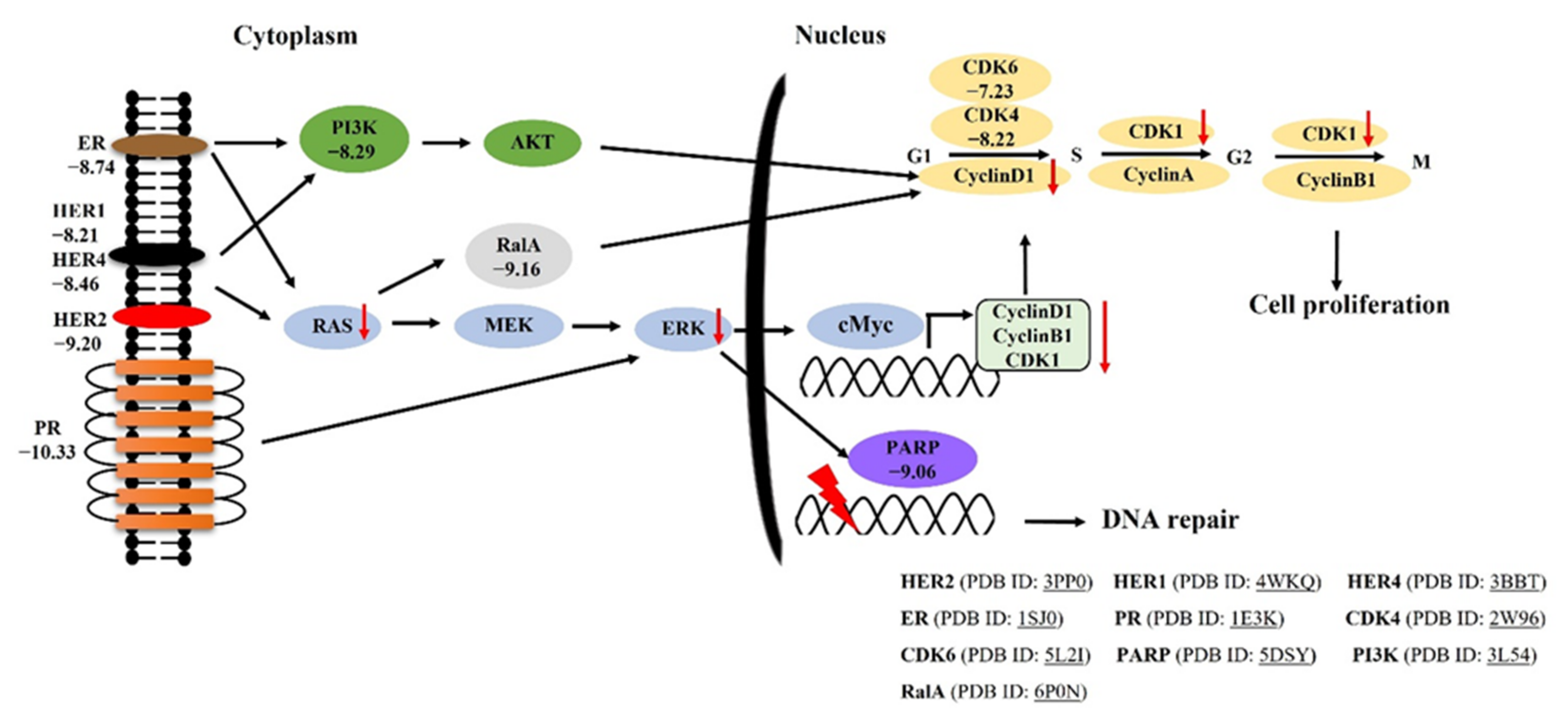
2.1. Molecular Docking of Trans-(−)-Kusunokinin and Trans-(+)-Kusunokinin with Breast Cancer Related-Candidate Proteins
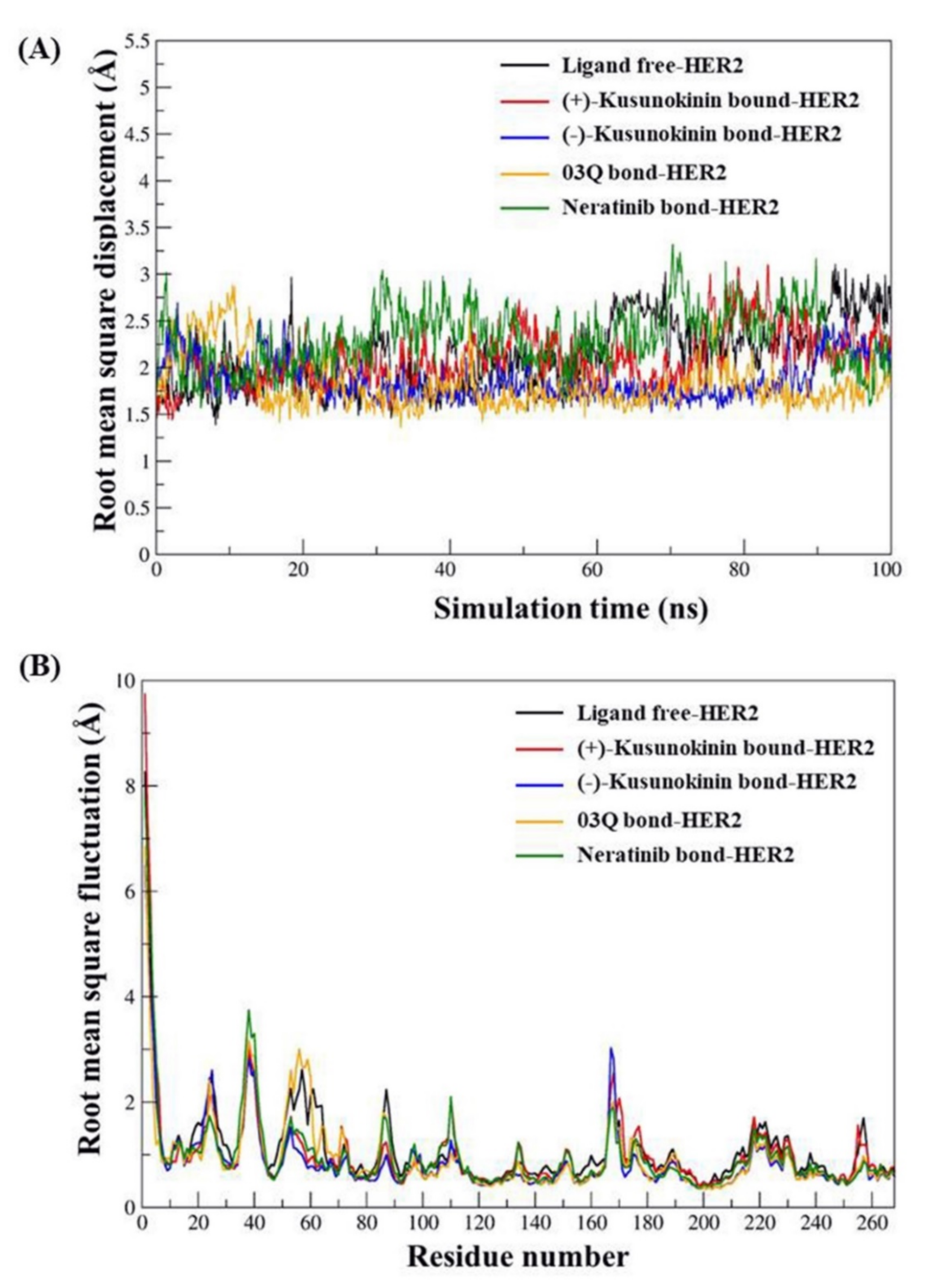
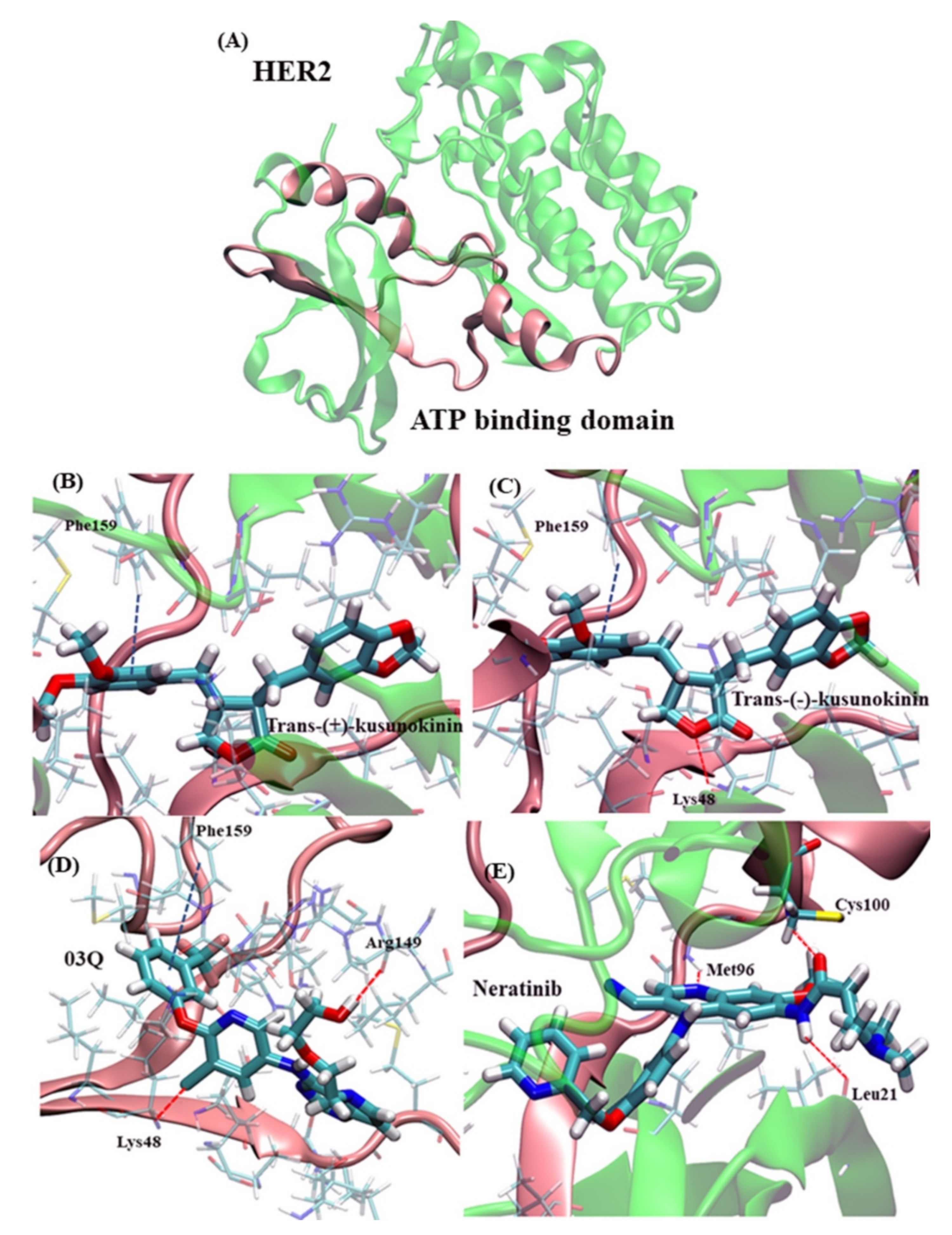
2.2. Atomistic Features of Trans-(−)-Kusunokinin and Trans-(+)-Kusunokinin and HER2
2.3. Cytotoxicity Effects of Synthetic (±)-Kusunokinin on Breast Cancer and Normal Cells
2.4. Inhibitory Effect of Synthetic (±)-Kusunokinin on Breast Cancer Cell Proliferation
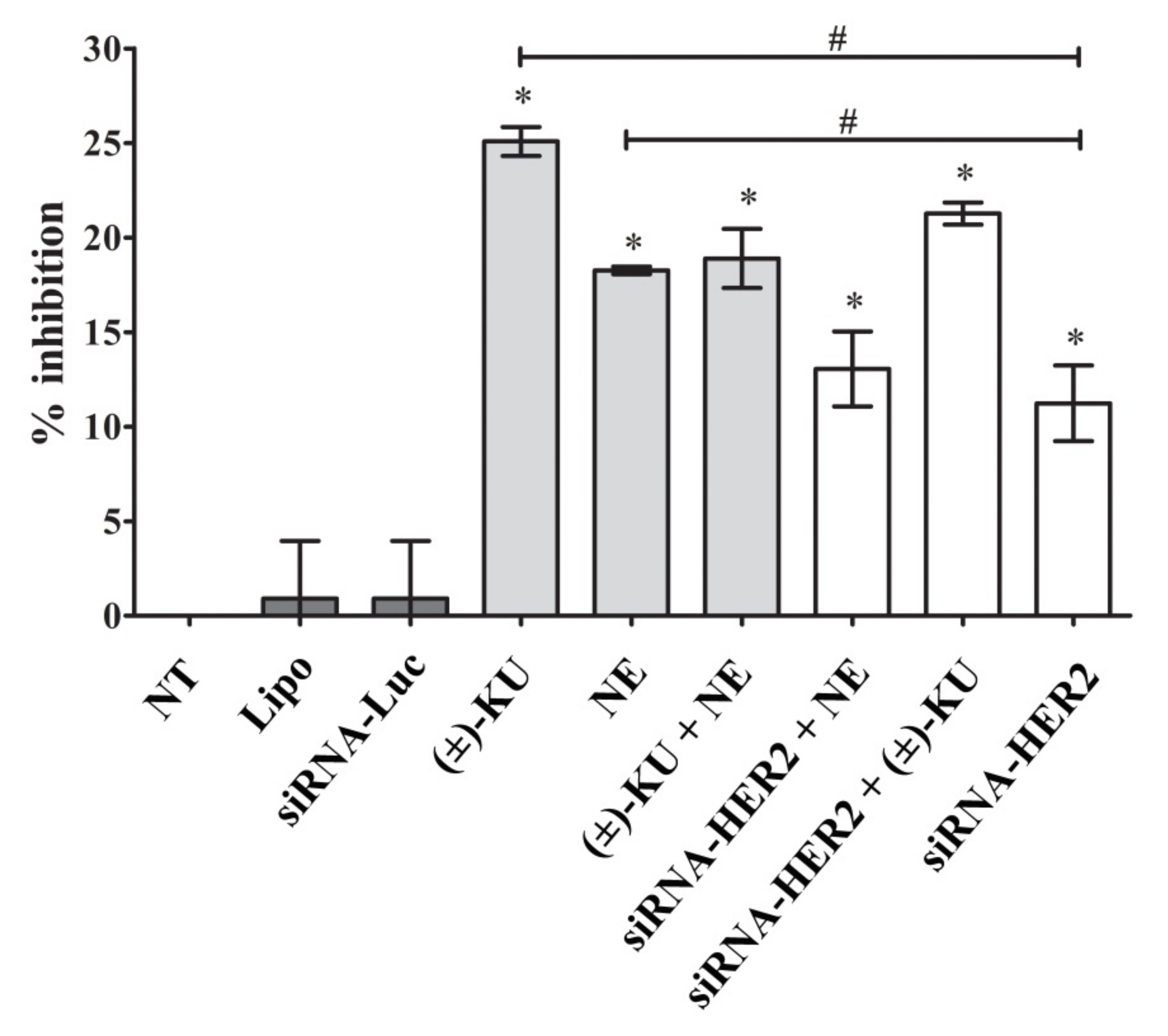
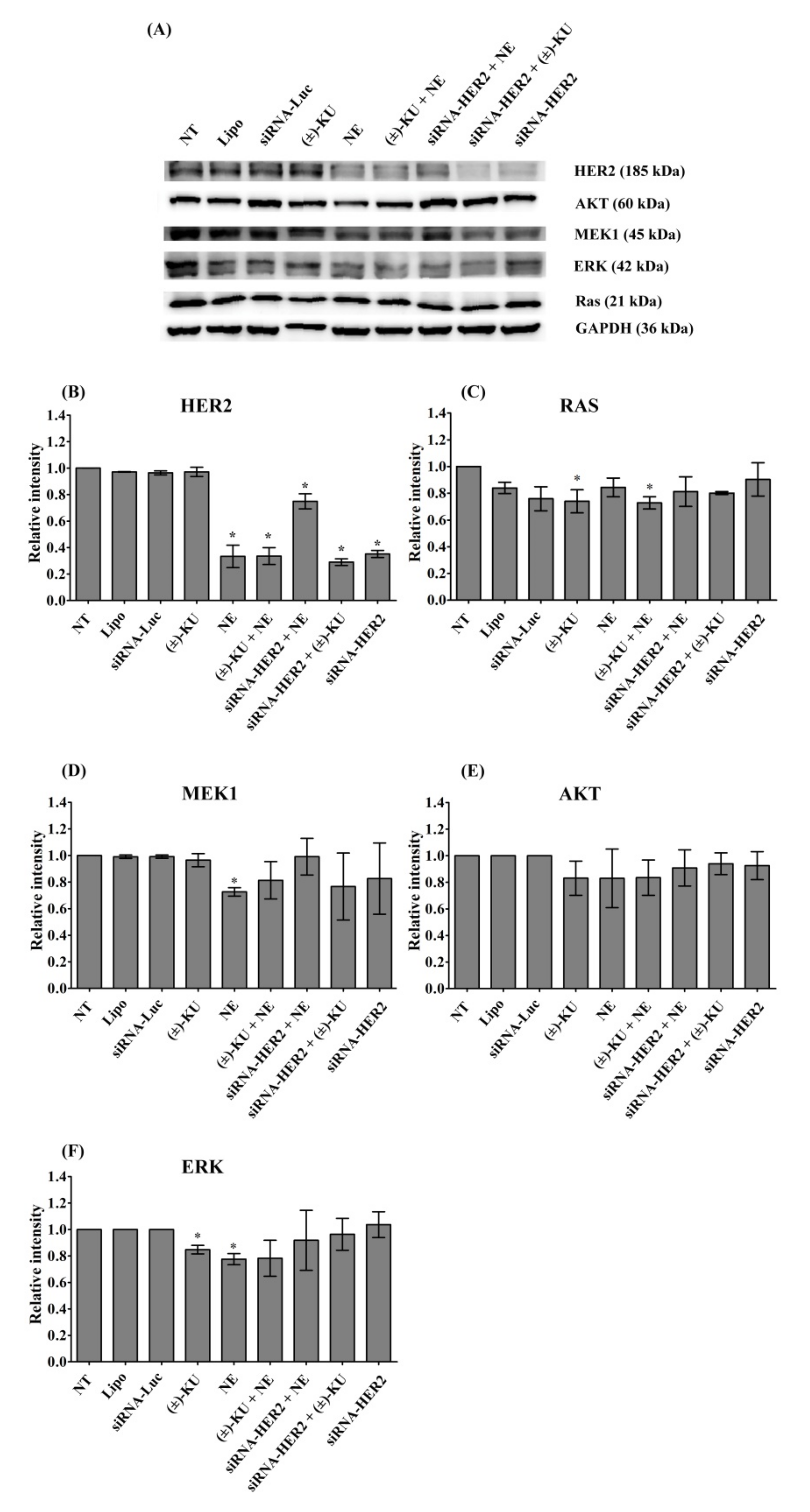
2.5. Synthetic (±)-Kusunokinin Inhibited Breast Cancer Cell Proliferation through the Suppression of RAS and ERK
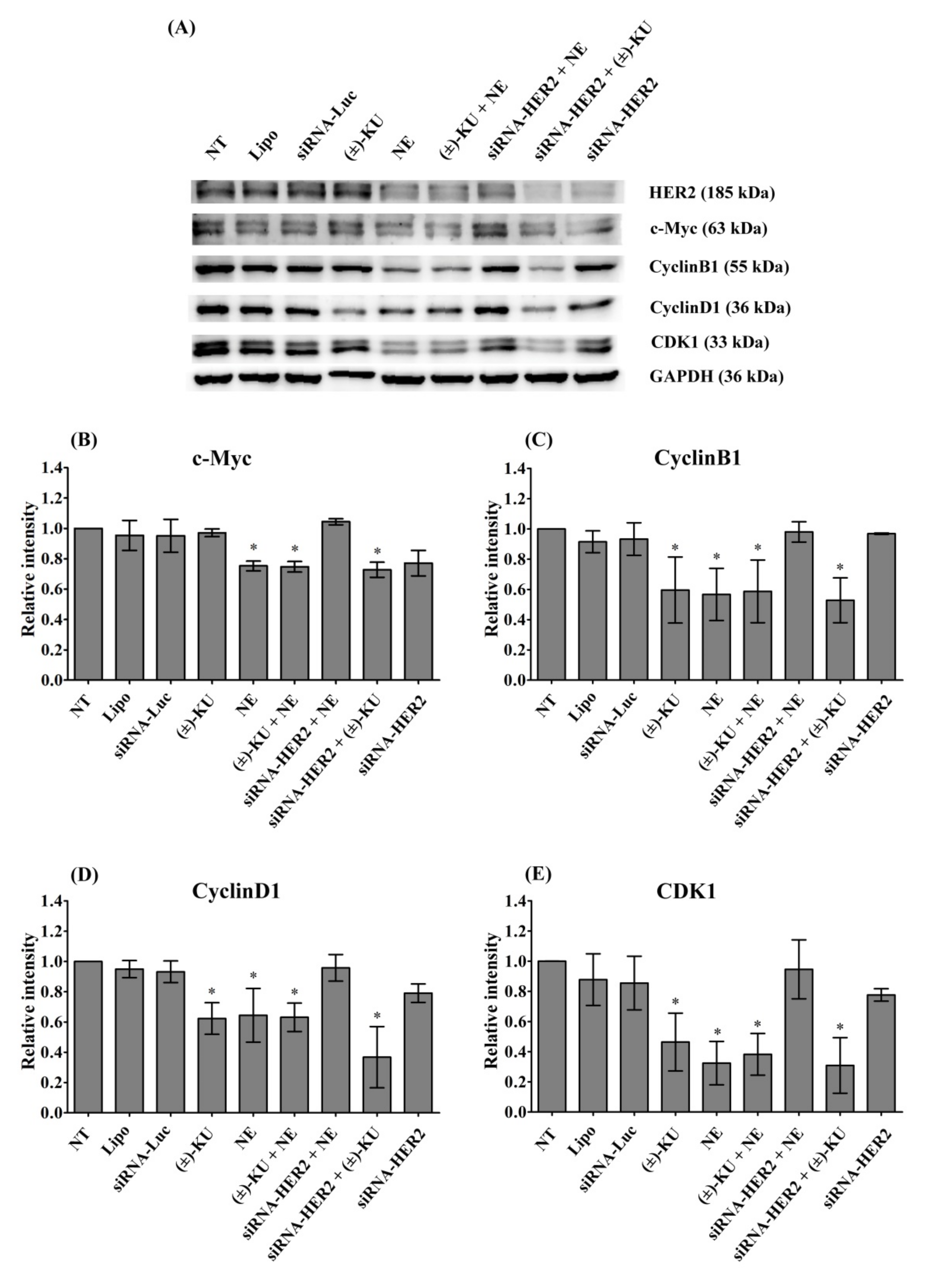
2.6. Synthetic (±)-Kusunokinin Decreased CyclinB1, CyclinD1 and CDK1; Down-Stream Proteins of HER2
3. Discussion
4. Materials and Methods
4.1. Compound Acquisition
4.2. Molecular Docking
4.3. Molecular Dynamics Simulation
4.4. Structural Analysis and Free Binding Energy Calculation
4.5. Cell Culture
4.6. Cytotoxicity Assay
4.7. In Vitro Transfection of Small Interfering RNA
4.8. Dye Exclusion Assay
4.9. Western Blot Analysis
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLO-BOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- McDonald, E.S.; Clark, A.S.; Tchou, J.; Zhang, P.; Freedman, G.M. Clinical diagnosis and management of breast cancer. J. Nucl. Med. 2016, 57, 9S–16S. [Google Scholar] [CrossRef] [Green Version]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Mori, R.; Nagao, Y. The efficacy of second-line hormone therapy for recurrence during adjuvant hormone therapy for breast cancer. Ther. Adv. Med. Oncol. 2014, 6, 36–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.H.; Hu, P.H.; Tu, J.H.; Yu, N.S. Luminal B breast cancer: Patterns of recurrence and clinical outcome. Oncotarget 2016, 7, 65024–65033. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Yu, X.; Li, S.; Tian, Y.; Liu, C. New perspectives for resistance to PARP inhibitors in triple-negative breast cancer. Front. Oncol. 2020, 10, 578095. [Google Scholar] [CrossRef]
- Binkhorst, L.; van Gelder, T.; Mathijssen, R.H.J. Individualization of tamoxifen treatment for breast carcinoma. Clin. Pharmacol. Ther. 2012, 92, 431–433. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that over-expresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef]
- Wieduwilt, M.J.; Moasser, M.M. The epidermal growth factor receptor family: Biology driving targeted therapeutics. Cell. Mol. Life Sci. 2008, 65, 1566–1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, B.; Fan, Y. CDK4/6 inhibition in early-stage breast cancer: How far is it from becoming standard of care? Lancet Oncol. 2021, 22, 159–160. [Google Scholar] [CrossRef]
- Curigliano, G. CDK4/6 inhibitors for HR+HER2+ early stage breast cancer—When to escalate treatment? Nat. Rev. Clin. Oncol. 2021, 18, 67–68. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, A.; Bertucci, A.; Bertucci, F. PARP inhibitors in the treatment of early breast cancer: The step beyond? Cancers 2020, 12, 1378. [Google Scholar] [CrossRef]
- Ellis, H.; Ma, C.X. PI3K inhibitors in breast cancer therapy. Curr. Oncol. Rep. 2019, 21, 110. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.; Nunes, M.R.; Stearns, V. CDK4/6 Inhibitors: Game changers in the management of hormone receptor–positive ad-vanced breast cancer? Oncology 2018, 32, 216–222. [Google Scholar] [PubMed]
- Zimmer, A.S.; Gillard, M.; Lipkowitz, S.; Lee, J.M. Update on PARP inhibitors in breast cancer. Curr. Treat. Options Oncol. 2018, 19, 21. [Google Scholar] [CrossRef]
- Juric, D.; Janku, F.; Rodón, J.; Burris, H.A.; Mayer, I.A.; Schuler, M.; Seggewiss-Bernhardt, R.; Gil-Martin, M.; Middleton, M.R.; Baselga, J.; et al. Alpelisib plus fulves-trant in PIK3CA-altered and PIK3CA-wild-type estrogen receptor-positive advanced breast cancer: A phase 1b clinical trial. JAMA Oncol. 2019, 5, e184475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, C.; Liu, D.; Li, L.; Wempe, M.F.; Guin, S.; Khanna, M.; Meier, J.; Hoffman, B.; Owens, C.; Wysoczynski, C.L.; et al. Discovery and characterization of small molecules that target the GTPase Ral. Nature 2014, 515, 443–447. [Google Scholar] [CrossRef] [Green Version]
- Sriwiriyajan, S.; Sukpondma, Y.; Srisawat, T.; Madla, S.; Graidist, P. (−)-Kusunokinin and piperloguminine from Piper nigrum: An alternative option to treat breast cancer. Biomed. Pharmacother. 2017, 92, 732–743. [Google Scholar] [CrossRef]
- Rattanaburee, T.; Thongpanchang, T.; Wongma, K.; Tedasen, A.; Sukpondma, Y.; Graidist, P. Anticancer activity of synthetic (±)-kusunokinin and its derivative (±)-bursehernin on human cancer cell lines. Biomed. Pharmacother. 2019, 117, 109115. [Google Scholar] [CrossRef]
- Tedasen, A.; Dokduang, S.; Sukpondma, Y.; Lailerd, N.; Madla, S.; Sriwiriyajan, S.; Rattanaburee, T.; Tipmanee, V.; Graidist, P. (−)-Kusunokinin inhibits breast cancer in N-nitrosomethylurea-induced mammary tumor rats. Eur. J. Pharmacol. 2020, 882, 173311. [Google Scholar] [CrossRef] [PubMed]
- Rattanaburee, T.; Tipmanee, V.; Tedasen, A.; Thongpanchang, T.; Graidist, P. Inhibition of CSF1R and AKT by (±)-kusunokinin hinders breast cancer cell proliferation. Biomed. Pharmacother. 2020, 129, 110361. [Google Scholar] [CrossRef] [PubMed]
- Tanawattanasuntorn, T.; Thongpanchang, T.; Rungrotmongkol, T.; Hanpaibool, C.; Graidist, P.; Tipmanee, V. (±)-Kusunokinin as a potential aldose reductase inhibitor: Equivalency observed via AKR1B1 dynamics simulation. ACS Omega 2021, 6, 606–614. [Google Scholar] [CrossRef]
- Feldinger, K.; Kong, A. Profile of neratinib and its potential in the treatment of breast cancer. Breast Cancer 2015, 7, 147–162. [Google Scholar] [CrossRef] [Green Version]
- Breslin, S.; Lowry, M.C.; O’Driscoll, L. Neratinib resistance and cross-resistance to other HER2-targeted drugs due to increased activity of metabolism enzyme cytochrome P4503A4. Br. J. Cancer 2017, 116, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Sudhan, D.R.; Guerrero-Zotano, A.; Won, H.; Gonzalez Ericsson, P.; Servetto, A.; Huerta-Rosario, M.; Ye, D.; Lee, K.M.; Formisano, L.; Guo, Y.; et al. Hyperactivation of TORC1 drives resistance to the pan-HER tyrosine kinase in-hibitor neratinib in HER2-mutant cancers. Cancer Cell 2020, 37, 183–199.e5. [Google Scholar] [CrossRef] [PubMed]
- Barcenas, C.H.; Hurvitz, S.A.; Di Palma, J.A.; Bose, R.; Chien, A.J.; Iannotti, N.; Marx, G.; Brufsky, A.; Litvak, A.; Ibrahim, E.; et al. Improved tolerability of neratinib in patients with HER2-positive early-stage breast cancer: The CONTROL trial. Ann. Oncol. 2020, 31, 1223–1230. [Google Scholar] [CrossRef]
- Mortimer, J.; Di Palma, J.; Schmid, K.; Ye, Y.; Jahanzeb, M. Patterns of occurrence and implications of neratinib-associated di-arrhea in patients with HER2-positive breast cancer: Analyses from the randomized phase III ExteNET trial. Breast Cancer Res. 2019, 21, 32. [Google Scholar] [CrossRef]
- Wissner, A.; Mansour, T.S. The development of HKI-272 and related compounds for the treatment of cancer. Arch. Pharm. 2008, 341, 465–477. [Google Scholar] [CrossRef]
- Anderson, W.F.; Chatterjee, N.; Ershler, W.B.; Brawley, O.W. Estrogen receptor breast cancer phenotypes in the surveillance, epidemiology, and end results database. Breast Cancer Res. Treat. 2002, 76, 27–36. [Google Scholar] [CrossRef]
- Colomer, R.; Beltran, M.; Dorcas, J.; Cortes-Funes, H.; Hornedo, J.; Valentin, V.; Vargas, C.; Mendiola, C.; Ciruelos, E. It is not time to stop progesterone receptor testing in breast cancer. J. Clin. Oncol. 2005, 23, 3868–3869. [Google Scholar] [CrossRef]
- Witton, C.J.; Reeves, J.R.; Going, J.J.; Cooke, T.G.; Bartlett, J.M. Expression of the HER1-4 family of receptor tyrosine kinases in breast cancer. J. Pathol. 2003, 200, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.L.; Hung, M.C. The role of HER2, EGFR, and other receptor tyrosine kinases in breast cancer. Cancer Metastasis Rev. 2016, 35, 575–588. [Google Scholar] [CrossRef] [Green Version]
- Maatta, J.A.; Sundvall, M.; Junttila, T.T.; Peri, L.; Laine, V.J.; Isola, J.; Egeblad, M.; Elenius, K. Proteolytic cleavage and phos-phorylation of a tumor-associated ErbB4 isoform promote ligand-independent survival and cancer cell growth. Mol. Biol. Cell 2006, 17, 67–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghoroghi, S.; Mary, B.; Larnicol, A.; Asokan, N.; Klein, A.; Osmani, N.; Busnelli, I.; Delalande, F.; Paul, N.; Halary, S.; et al. Ral GTPases promote breast cancer metastasis by controlling biogenesis and organ targeting of exosomes. eLife 2021, 10, e61539. [Google Scholar] [CrossRef] [PubMed]
- Aertgeerts, K.; Skene, R.; Yano, J.; Sang, B.C.; Zou, H.; Snell, G.; Jennings, A.; Iwamoto, K.; Habuka, N.; Hirokawa, A.; et al. Structural analysis of the mechanism of inhibition and allosteric activation of the kinase domain of HER2 protein. J. Biol. Chem. 2011, 286, 18756–18765. [Google Scholar] [CrossRef] [Green Version]
- Saeloh, D.; Wenzel, M.; Rungrotmongkol, T.; Hamoen, L.W.; Tipmanee, V.; Voravuthikunchai, S.P. Effects of rhodomyrtone on gram-positive bacterial tubulin homologue FtsZ. PeerJ 2017, 5, e2962. [Google Scholar] [CrossRef] [PubMed]
- Trowe, T.; Boukouvala, S.; Calkins, K.; Cutler, R.E.; Fong, R.; Funke, R.; Gendreau, S.B.; Kim, Y.D.; Miller, N.; Woolfrey, J.R.; et al. EXEL-7647 Inhibits mutant forms of ErbB2 as-sociated with lapatinib resistance and neoplastic transformation. Clin. Cancer Res. 2008, 14, 2465–2475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awada, A.; Colomer, R.; Inoue, K.; Bondarenko, I.; Badwe, R.A.; Demetriou, G.; Lee, S.C.; Mehta, A.O.; Kim, S.B.; Bachelot, T.; et al. Neratinib plus paclitaxel vs trastuzumab plus paclitaxel in previously untreated metastatic ERBB2-positive breast cancer: The NEfERT-T randomized clinical trial. JAMA Oncol. 2016, 2, 1557–1564. [Google Scholar] [CrossRef] [PubMed]
- Kulukian, A.; Lee, P.; Taylor, J.; Rosler, R.; de Vries, P.; Watson, D.; Forero-Torres, A.; Peterson, S. Preclinical activity of HER2-selective tyrosine kinase inhibitor tucatinib as a single agent or in combination with trastuzumab or docetaxel in solid tumor models. Mol. Cancer Ther. 2020, 19, 976–987. [Google Scholar] [CrossRef] [Green Version]
- Schlam, I.; Swain, S.M. HER2-positive breast cancer and tyrosine kinase inhibitors: The time is now. NPJ Breast Cancer 2021, 7, 56. [Google Scholar] [CrossRef]
- Bose, P.; Ozer, H. Neratinib: An oral, irreversible dual EGFR/HER2 inhibitor for breast and non-small cell lung cancer. Expert Opin. Investig. Drugs 2009, 18, 1735–1751. [Google Scholar] [CrossRef] [PubMed]
- Rabindran, S.K.; Discafani, C.M.; Rosfjord, E.C.; Baxter, M.; Floyd, M.B.; Golas, J.; Hallett, W.A.; Johnson, B.D.; Nilakantan, R.; Overbeek, E.; et al. Antitumor activity of HKI-272, an orally active, irreversible inhibitor of the HER-2 tyrosine kinase. Cancer Res. 2004, 64, 3958–3965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, F.; Ouyang, Q.; Li, W.; Jiang, Z.; Tong, Z.; Liu, Y.; Li, H.; Yu, S.; Feng, J.; Wang, S.; et al. Pyrotinib or lapatinib combined with capecitabine in HER2-positive metastatic breast cancer with prior taxanes, anthracyclines, and/or trastuzumab: A randomized, phase II study. J. Clin. Oncol. 2019, 37, 2610–2619. [Google Scholar] [CrossRef]
- Ishikawa, T.; Ichikawa, Y.; Shimizu, D.; Sasaki, T.; Tanabe, M.; Chishima, T.; Takabe, K.; Endo, I. The role of HER-2 in breast cancer. J. Surg. Sci. 2014, 2, 4–9. [Google Scholar]
- Moore, K.M.; Thomas, G.J.; Duffy, S.W.; Warwick, J.; Gabe, R.; Chou, P.; Ellis, I.O.; Green, A.R.; Haider, S.; Brouilette, K.; et al. Therapeutic targeting of integrin al-phavbeta6 in breast cancer. J. Natl. Cancer Inst. 2014, 106, dju169. [Google Scholar] [CrossRef] [Green Version]
- Witkiewicz, A.K.; Cox, D.; Knudsen, E.S. CDK4/6 inhibition provides a potent adjunct to Her2-targeted therapies in preclinical breast cancer models. Genes Cancer 2014, 5, 261–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choong, G.M.; Cullen, G.D.; O’Sullivan, C.C. Evolving standards of care and new challenges in the management of HER2-positive breast cancer. CA Cancer J. Clin. 2020, 70, 355–374. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, C.N. Epidermal growth factor receptor in non-small cell lung cancer. Transl. Lung Cancer Res. 2015, 4, 110–118. [Google Scholar] [CrossRef]
- Sherr, C.J.; Beach, D.; Shapiro, G.I. Targeting CDK4 and CDK6: From discovery to therapy. Cancer Discov. 2016, 6, 353–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zajac-Kaye, M. Myc oncogene: A key component in cell cycle regulation and its implication for lung cancer. Lung Cancer 2001, 34, S43–S46. [Google Scholar] [CrossRef]
- Ding, L.; Cao, J.; Lin, W.; Chen, H.; Xiong, X.; Ao, H.; Yu, M.; Lin, J.; Cui, Q. The roles of cyclin-dependent kinases in cell-cycle progression and therapeutic strategies in human breast cancer. Int. J. Mol. Sci. 2020, 21, 1960. [Google Scholar] [CrossRef] [Green Version]
- Canonici, A.; Gijsen, M.; Mullooly, M.; Bennett, R.; Bouguern, N.; Pedersen, K.; O’Brien, N.A.; Roxanis, I.; Li, J.L.; Bridge, E.; et al. Neratinib overcomes trastuzumab re-sistance in HER2 amplified breast cancer. Oncotarget 2013, 4, 1592–1605. [Google Scholar] [CrossRef] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian, Inc.: Walling-ford, CT, USA, 2016. [Google Scholar]
- Case, D.; Babin, V.; Berryman, J.; Betz, R.; Cai, Q.; Cerutti, D.; Cheatham III, T.; Darden, T.; Duke, R.; Gohlke, H. AMBER 16, 2015; University of California: San Francisco, CA, USA, 2015. [Google Scholar]
- Berendsen, H.J.; Postma, J.V.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Homeyer, N.; Gohlke, H. Free energy calculations by the molecular mechanics Poisson- Boltzmann surface area method. Mol. Inf. 2012, 31, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Jewboonchu, J.; Saetang, J.; Saeloh, D.; Siriyong, T.; Rungrotmongkol, T.; Voravuthikunchai, S.P.; Tipmanee, V. Atomistic in-sight and modeled elucidation of conessine towards Pseudomonas aeruginosa efflux pump. J. Biol. Struct. Dyn. 2020, 2020, 1828169. [Google Scholar] [CrossRef]
- Sriwiriyajan, S.; Ninpesh, T.; Sukpondma, Y.; Nasomyon, T.; Graidist, P. Cytotoxicity Screening of Plants of Genus Piper in Breast Cancer Cell Lines. Trop. J. Pharm. Res. 2014, 13, 921. [Google Scholar] [CrossRef] [Green Version]
- Graidist, P.; Phongdara, A.; Fujise, K. Antiapoptotic protein partners fortilin and MCL1 independently protect cells from 5-fluorouracil-induced cytotoxicity. J. Biol. Chem. 2004, 279, 40868–40875. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Protein | Known Inhibitor | Docking Score (kcal/mol) | ||
|---|---|---|---|---|
| Inhibitor | (−)-Kus 1 | (+)-Kus 2 | ||
| Human epidermal growth factor receptor 2 (HER2) | 03Q Neratinib | −9.13 −8.88 | −9.20 −9.20 | −9.16 −9.16 |
| Human epidermal growth factor receptor 1 (HER1) | Gefitinib | −7.69 | −8.21 | −8.03 |
| Human epidermal growth factor receptor 4 (HER4) | Lapatinib | −10.37 | −8.46 | −8.64 |
| Estrogen receptor (ER) | E4D | −13.55 | −8.74 | −8.87 |
| Progesterone receptor (PR) | R18 | −11.25 | −10.33 | −10.02 |
| Cyclin-dependent kinases 4 (CDK4) | Palbociclib | −9.02 | −8.22 | −9.09 |
| Cyclin-dependent kinases 6 (CDK6) | LQQ | −9.12 | −7.23 | −8.31 |
| Poly (ADP-ribose) polymerase (PARP) | UHB | −11.27 | −9.06 | −9.13 |
| Phosphoinositide−3 kinase (PI3K) | LXX | −8.46 | −8.29 | −9.15 |
| Ras-related protein Ral-A (RalA) | NLS | −9.64 | −9.16 | −9.33 |
| Inhibitor | MM/GBSA(kcal/mol) | MM/PBSA(kcal/mol) |
|---|---|---|
| Trans-(+)-kusunokinin | −39.03 ± 0.14 | −29.18 ± 0.18 |
| Trans-(−)-Kusunokinin | −44.06 ± 0.14 | −29.79 ± 0.15 |
| 03Q | −54.28 ± 0.17 | −40.78 ± 0.22 |
| Neratinib | −50.44 ± 0.18 | −40.75 ± 0.21 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rattanaburee, T.; Tanawattanasuntorn, T.; Thongpanchang, T.; Tipmanee, V.; Graidist, P. Trans-(−)-Kusunokinin: A Potential Anticancer Lignan Compound against HER2 in Breast Cancer Cell Lines? Molecules 2021, 26, 4537. https://doi.org/10.3390/molecules26154537
Rattanaburee T, Tanawattanasuntorn T, Thongpanchang T, Tipmanee V, Graidist P. Trans-(−)-Kusunokinin: A Potential Anticancer Lignan Compound against HER2 in Breast Cancer Cell Lines? Molecules. 2021; 26(15):4537. https://doi.org/10.3390/molecules26154537
Chicago/Turabian StyleRattanaburee, Thidarath, Tanotnon Tanawattanasuntorn, Tienthong Thongpanchang, Varomyalin Tipmanee, and Potchanapond Graidist. 2021. "Trans-(−)-Kusunokinin: A Potential Anticancer Lignan Compound against HER2 in Breast Cancer Cell Lines?" Molecules 26, no. 15: 4537. https://doi.org/10.3390/molecules26154537