
N-Skatyltryptamines—Dual 5-HT6R/D2R Ligands with Antipsychotic and Procognitive Potential

 , , ,
, , ,  , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Results
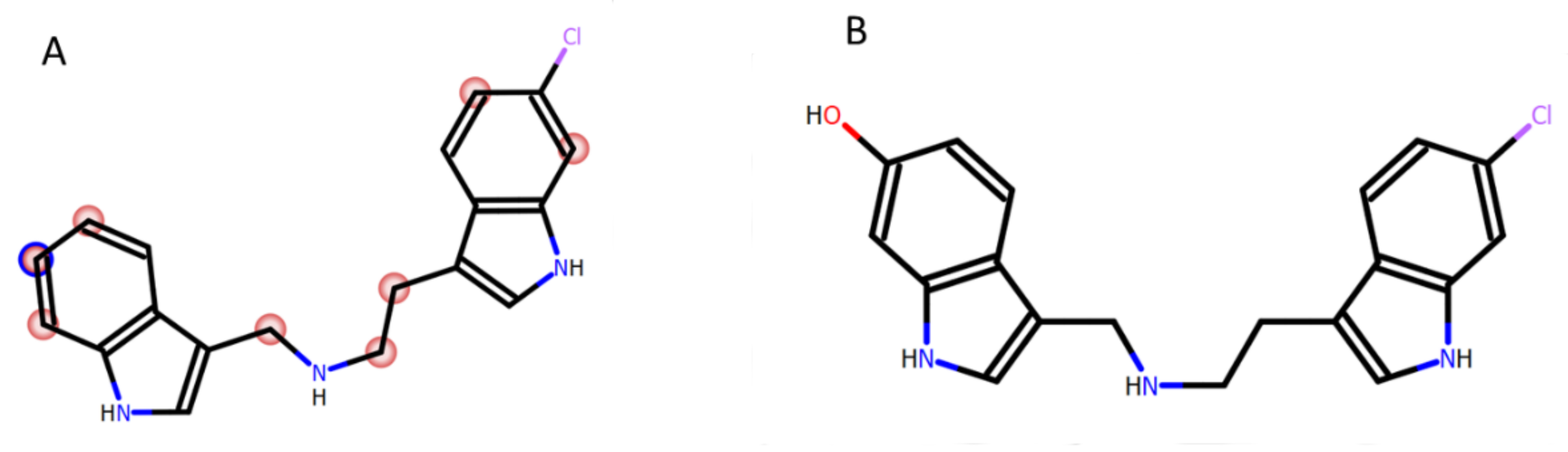
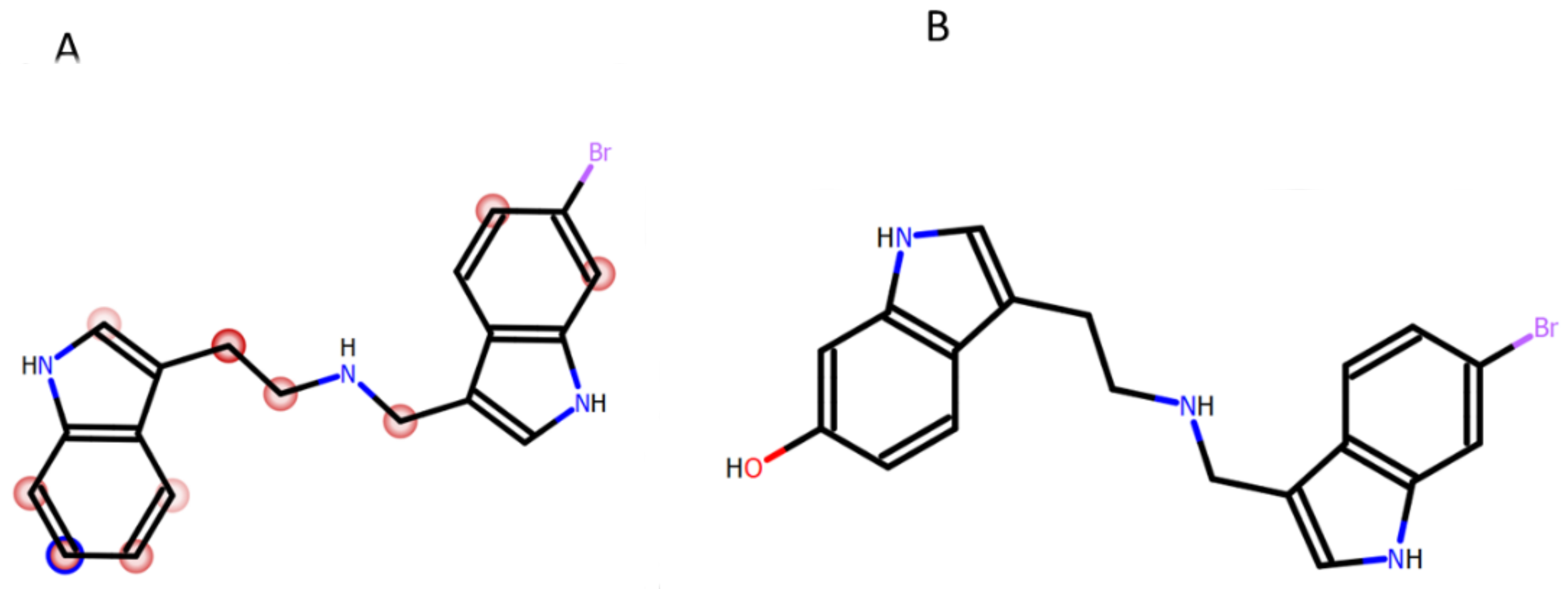
2.1. Chemistry
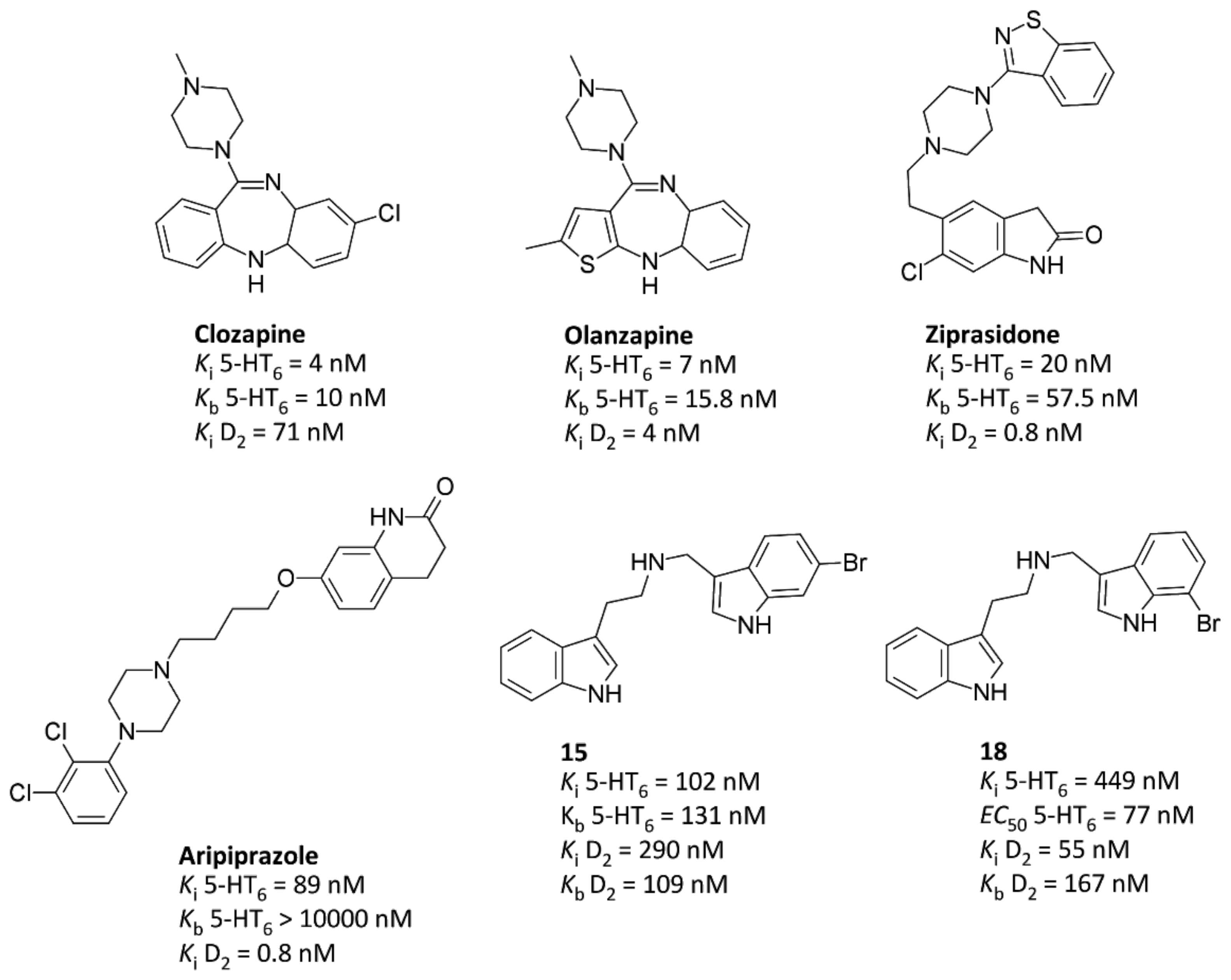
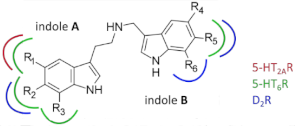
2.2. Structure–Affinity Relationship
2.3. Functional Assays toward 5-HT6R and D2R
2.4. Molecular Modeling
2.5. In Vitro ADMETox Studies
2.5.1. Permeability Assay
2.5.2. Metabolic Stability
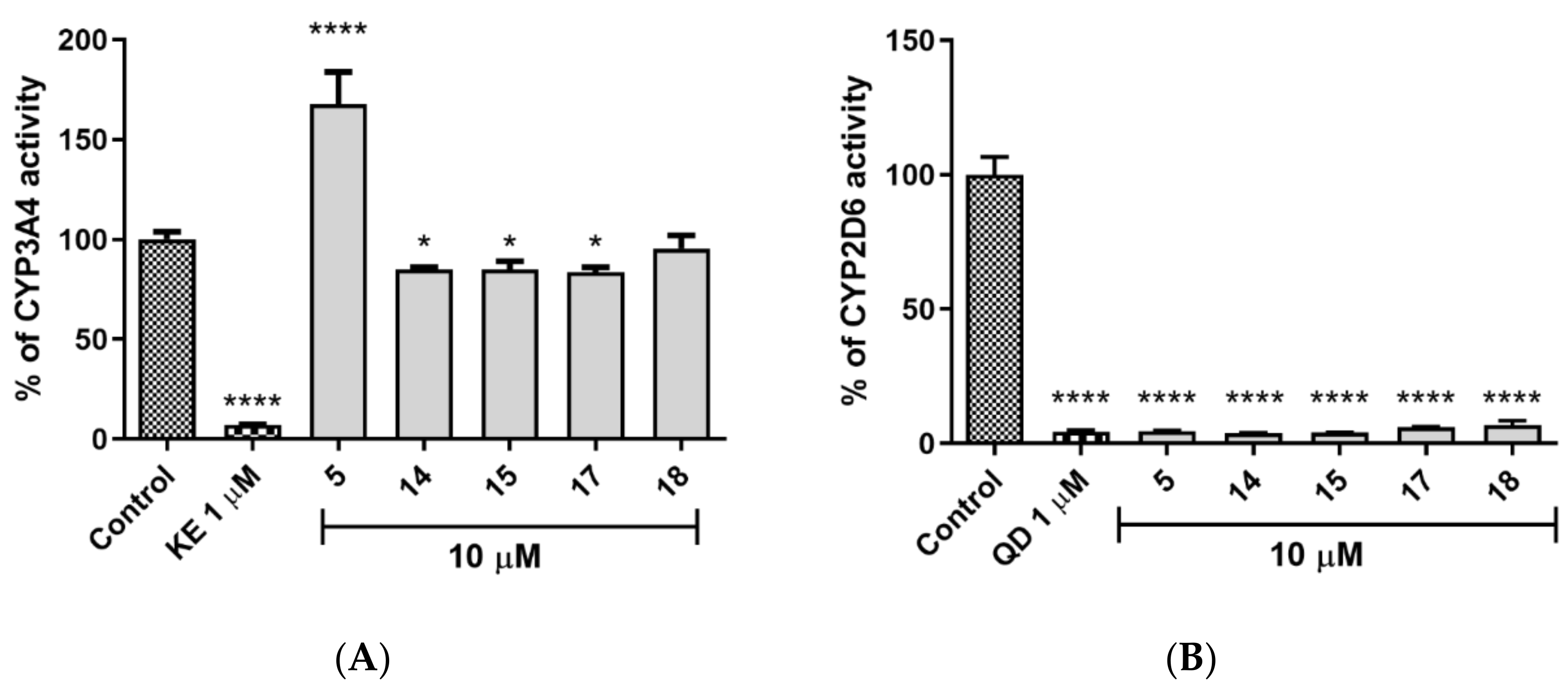
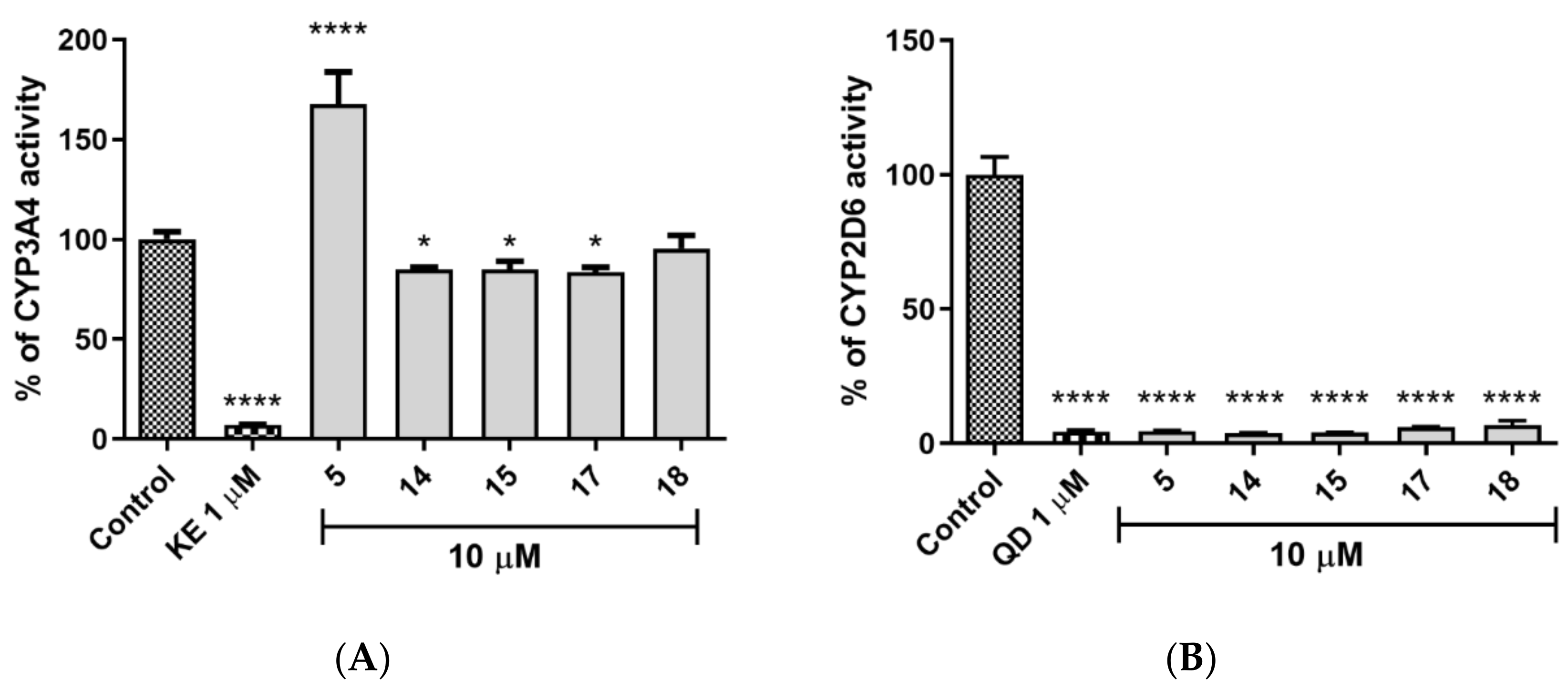
2.5.3. CYP450 Inhibition
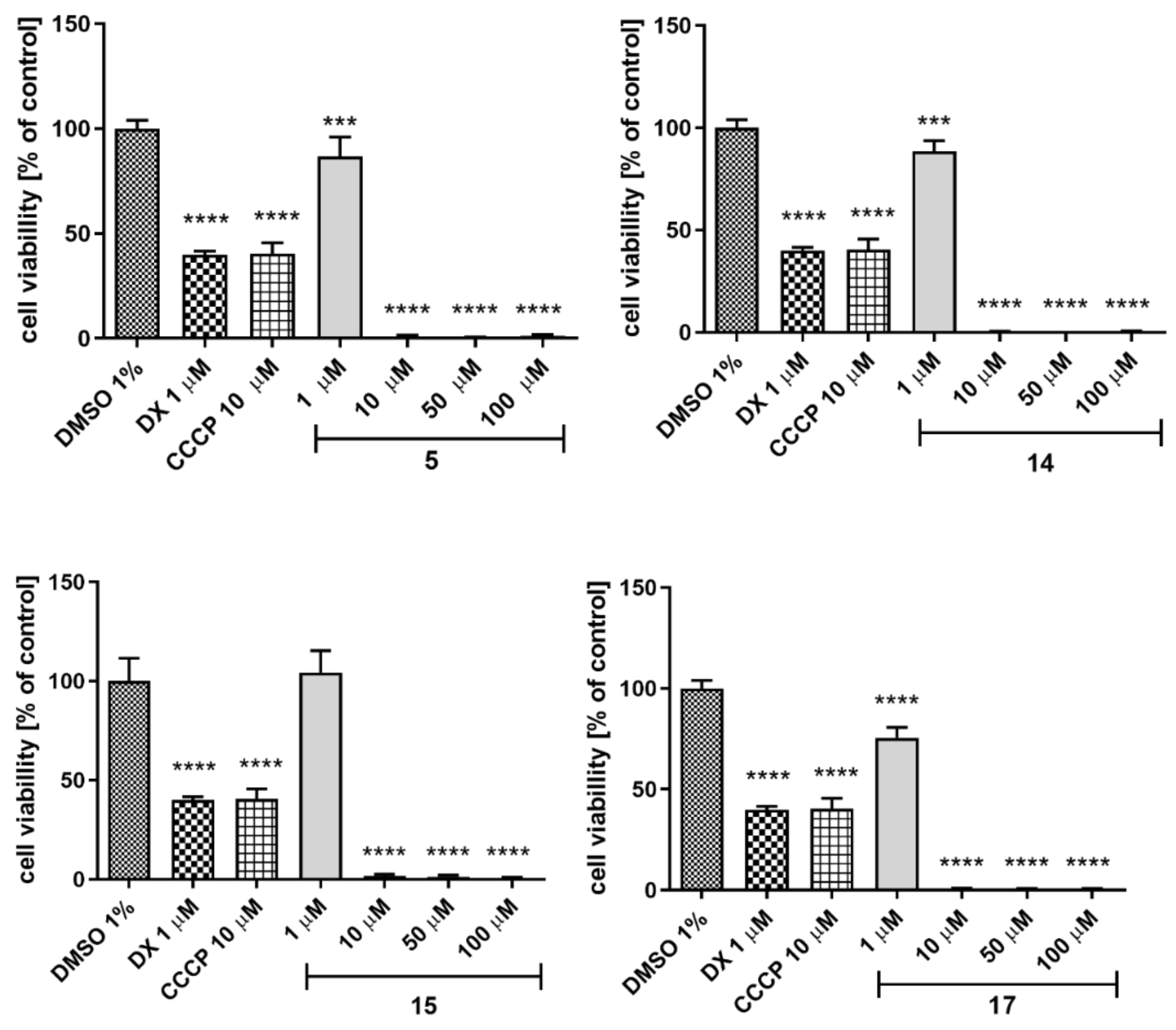
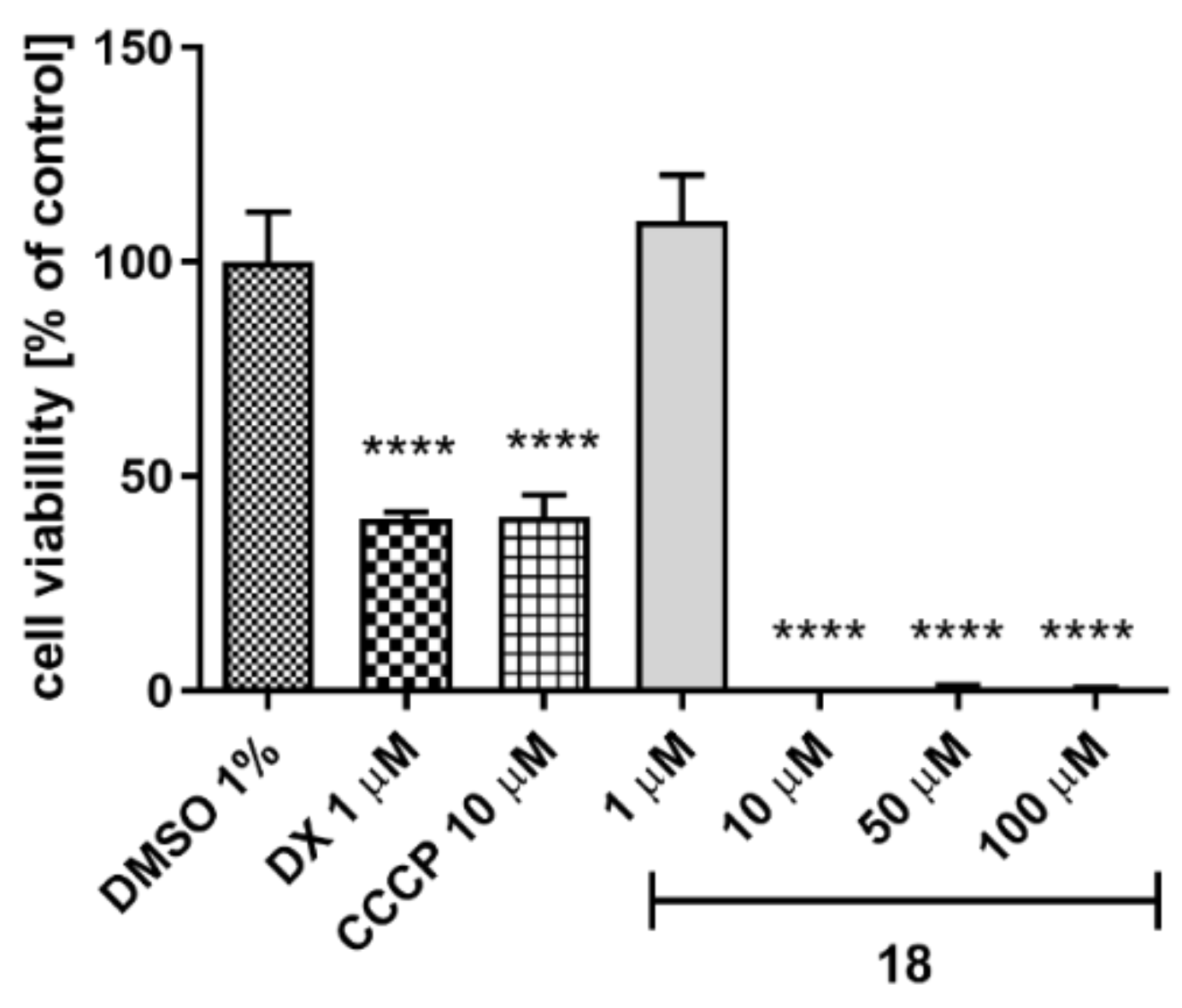
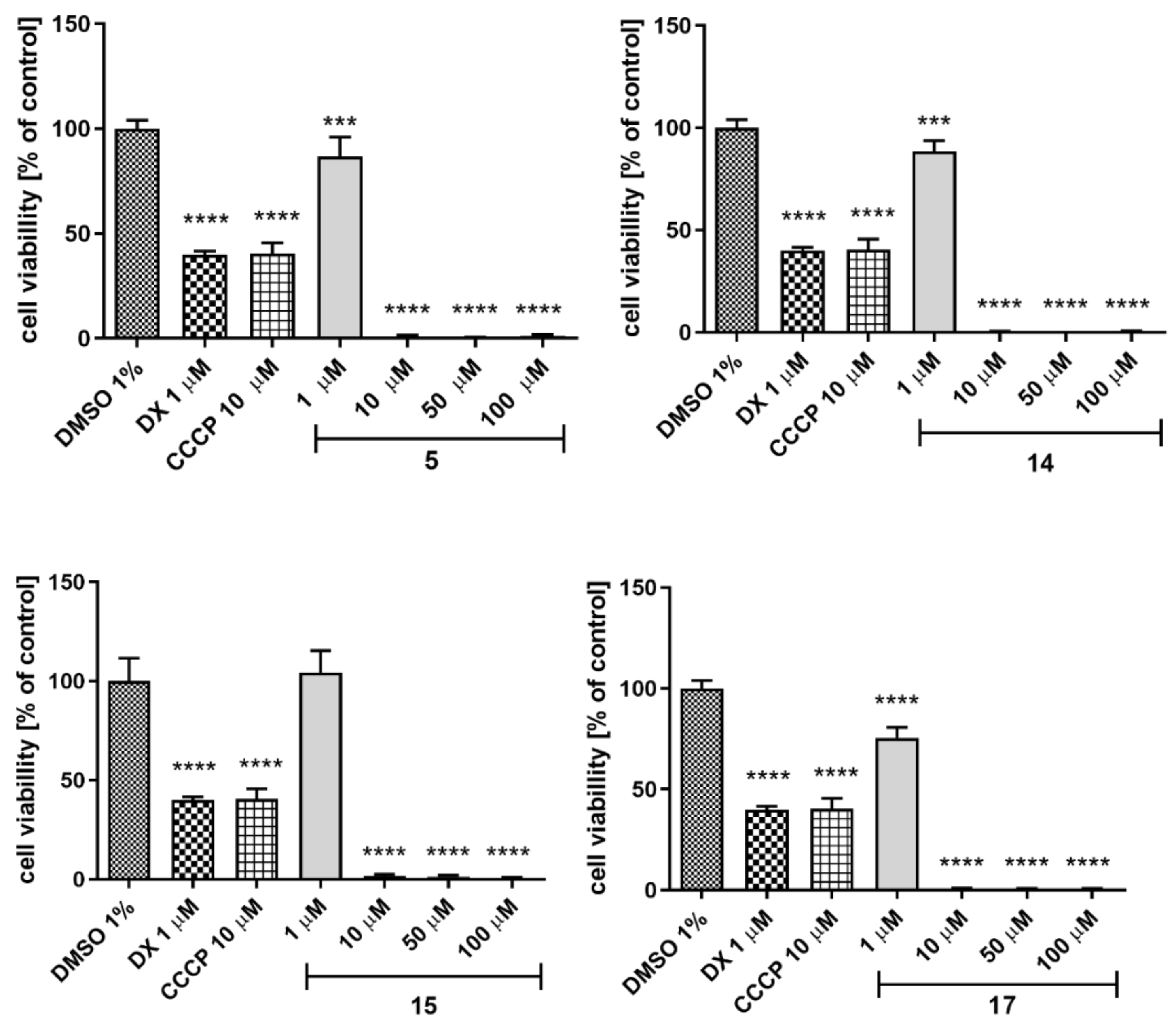
2.5.4. Hepatotoxicity
2.6. In Vivo Behavioral Tests
2.6.1. MK-801-Induced Hyperactivity in Mice
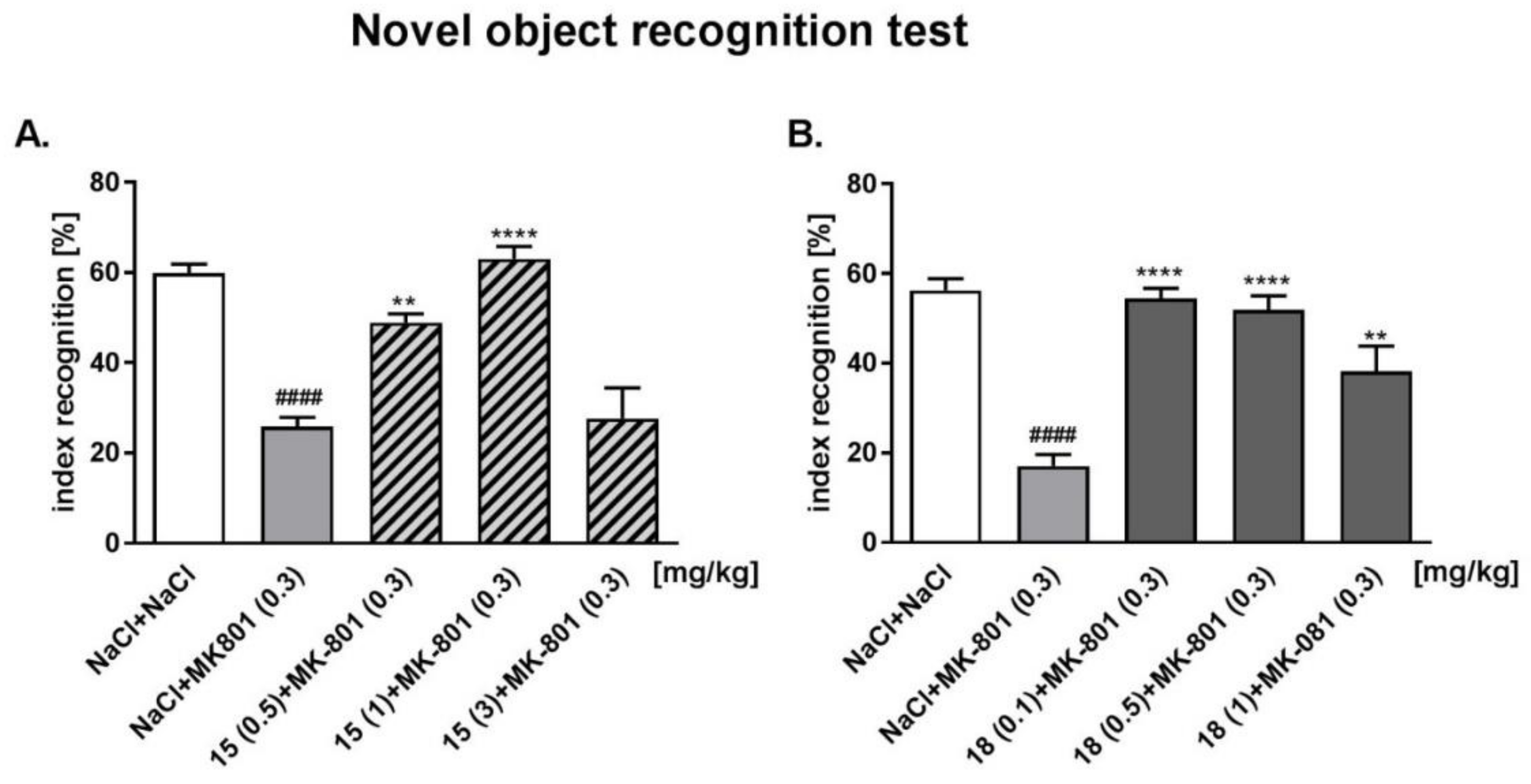
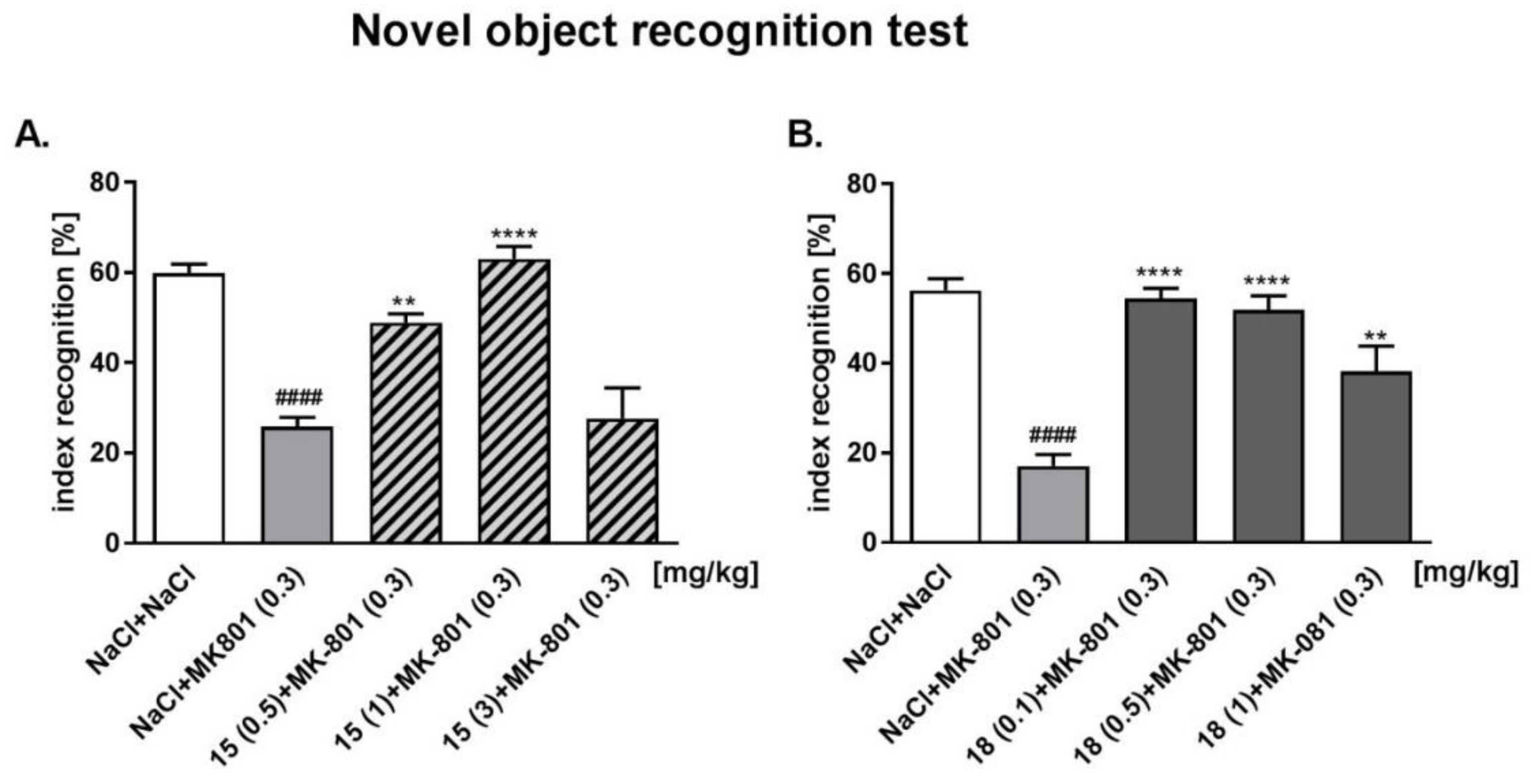
2.6.2. Novel Object Recognition (NOR) Test
2.6.3. Effect of Compound 15 and 18 on Spontaneous Activity of Mice
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.2. In Vitro Pharmacology
4.2.1. Radioligand Binding Assay
4.2.2. D2R Functional Assay
4.2.3. 5-HT6R Functional Assays
4.3. Molecular Modeling
4.4. In Vitro ADMETox Studies
4.4.1. PAMPA
4.4.2. Metabolic Stability
4.4.3. CYP450 Inhibition
4.4.4. Hepatotoxicity Assay
4.5. Behavioral Tests
4.5.1. Novel Object Recognition Test
4.5.2. MK-801-Induced Hyperactivity
4.5.3. Locomotor Activity of Mice
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Sample Availability
References
- Kohen, R.; Metcalf, M.A.; Khan, N.; Druck, T.; Huebner, K.; Lachowicz, J.E.; Meltzer, H.Y.; Sibley, D.R.; Roth, B.L.; Hamblin, M.W. Cloning, characterization, and chromosomal localization of a human 5-HT6 serotonin receptor. J. Neurochem. 1996, 66, 47–56. [Google Scholar] [CrossRef]
- Bryan, L.B.; Craigo, S.C.; Choudhary, M.S.; Uluer, A.; Monsma, F.J.; Shen, Y.; Meltzer, H.Y.; Sibley, D.R. Binding of typical and atypical antipsychotic agents to 5-hydroxytryptamine-6 and 5-hydroxytryptamine-7 receptors. J. Pharmacol. Exp. Ther. 1994, 268, 1403–1410. [Google Scholar]
- Glatt, C.E.; Snowman, A.M.; Sibley, D.R.; Snyder, S.H. Clozapine: Selective labeling of sites resembling 5HT6 serotonin receptors may reflect psychoactive profile. Mol. Med. 1995, 1, 398–406. [Google Scholar] [CrossRef] [Green Version]
- Schechter, L.E.; Lin, Q.; Smith, D.L.; Zhang, G.; Shan, Q.; Platt, B.; Brandt, M.R.; Dawson, L.A.; Cole, D.; Bernotas, R. Neuropharmacological profile of novel and selective 5-HT6 receptor agonists: WAY-181187 and WAY-208466. Neuropsychopharmacology 2008, 33, 1323–1335. [Google Scholar] [CrossRef]
- Bourson, A.; Borroni, E.; Austin, R.H.; Monsma, F.J.; Sleight, A.J. Determination of the role of the 5-HT6 receptor in the rat brain: A study using antisense oligonucleotides. J. Pharmacol. Exp. Ther. 1995, 274, 173–180. [Google Scholar]
- Axovant Announces Negative Topline Results of Intepirdine Phase 3 MINDSET Trial in Alzheimer’s Disease. Available online: http://investors.axovant.com/node/7286/pdf (accessed on 26 September 2017).
- Fullerton, T.; Binneman, B.; David, W.; Delnomdedieu, M.; Kupiec, J.; Lockwood, P.; Mancuso, J.; Miceli, J.; Bell, J. A Phase 2 clinical trial of PF-05212377 (SAM-760) in subjects with mild to moderate Alzheimer’s disease with existing neuropsychiatric symptoms on a stable daily dose of donepezil. Alzheimer’s Res. Ther. 2018, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Atri, A.; Frölich, L.; Ballard, C.; Tariot, P.N.; Molinuevo, J.L.; Boneva, N.; Windfeld, K.; Raket, L.L.; Cummings, J.L. Effect of idalopirdine as adjunct to cholinesterase inhibitors on change in cognition in patients with Alzheimer disease three randomized clinical trials. J. Am. Med. Assoc. 2018, 319, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Nirogi, R.; Mudigonda, K.; Bhyrapuneni, G.; Muddana, N.R.; Goyal, V.K.; Pandey, S.K.; Palacharla, R.C. Safety, Tolerability and pharmacokinetics of the serotonin 5-HT6 receptor antagonist, SUVN-502, in healthy young adults and elderly subjects. Clin. Drug Investig. 2018, 38, 401–415. [Google Scholar] [CrossRef] [PubMed]
- Canale, V.; Grychowska, K.; Kurczab, R.; Ryng, M.; Keeri, A.R.; Satała, G.; Olejarz-Maciej, A.; Koczurkiewicz, P.; Drop, M.; Blicharz, K.; et al. A dual-acting 5-HT6 receptor inverse agonist/MAO-B inhibitor displays glioprotective and pro-cognitive properties. Eur. J. Med. Chem. 2020, 208, 112765. [Google Scholar] [CrossRef] [PubMed]
- Grychowska, K.; Chaumont-Dubel, S.; Kurczab, R.; Koczurkiewicz, P.; Deville, C.; Krawczyk, M.; Pietruś, W.; Satała, G.; Buda, S.; Piska, K.; et al. Dual 5-HT6 and D3 receptor antagonists in a group of 1H-pyrrolo[3,2- c]quinolines with neuroprotective and procognitive activity. ACS Chem. Neurosci. 2019, 10, 3183–3196. [Google Scholar] [CrossRef]
- Staroń, J.; Kurczab, R.; Warszycki, D.; Satała, G.; Krawczyk, M.; Bugno, R.; Lenda, T.; Popik, P.; Hogendorf, A.S.; Hogendorf, A.; et al. Virtual screening-driven discovery of dual 5-HT6/5-HT2A receptor ligands with pro-cognitive properties. Eur. J. Med. Chem. 2019, 185, 111857. [Google Scholar] [CrossRef]
- Marcinkowska, M.; Bucki, A.; Panek, D.; Siwek, A.; Fajkis, N.; Bednarski, M.; Zygmunt, M.; Godyń, J.; Del Rio Valdivieso, A.; Kotańska, M.; et al. Anti-Alzheimer’s multitarget-directed ligands with serotonin 5-HT6 antagonist, butyrylcholinesterase inhibitory, and antioxidant activity. Arch. Pharm. 2019, 352, 1–10. [Google Scholar] [CrossRef]
- Hatat, B.; Yahiaoui, S.; Lecoutey, C.; Davis, A.; Freret, T.; Boulouard, M.; Claeysen, S.; Rochais, C.; Dallemagne, P. A novel in vivo anti-amnesic agent, specially designed to express both acetylcholinesterase (AChE) inhibitory, serotonergic subtype 4 receptor (5-HT4R) agonist and serotonergic subtype 6 receptor (5-HT6R) inverse agonist activities, with a potential inter. Front. Aging Neurosci. 2019, 11, 1–14. [Google Scholar] [CrossRef]
- Toublet, F.X.; Lalut, J.; Hatat, B.; Lecoutey, C.; Davis, A.; Since, M.; Corvaisier, S.; Freret, T.; Sopková-de Oliveira Santos, J.; Claeysen, S.; et al. Pleiotropic prodrugs: Design of a dual butyrylcholinesterase inhibitor and 5-HT6 receptor antagonist with therapeutic interest in Alzheimer’s disease. Eur. J. Med. Chem. 2021, 210, 113059. [Google Scholar] [CrossRef] [PubMed]
- Zajdel, P.; Kos, T.; Marciniec, K.; Satała, G.; Canale, V.; Kamiński, K.; Hołuj, M.; Lenda, T.; Koralewski, R.; Bednarski, M. Novel multi-target azinesulfonamides of cyclic amine derivatives as potential antipsychotics with pro-social and pro-cognitive effects. Eur. J. Med. Chem. 2018, 145, 790–804. [Google Scholar] [CrossRef] [PubMed]
- Morozova, M.A.; Lepilkina, T.A.; Rupchev, G.E.; Beniashvily, A.G.; Burminskiy, D.S.; Potanin, S.S.; Bondarenko, E.V.; Kazey, V.I.; Lavrovsky, Y.; Ivachtchenko, A.V. Add-on clinical effects of selective antagonist of 5HT6 receptors AVN-211 (CD-008-0173) in patients with schizophrenia stabilized on antipsychotic treatment: Pilot study. CNS Spectr. 2014, 19, 316–323. [Google Scholar] [CrossRef] [Green Version]
- Woods, S.; Clarke, N.N.; Layfield, R.; Fone, K.C.F. 5-HT6 receptor agonists and antagonists enhance learning and memory in a conditioned emotion response paradigm by modulation of cholinergic and glutamatergic mechanisms. Br. J. Pharmacol. 2012, 167, 436–449. [Google Scholar] [CrossRef] [Green Version]
- Meneses, A. Memory formation and memory alterations: 5-HT6 and 5-HT7 receptors, novel alternative. Rev. Neurosci. 2014, 25, 325–356. [Google Scholar] [CrossRef]
- Rychtyk, J.; Partyka, A.; Gdula-Argasińska, J.; Mysłowska, K.; Wilczyńska, N.; Jastrzębska-Więsek, M.; Wesołowska, A. 5-HT6 receptor agonist and antagonist improve memory impairments and hippocampal BDNF signaling alterations induced by MK-801. Brain Res. 2019, 1722, 146375. [Google Scholar] [CrossRef] [PubMed]
- Goff, D.; Coyle, J. The emerging role of glutamate in the pathophysiology and treatment of schizophrenia. Am. J. Psychiatry 1983, 75, 1367–1377. [Google Scholar] [CrossRef] [PubMed]
- Nikiforuk, A. Assessment of cognitive functions in animal models of schizophrenia. Pharmacol. Rep. 2018, 70, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Bubeníková-Valešová, V.; Horáček, J.; Vrajová, M.; Höschl, C. Models of schizophrenia in humans and animals based on inhibition of NMDA receptors. Neurosci. Biobehav. Rev. 2008, 32, 1014–1023. [Google Scholar] [CrossRef] [PubMed]
- Arnt, J.; Bang-Andersen, B.; Grayson, B.; Bymaster, F.P.; Cohen, M.P.; Delapp, N.W.; Giethlen, B.; Kreilgaard, M.; McKinzie, D.L.; Neill, J.C. Lu AE58054, a 5-HT6 antagonist, reverses cognitive impairment induced by subchronic phencyclidine in a novel object recognition test in rats. Int. J. Neuropsychopharmacol. 2010, 13, 1021–1033. [Google Scholar] [CrossRef] [PubMed]
- Manahan-Vaughan, D.; von Haebler, D.; Winter, C.; Juckel, G.; Heinemann, U. A single application of MK801 causes symptoms of acute psychosis, deficits in spatial semory and impairment of synaptic plasticity in rats. Hippocampus 2008, 18, 125–134. [Google Scholar] [CrossRef]
- Nichols, D.E.; Sassano, M.F.; Halberstadt, A.L.; Klein, L.M.; Brandt, S.D.; Elliott, S.P.; Fiedler, W.J. N-Benzyl-5-methoxytryptamines as Potent serotonin 5-HT2 receptor family agonists and comparison with a series of phenethylamine analogues. ACS Chem. Neurosci. 2015, 6, 1165–1175. [Google Scholar] [CrossRef]
- Yamada, F.; Hashimme, T.; Somei, M. Simple one step syntheses of indole-3-acetonitriles from indole-3-carboxaldehydes. Heterocycles 1998, 47, 509–516. [Google Scholar]
- Prior, T.I.; Chue, P.S.; Tibbo, P.; Baker, G.B. Drug metabolism and atypical antipsychotics. Eur. Neuropsychopharmacol. 1999, 9, 301–309. [Google Scholar] [CrossRef]
- Glennon, R.A.; Dukat, M.; El-Bermawy, M.; Law, H.; De Los Angeles, J.; Teitler, M.; King, A.; Herrick-Davis, K. Influence of amine substituents on 5-HT2A versus 5-HT2C binding of phenylalkyl- and indolylalkylamines. J. Med. Chem. 1994, 37, 1929–1935. [Google Scholar] [CrossRef] [PubMed]
- Nichols, D.E.; Frescas, S.P.; Chemel, B.R.; Rehder, K.S.; Zhong, D.; Lewin, A.H. High specific activity tritium-labeled N-(2-methoxybenzyl)-2,5-dimethoxy-4-iodophenethylamine (INBMeO): A high-affinity 5-HT2A receptor-selective agonist radioligand. Bioorg. Med. Chem. 2008, 16, 6116–6123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupuis, D.S.; la Cour, C.M.; Chaput, C.; Verrièle, L.; Lavielle, G.; Millan, M.J. Actions of novel agonists, antagonists and antipsychotic agents at recombinant rat 5-HT6 receptors: A comparative study of coupling to Gαs. Eur. J. Pharmacol. 2008, 588, 170–177. [Google Scholar] [CrossRef]
- O’Neill, M.F.; Shaw, G. Comparison of dopamine receptor antagonists on hyperlocomotion induced by cocaine, amphetamine, MK-801 and the dopamine D1 agonist C-APB in mice. Psychopharmacology 1999, 145, 237–250. [Google Scholar] [CrossRef]
- Torrisi, S.A.; Lavanco, G.; Maurel, O.M.; Gulisano, W.; Laudani, S.; Geraci, F.; Grasso, M.; Barbagallo, C.; Caraci, F.; Bucolo, C.; et al. A novel arousal-based individual screening reveals susceptibility and resilience to PTSD-like phenotypes in mice. Neurobiol. Stress 2021, 14, 100286. [Google Scholar] [CrossRef] [PubMed]
- Mkrtchian, A.; Evans, J.W.; Kraus, C.; Yuan, P.; Kadriu, B.; Nugent, A.C.; Roiser, J.P.; Zarate, C.A. Ketamine modulates fronto-striatal circuitry in depressed and healthy individuals. Mol. Psychiatry 2020. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, M.J. 5-HT6 receptors and Alzheimer’ s disease. Alzheimer’s Res. Ther. 2013, 5, 1–8. [Google Scholar]
- Yung-Chi, C.; Prusoff, W.H. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef]
- Łażewska, D.; Kurczab, R.; Więcek, M.; Kamińska, K.; Satała, G.; Jastrzębska-Więsek, M.; Partyka, A.; Bojarski, A.J.; Wesołowska, A.; Kieć-Kononowicz, K. The computer-aided discovery of novel family of the 5-HT6 serotonin receptor ligands among derivatives of 4-benzyl-1,3,5-triazine. Eur. J. Med. Chem. 2017, 135, 117–124. [Google Scholar] [CrossRef]
- Latacz, G.; Hogendorf, A.S.; Hogendorf, A.; Lubelska, A.; Wierońska, J.M.; Woźniak, M.; Cieślik, P.; Kieć-Kononowicz, K.; Handzlik, J.; Bojarski, A.J. Search for a 5-CT alternative. In vitro and in vivo evaluation of novel pharmacological tools: 3-(1-alkyl-1H-imidazol-5-yl)-1H-indole-5-carboxamides, low-basicity 5-HT7 receptor agonists. MedChemComm 2018, 9, 1882–1890. [Google Scholar] [CrossRef]
- Latacz, G.; Lubelska, A.; Jastrzębska-Więsek, M.; Partyka, A.; Marć, M.A.; Satała, G.; Wilczyńska, D.; Kotańska, M.; Więcek, M.; Kamińska, K. The 1,3,5-triazine derivatives as innovative chemical family of 5-HT6 serotonin receptor agents with therapeutic perspectives for cognitive impairment. Int. J. Mol. Sci. 2019, 20, 3420. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Murawski, A.; Patel, K.; Crespi, C.L.; Balimane, P.V. A novel design of artificial membrane for improving the PAMPA model. Pharm. Res. 2008, 25, 1511–1520. [Google Scholar] [CrossRef]
- Latacz, G.; Lubelska, A.; Jastrzębska-Więsek, M.; Partyka, A.; Sobiło, A.; Olejarz, A.; Kucwaj-Brysz, K.; Satała, G.; Bojarski, A.J.; Wesołowska, A. In the search for a lead structure among series of potent and selective hydantoin 5-HT7R agents: The drug-likeness in vitro study. Chem. Biol. Drug Des. 2017, 90, 1295–1306. [Google Scholar] [CrossRef]
- Latacz, G.; Lubelska, A.; Jastrzębska-Więsek, M.; Partyka, A.; Kucwaj-Brysz, K.; Wesołowska, A.; Kieć-Kononowicz, K.; Handzlik, J. MF-8, a novel promising arylpiperazine-hydantoin based 5-HT7 receptor antagonist: In vitro drug-likeness studies and in vivo pharmacological evaluation. Bioorg. Med. Chem. Lett. 2018, 28, 878–883. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.; Hansson, S.; Carlsson, A.; Carlsson, M.L. Differential effects of the N-methyl-d-aspartate receptor antagonist MK-801 on different stages of object recognition memory in mice. Neuroscience 2007, 149, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Cieślik, P.; Woźniak, M.; Kaczorowska, K.; Brański, P.; Burnat, G.; Chocyk, A.; Bobula, B.; Gruca, P.; Litwa, E.; Pałucha-Poniewiera, A. Negative allosteric modulators of mGlu7 receptor as putative antipsychotic drugs. Front. Mol. Neurosci. 2018, 11, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Rorick-Kehn, L.M.; Johnson, B.G.; Knitowski, K.M.; Salhoff, C.R.; Witkin, J.M.; Perry, K.W.; Griffey, K.I.; Tizzano, J.P.; Monn, J.A.; McKinzie, D.L.; et al. In vivo pharmacological characterization of the structurally novel, potent, selective mGlu2/3 receptor agonist LY404039 in animal models of psychiatric disorders. Psychopharmacology 2007, 193, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Rorick-Kehn, L.M.; Johnson, B.G.; Burkey, J.L.; Wright, R.A.; Calligaro, D.O.; Marek, G.J.; Nisenbaum, E.S.; Catlow, J.T.; Kingston, A.E.; Giera, D.D. Pharmacological and pharmacokinetic properties of a structurally novel, potent, and selective metabotropic glutamate 2/3 receptor agonist: In vitro characterization of agonist (−)-(1R,4S,5S,6S)-4-amino-2-sulfonylbicyclo[3.1.0]- hexane-4,6-dicarboxylic aci. J. Pharmacol. Exp. Ther. 2007, 321, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Wierońska, J.M.; Stachowicz, K.; Acher, F.; Lech, T.; Pilc, A. Opposing efficacy of group III mGlu receptor activators, LSP1-2111 and AMN082, in animal models of positive symptoms of schizophrenia. Psychopharmacology 2012, 220, 481–494. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| pKi [nM] | |||||||||||
| ID | R1 | R2 | R3 | R4 | R5 | R6 | 5-HT1A | 5-HT2A | 5-HT6 | 5-HT7 | D2 |
| Trp | H | H | H | - | - | - | 7.16 | 6.51 | 6.95 | 7.22 | <5.00 |
| 5-MeOTrp | MeO | H | H | - | - | - | 9.05 | 7.35 | 7.92 | 6.32 | 6.18 |
| 5-Cl-Trp | Cl | H | H | - | - | - | 8.80 | 7.50 | 8.42 | 8.80 | 6.61 |
| 1 | H | H | H | H | H | H | 6.82 | 8.70 | 7.76 | 5.54 | 7.22 |
| 2 | Me | H | H | H | H | H | 7.16 | 7.25 | 7.89 | 6.53 | <5.00 |
| 3 | OMe | H | H | H | H | H | 7.81 | 8.08 | 7.93 | 7.25 | 6.49 |
| 4 | Cl | H | H | H | H | H | n.d. | n.d. | 7.97 | n.d. | 6.61 |
| 5 | H | Cl | H | H | H | H | 6.48 | 6.93 | 7.78 | 5.80 | 7.81 |
| 6 | H | H | F | H | H | H | 6.56 | 7.42 | 7.77 | 5.99 | 6.53 |
| 7 | H | H | Cl | H | H | H | 6.29 | 7.20 | 7.88 | 5.60 | 5.93 |
| 8 | H | H | Br | H | H | H | 6.22 | 7.11 | 7.79 | 5.69 | 6.52 |
| 9 | H | H | H | OH | H | H | 6.54 | 6.71 | 6.65 | 6.29 | 6.12 |
| 10 | H | H | H | F | H | H | 7.31 | 6.46 | 7.39 | 6.32 | 6.07 |
| 11 | H | H | H | Cl | H | H | 7.17 | 6.49 | 7.29 | 6.42 | 6.68 |
| 12 | H | H | H | I | H | H | 7.31 | 6.63 | 7.34 | 6.29 | 6.68 |
| 13 | H | H | H | H | F | H | 6.87 | 6.59 | 7.65 | 5.74 | 7.52 |
| 14 | H | H | H | H | Cl | H | 6.97 | 7.33 | 8.05 | 5.80 | 7.91 |
| 15 | H | H | H | H | Br | H | 7.01 | 6.39 | 7.99 | 6.33 | 7.54 |
| 16 | H | H | H | H | H | F | 6.25 | 6.91 | 7.50 | 5.89 | 8.04 |
| 17 | H | H | H | H | H | Cl | 7.55 | 6.41 | 7.44 | 6.97 | 8.29 |
| 18 | H | H | H | H | H | Br | 7.52 | 6.40 | 7.35 | 6.88 | 8.26 |
| 19 | H | H | H | H | H | OBn | 7.13 | 6.64 | 7.52 | 5.95 | 6.39 |
| 20 | OMe | H | H | H | H | Cl | 8.10 | 7.51 | 8.01 | 7.53 | 6.63 |
| 21 | OMe | H | H | H | Cl | H | 7.73 | 7.76 | 8.28 | 7.09 | 6.81 |
| 22 | H | Cl | H | H | H | Cl | 6.25 | 6.39 | 7.44 | 5.88 | 6.30 |
| ID | R1 | R2 | R3 | R4 | R5 | R6 | 5-HT6 EC50 | 5-HT6 Kb | D2 Kb |
|---|---|---|---|---|---|---|---|---|---|
| [nM] | |||||||||
| 1 | H | H | H | H | H | H | n.d. | 151 | n.d. |
| 5 | H | Cl | H | H | H | H | 157 | 136 | 104 |
| 13 | H | H | H | H | F | H | n.d. | 30 | 91 |
| 14 | H | H | H | H | Cl | H | n.d. | 112 | 290 |
| 15 | H | H | H | H | Br | H | n.d. | 131 | 109 |
| 17 | H | H | H | H | H | Cl | n.d. | 234 | 215 |
| 18 | H | H | H | H | H | Br | 77 | 10,000 | 167 |
| 21 | OMe | H | H | H | H | Cl | 22 | 10,000 | n.d. |
| 22 | OMe | H | H | H | Cl | H | 26 | n.d. | n.d. |
| Compound | PAMPA Pe * [10−6 cm/s] ± SD |
|---|---|
| Norfloxacin | 0.56 ± 0.13 |
| Caffeine | 15.1 ± 0.40 |
| 5 | 7.09 ± 1.24 |
| 14 | 8.34 ± 1.33 |
| 15 | 2.42 ± 0.92 |
| 17 | 8.42 ± 0.27 |
| 18 | 3.90 ± 0.56 |
| Compound | Molecular Mass (m/z) | Retention Time (min) | Molecular Mass of the Metabolite (m/z) | Metabolic Pathway |
|---|---|---|---|---|
| 5 | 324.25 | 4.17 | M1 340.26 | hydroxylation |
| 14 | 324.31 | 4.054.38 | M1 340.26 M2 340.33 | hydroxylation hydroxylation |
| 15 | 370.13 | 4.124.34 | M1 386.15 M2 384.09 | hydroxylation hydroxylation |
| 17 | 324.25 | 3.954.27 | M1 340.26 M2 340.26 | hydroxylation hydroxylation |
| 18 | 368.14 | 4.004.27 | M1 384.09 M2 386.08 | hydroxylation hydroxylation |
| Compounds | Doses (mg/kg) | Ambulation Scores ± SEM |
|---|---|---|
| NaCl | - | 100.0 ± 11.81 |
| 15 | 0.5 | 134.9 ± 11.13 |
| 15 | 1 | 124.7 ± 21.75 |
| 15 | 3 | 91.8 ± 24.61 |
| NaCl | - | 100.0 ± 8.95 |
| 18 | 0.05 | 113.6 ± 34.00 |
| 18 | 0.1 | 131.8 ± 19.50 |
| 18 | 0.5 | 116.7 ± 13.23 |
| 18 | 1 | 85.31 ± 15.86 |
| 18 | 3 | 87.79 ± 20.89 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hogendorf, A.; Hogendorf, A.S.; Kurczab, R.; Satała, G.; Szewczyk, B.; Cieślik, P.; Latacz, G.; Handzlik, J.; Lenda, T.; Kaczorowska, K.; et al. N-Skatyltryptamines—Dual 5-HT6R/D2R Ligands with Antipsychotic and Procognitive Potential. Molecules 2021, 26, 4605. https://doi.org/10.3390/molecules26154605
Hogendorf A, Hogendorf AS, Kurczab R, Satała G, Szewczyk B, Cieślik P, Latacz G, Handzlik J, Lenda T, Kaczorowska K, et al. N-Skatyltryptamines—Dual 5-HT6R/D2R Ligands with Antipsychotic and Procognitive Potential. Molecules. 2021; 26(15):4605. https://doi.org/10.3390/molecules26154605
Chicago/Turabian StyleHogendorf, Agata, Adam S. Hogendorf, Rafał Kurczab, Grzegorz Satała, Bernadeta Szewczyk, Paulina Cieślik, Gniewomir Latacz, Jadwiga Handzlik, Tomasz Lenda, Katarzyna Kaczorowska, and et al. 2021. "N-Skatyltryptamines—Dual 5-HT6R/D2R Ligands with Antipsychotic and Procognitive Potential" Molecules 26, no. 15: 4605. https://doi.org/10.3390/molecules26154605