
Electronic Circular Dichroism Spectra of DNA Quadruple Helices Studied by Molecular Dynamics Simulations and Excitonic Calculations including Charge Transfer States


 , , and
, , and
Abstract
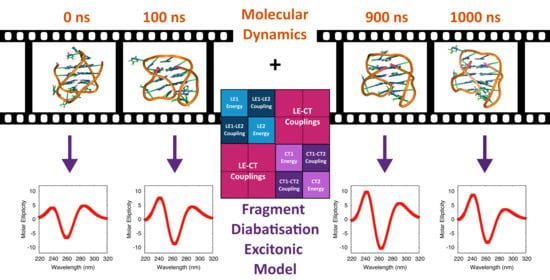
:
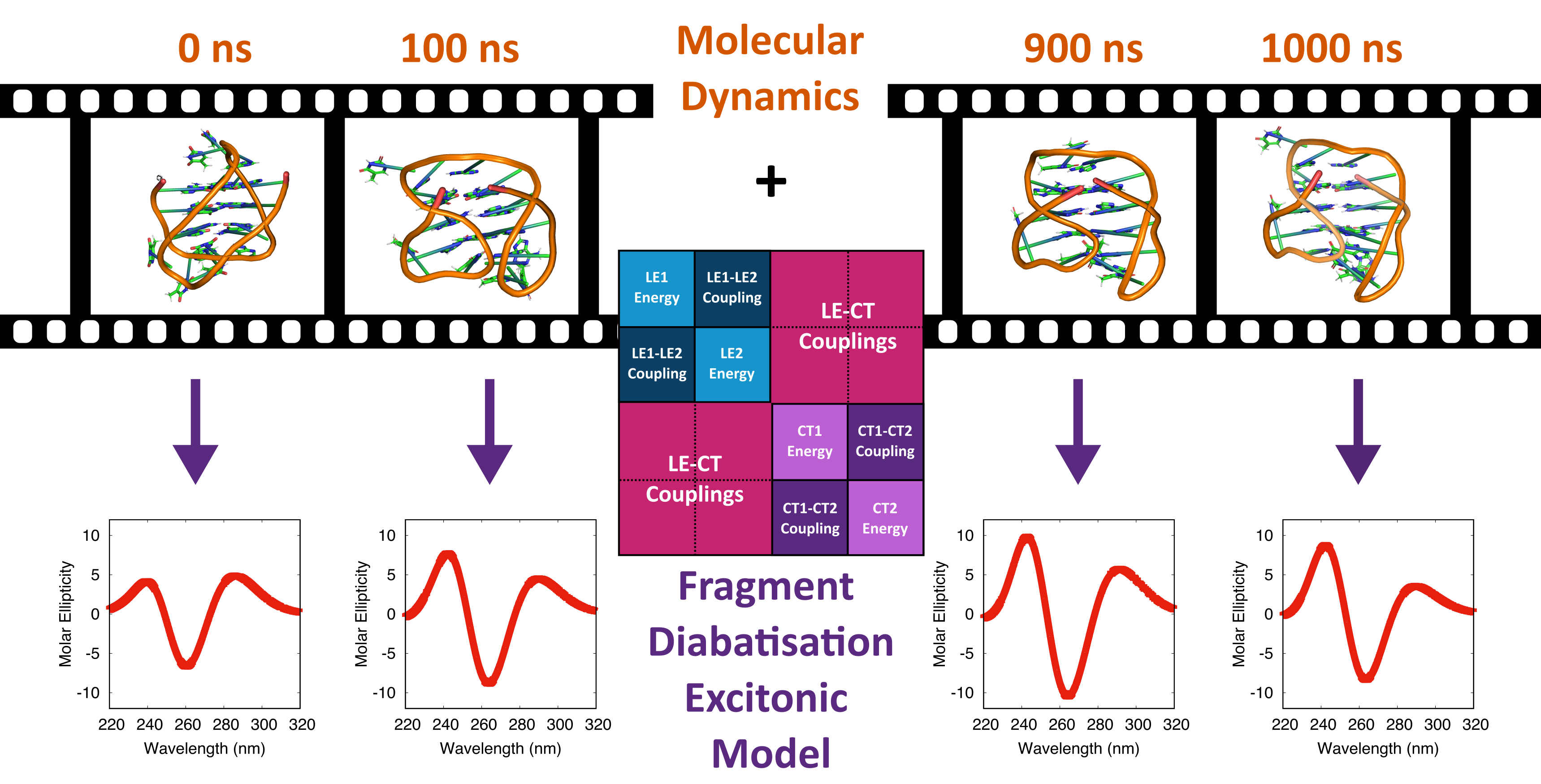{kind=link}
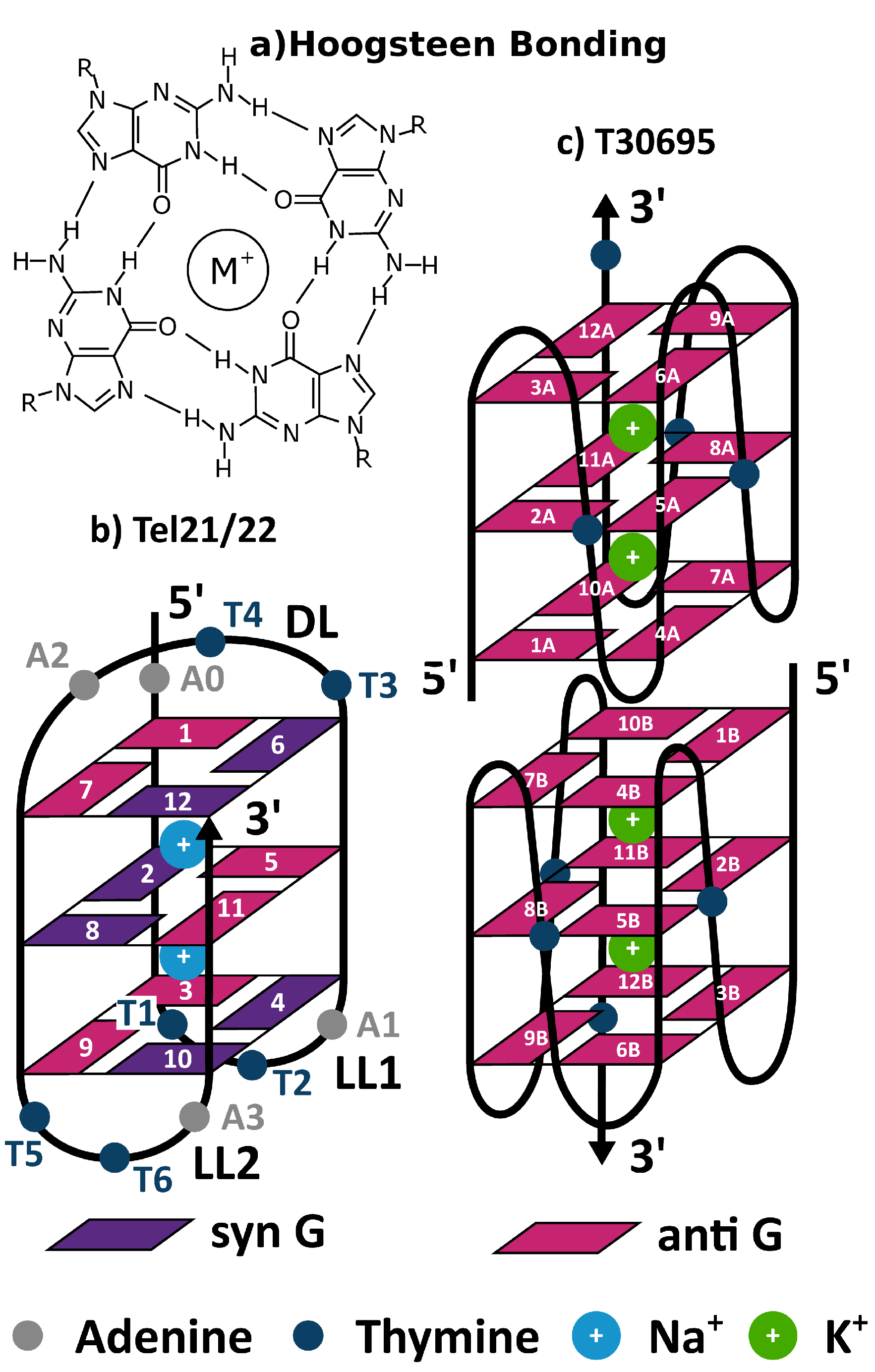{kind=link}
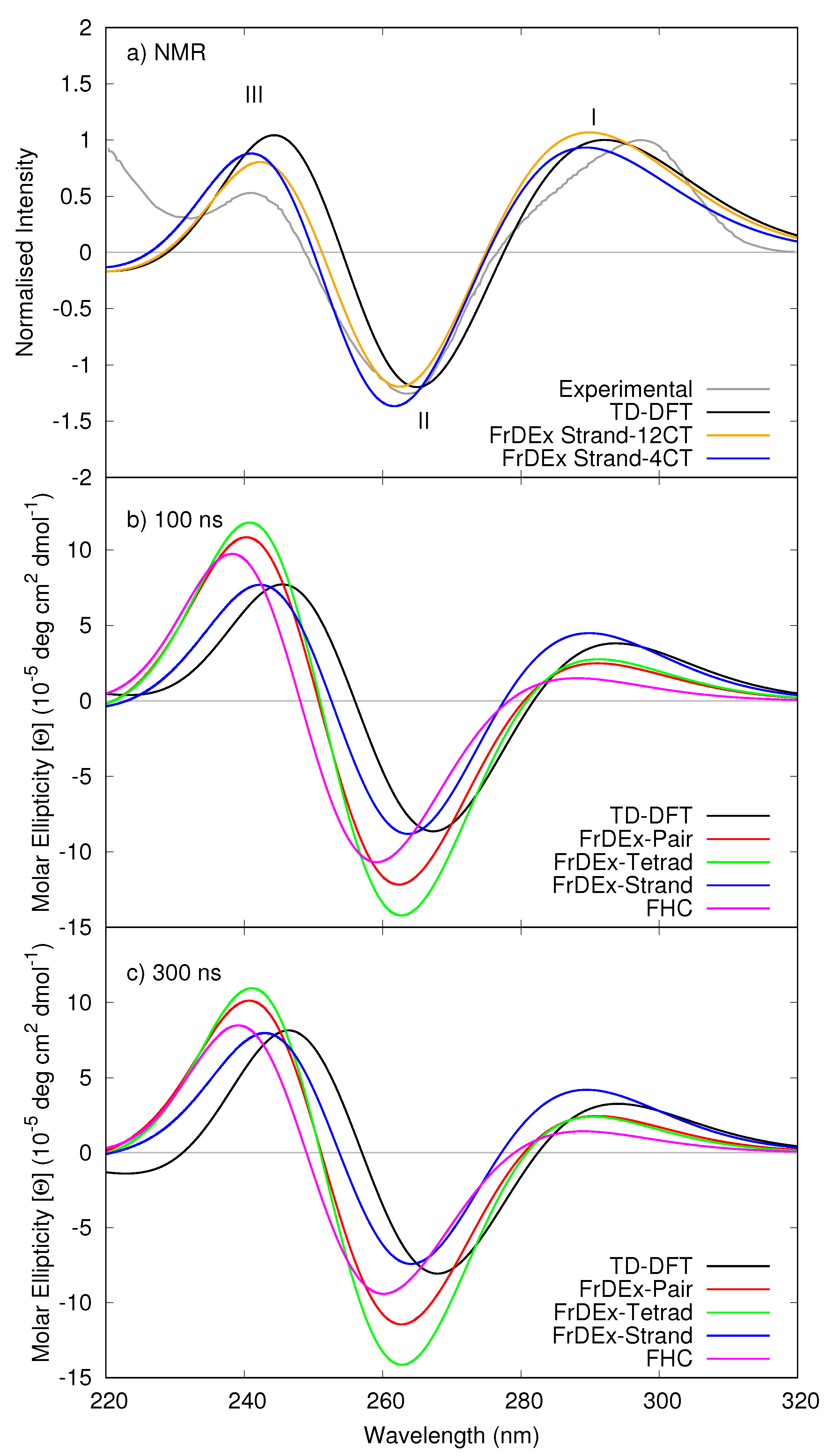{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Methods
Computational Details
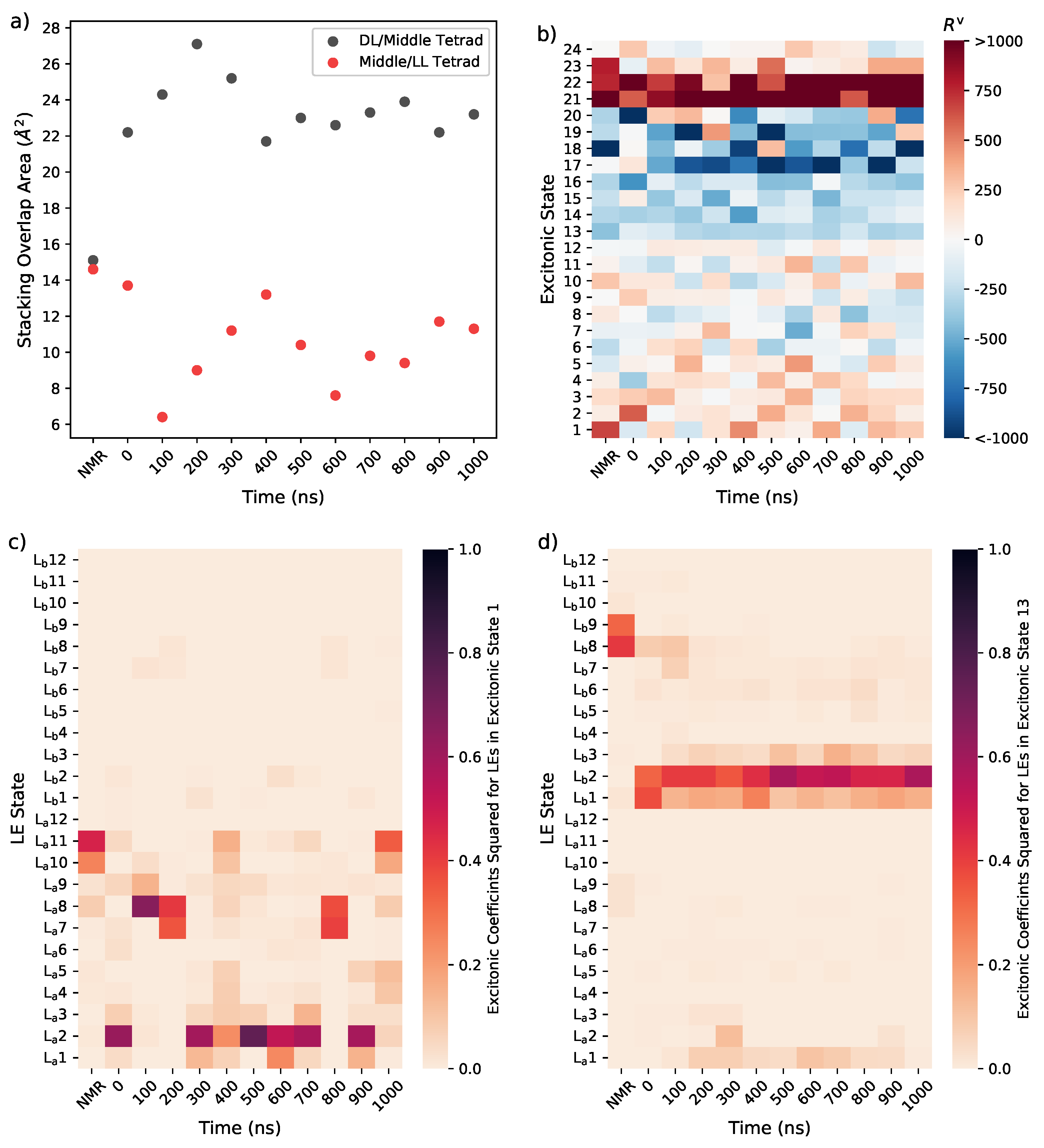
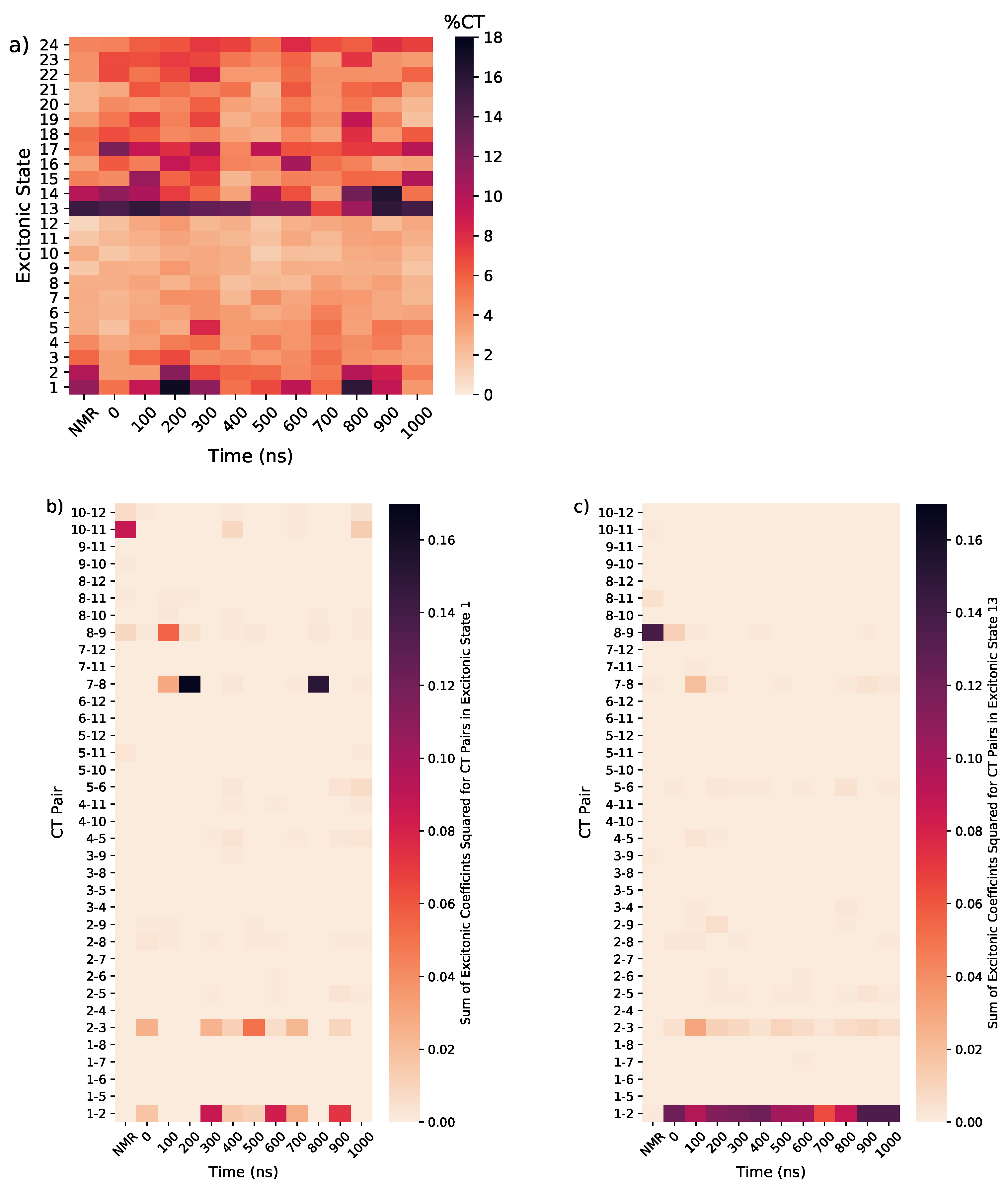
3. Results
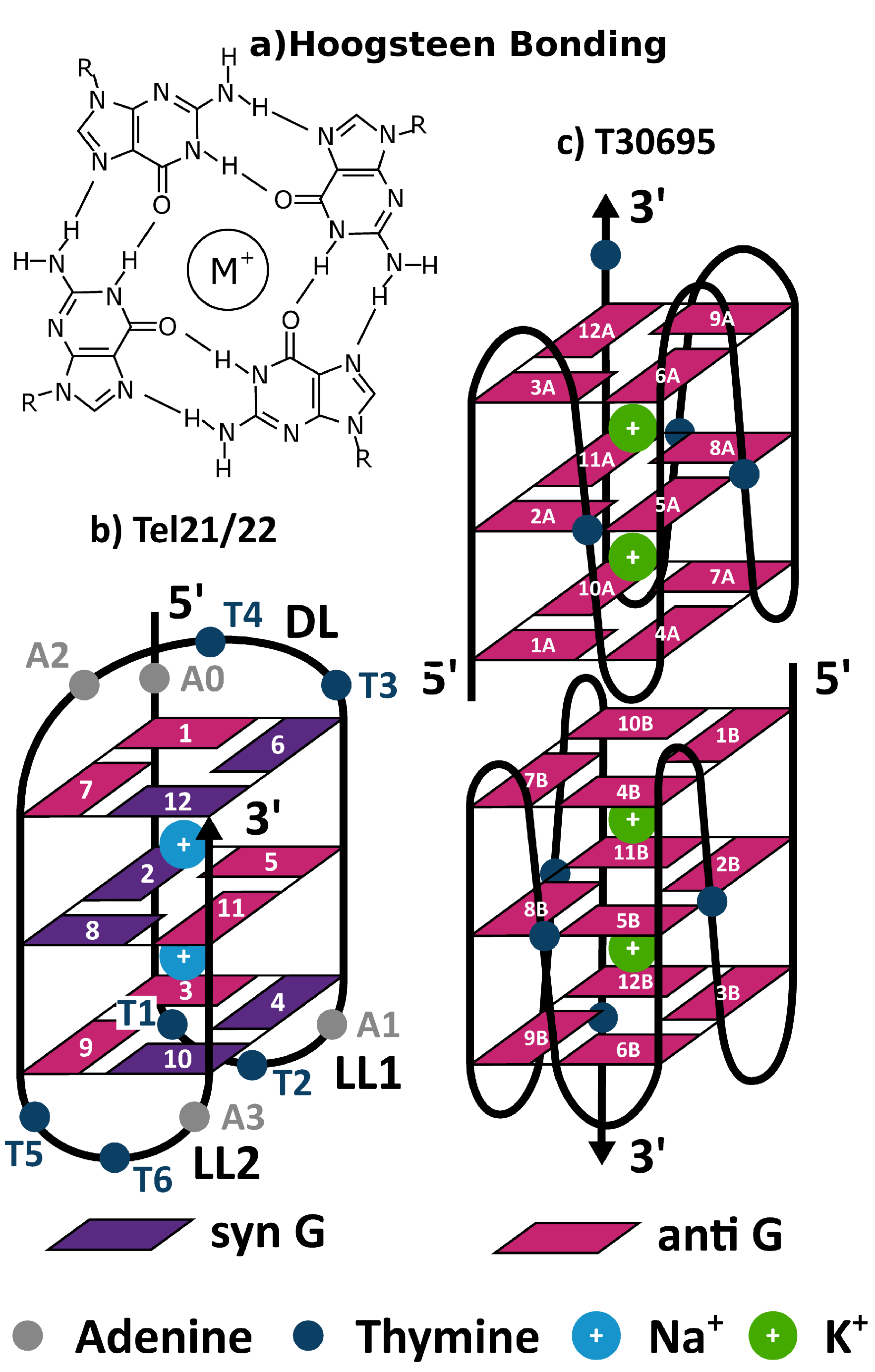
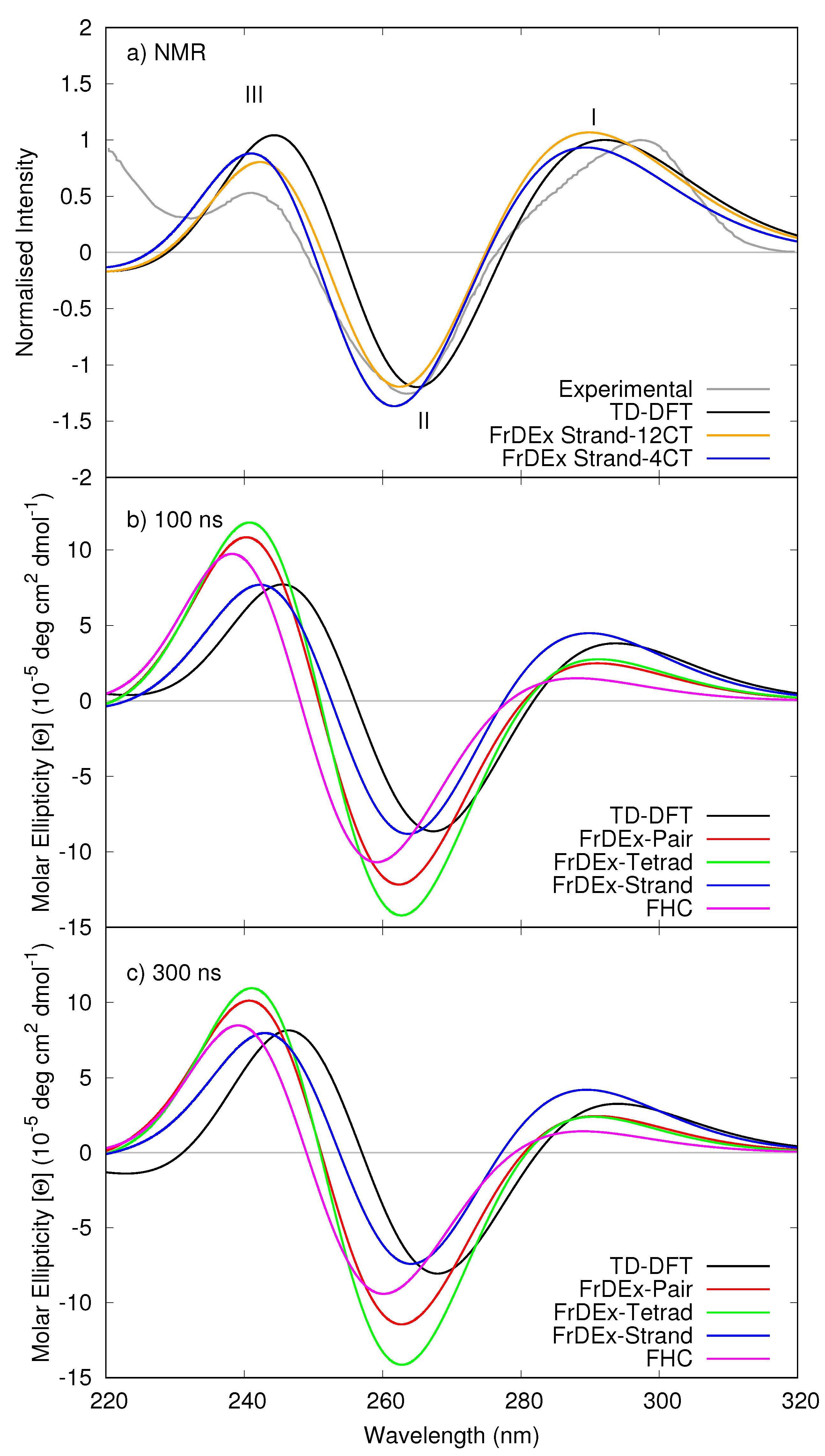
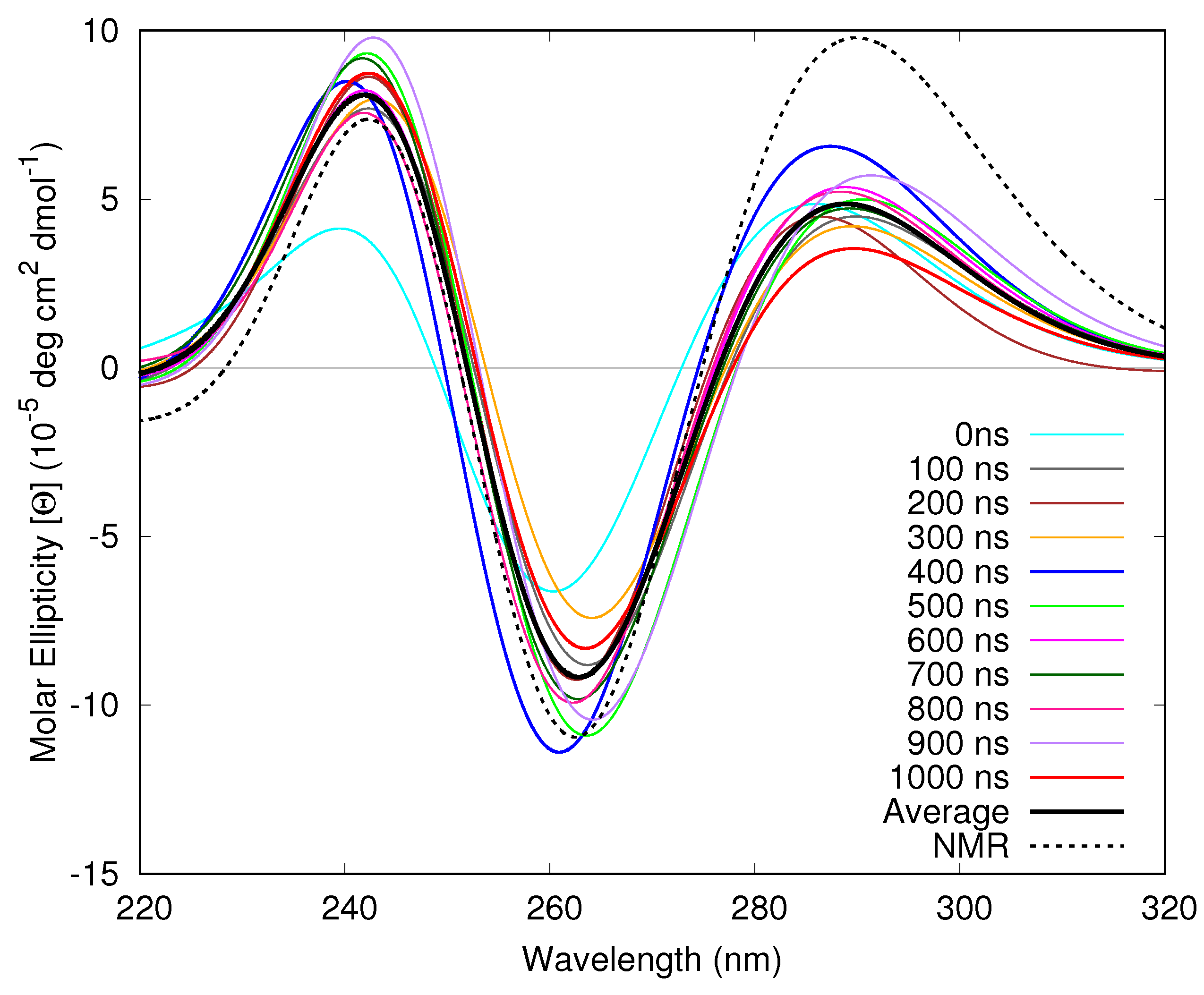
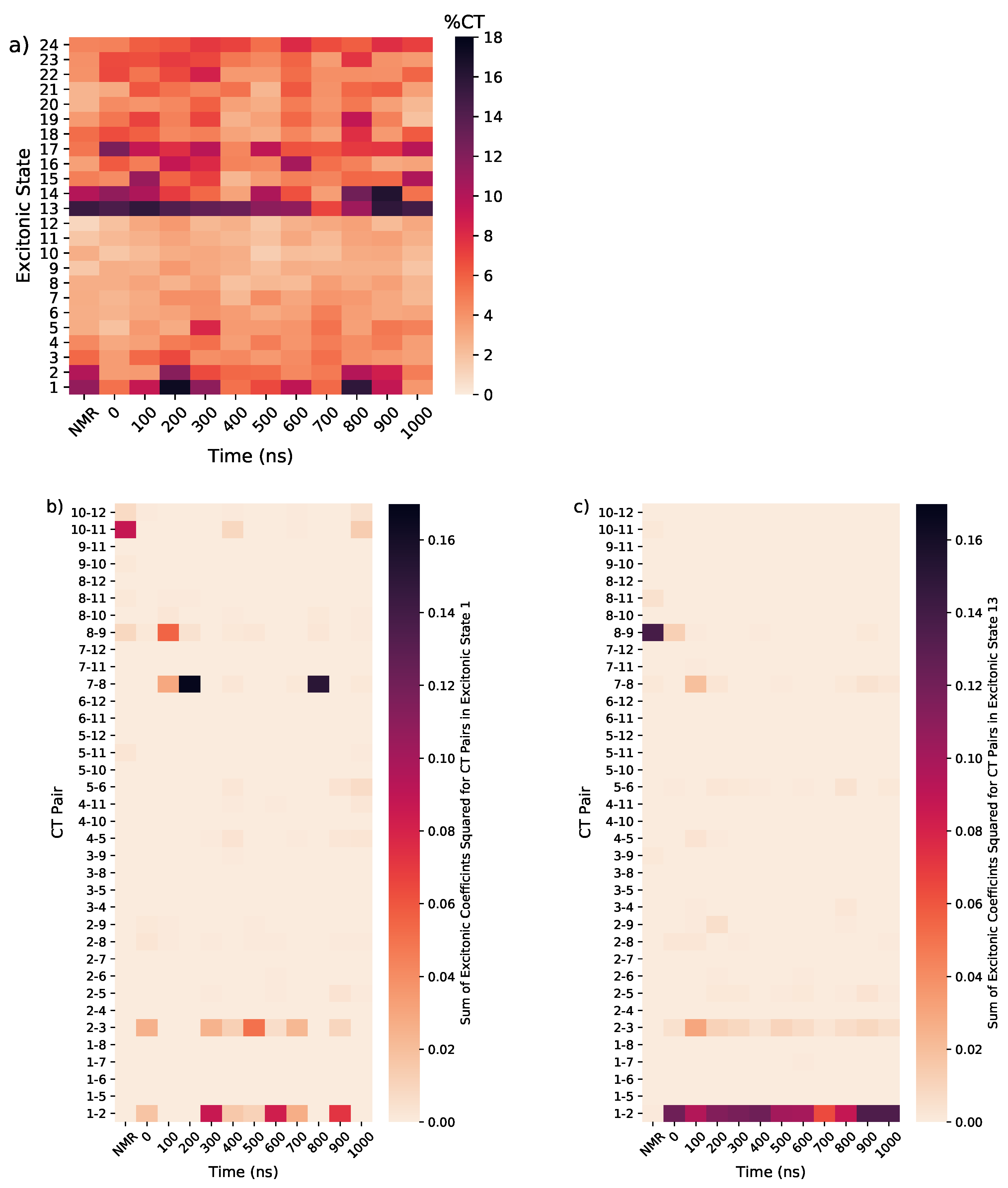
3.1. Tel21
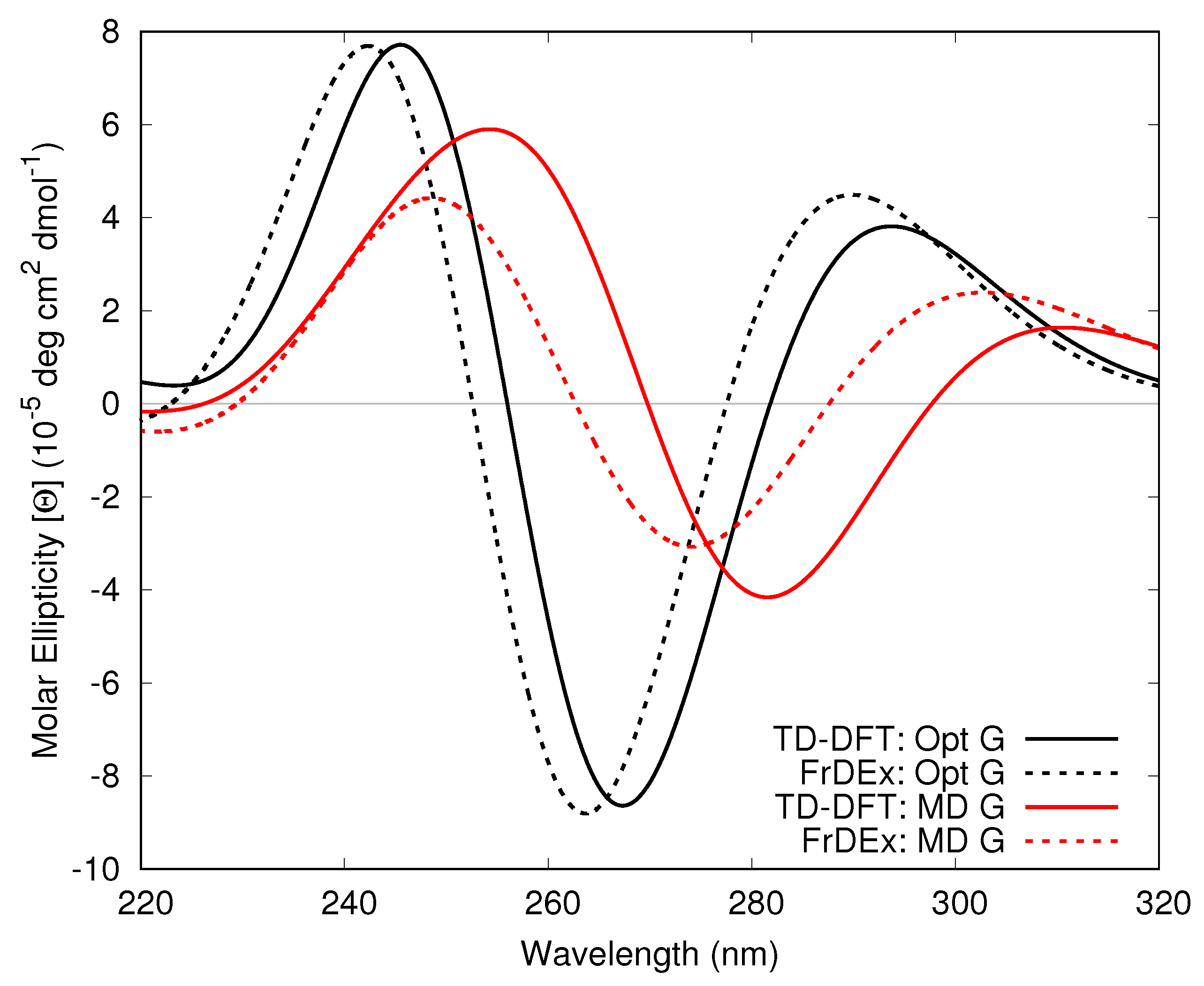
3.1.1. Assessment of the FrDEx Options
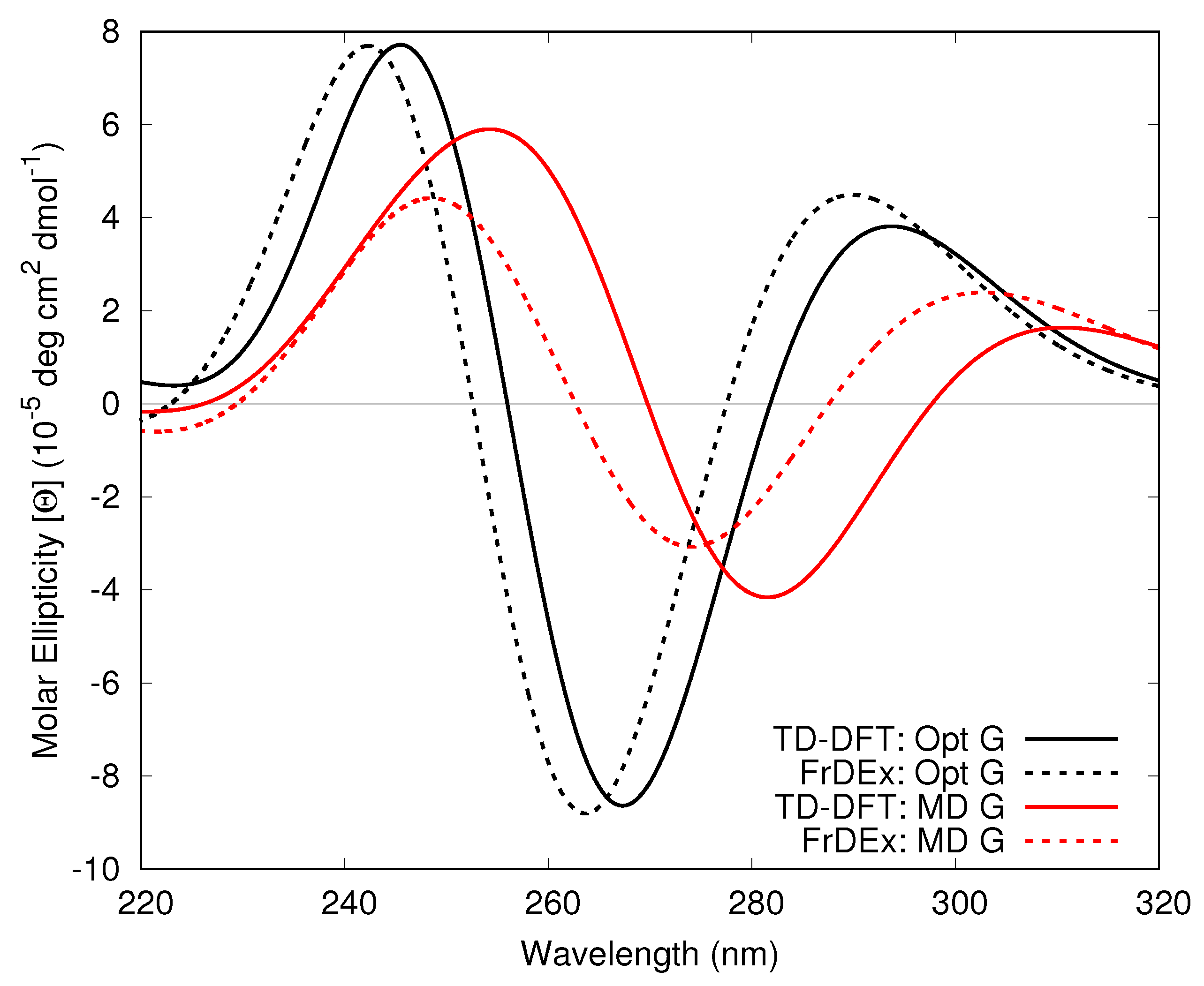
3.1.2. Guanine Internal Geometry
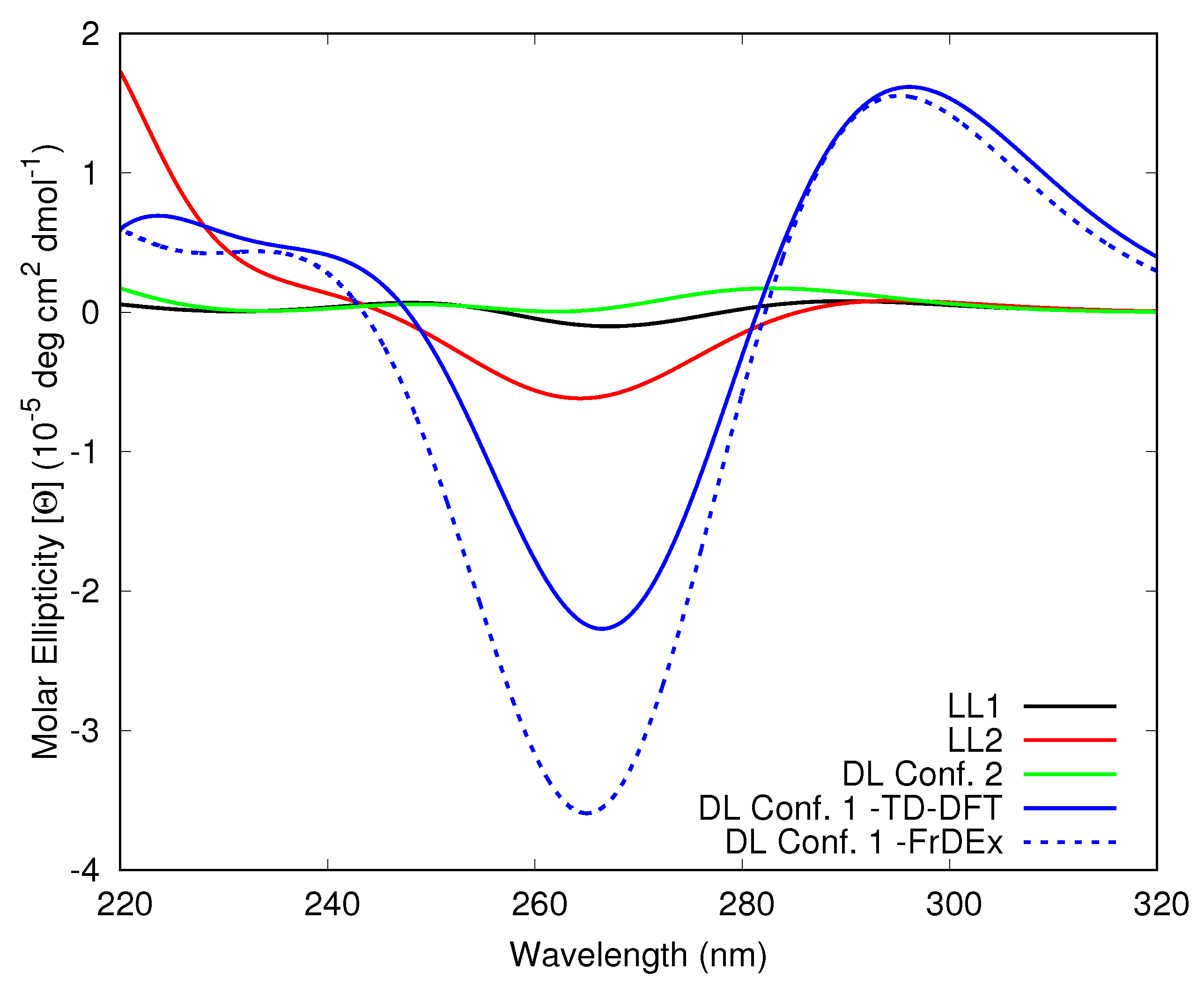
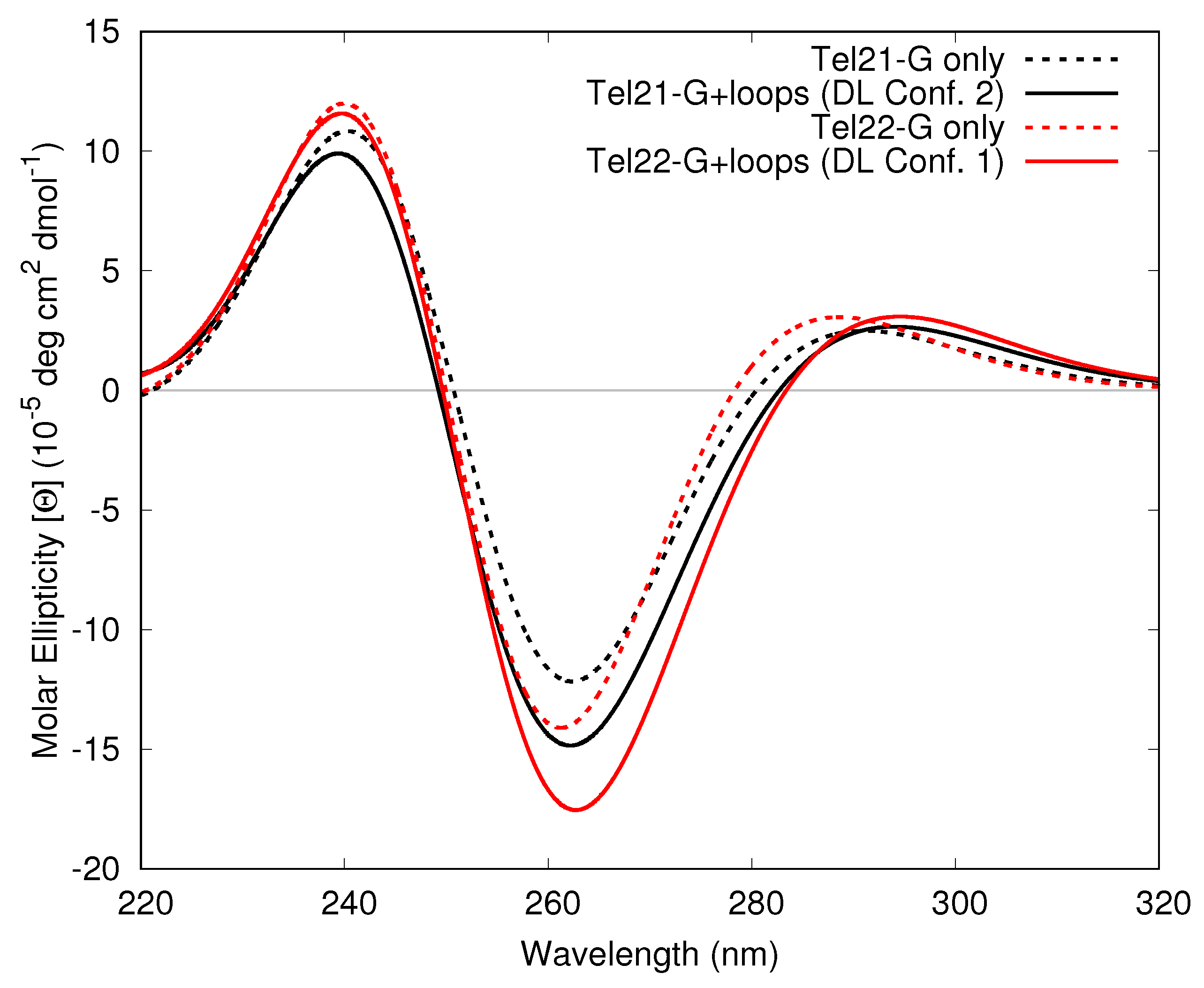
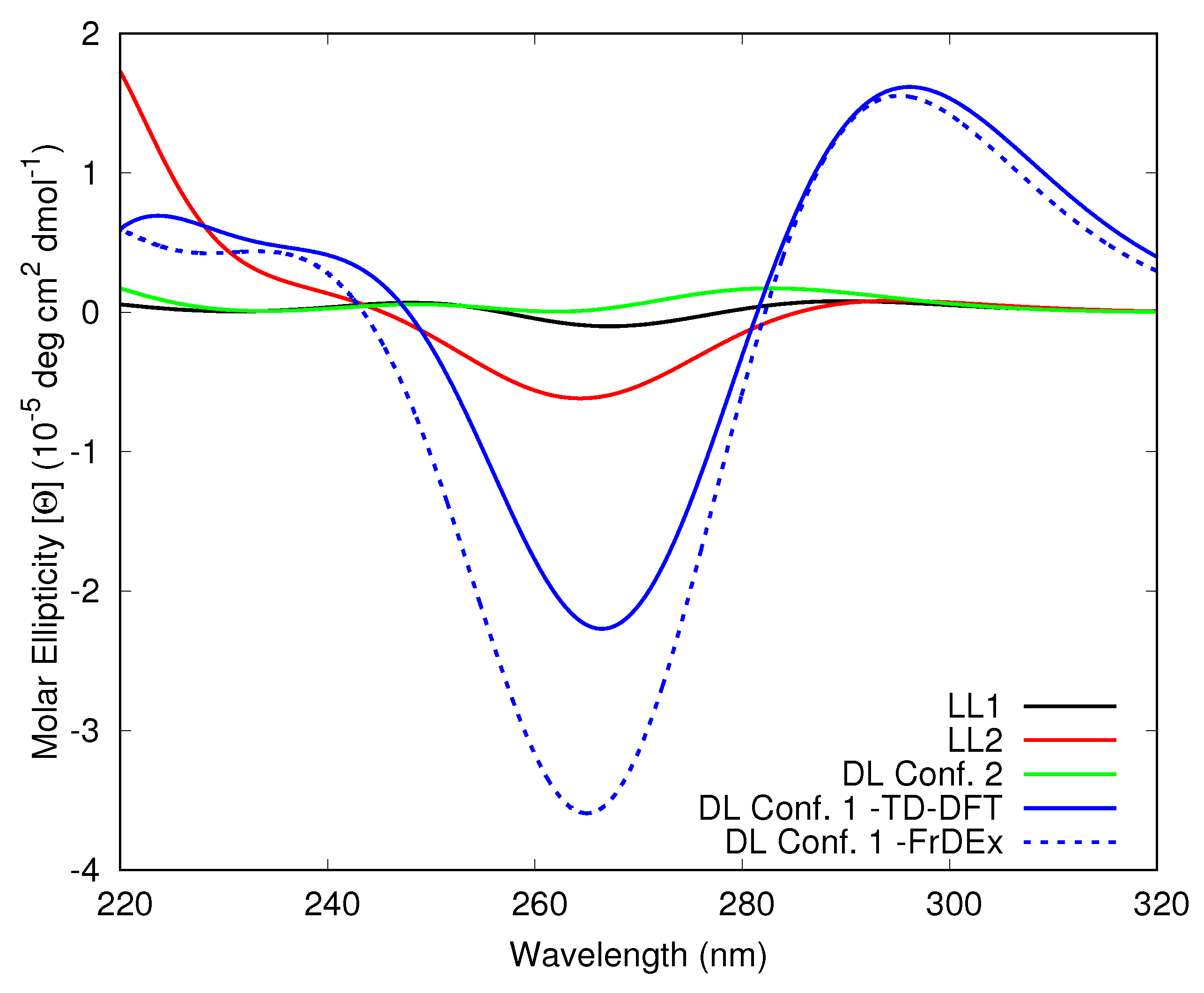
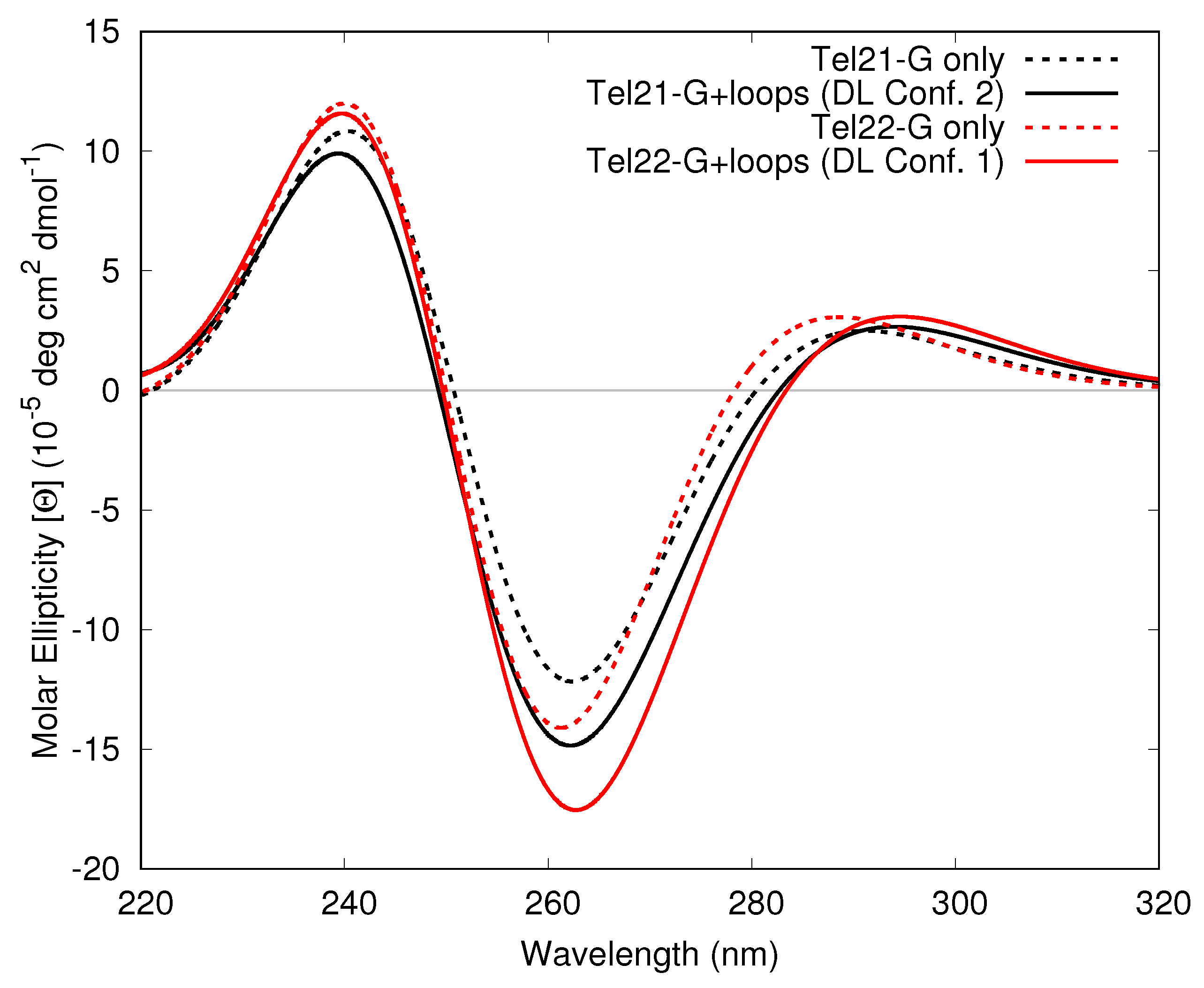
3.1.3. Including Loop Bases
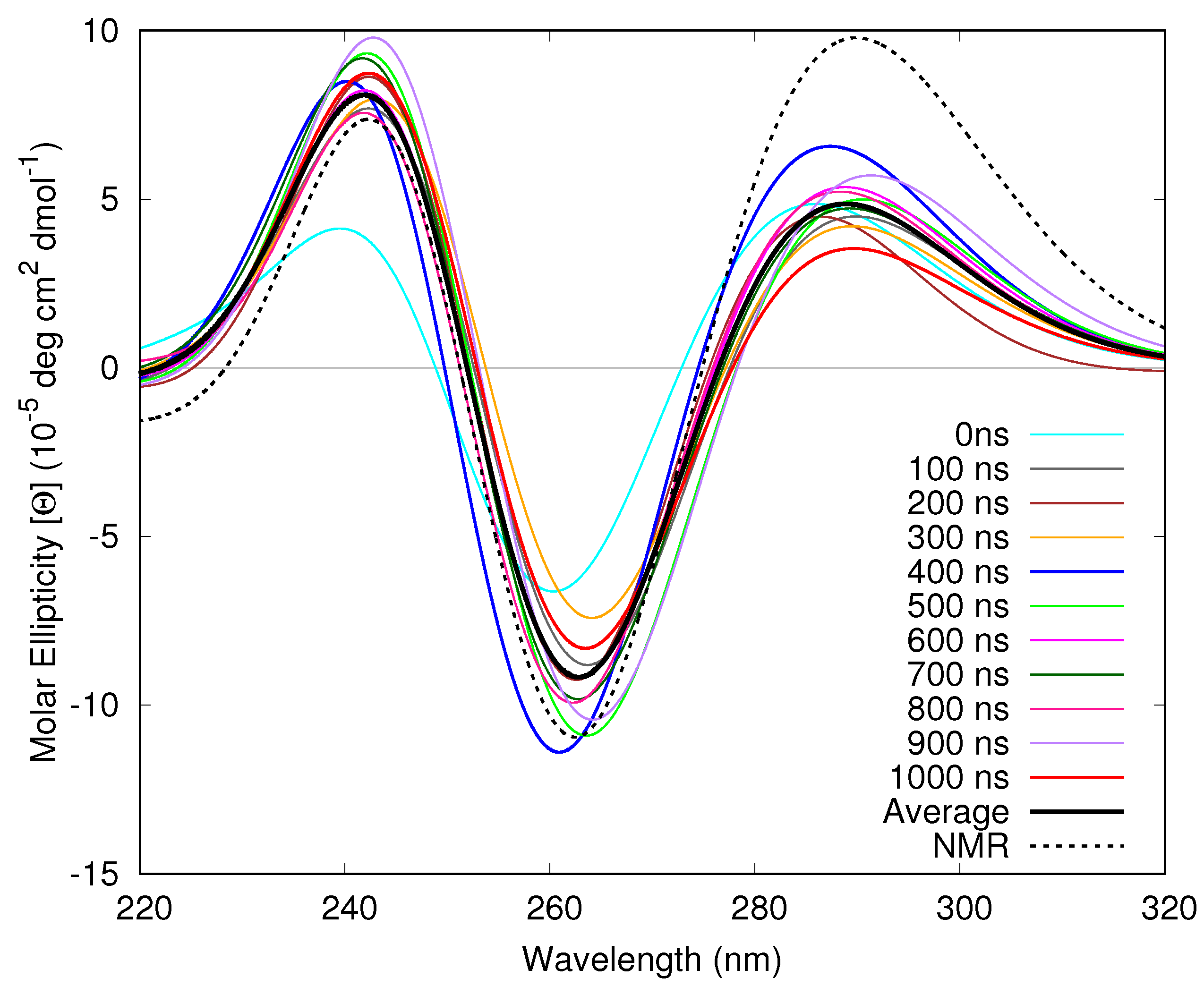
3.1.4. Considering Thermal Fluctuations
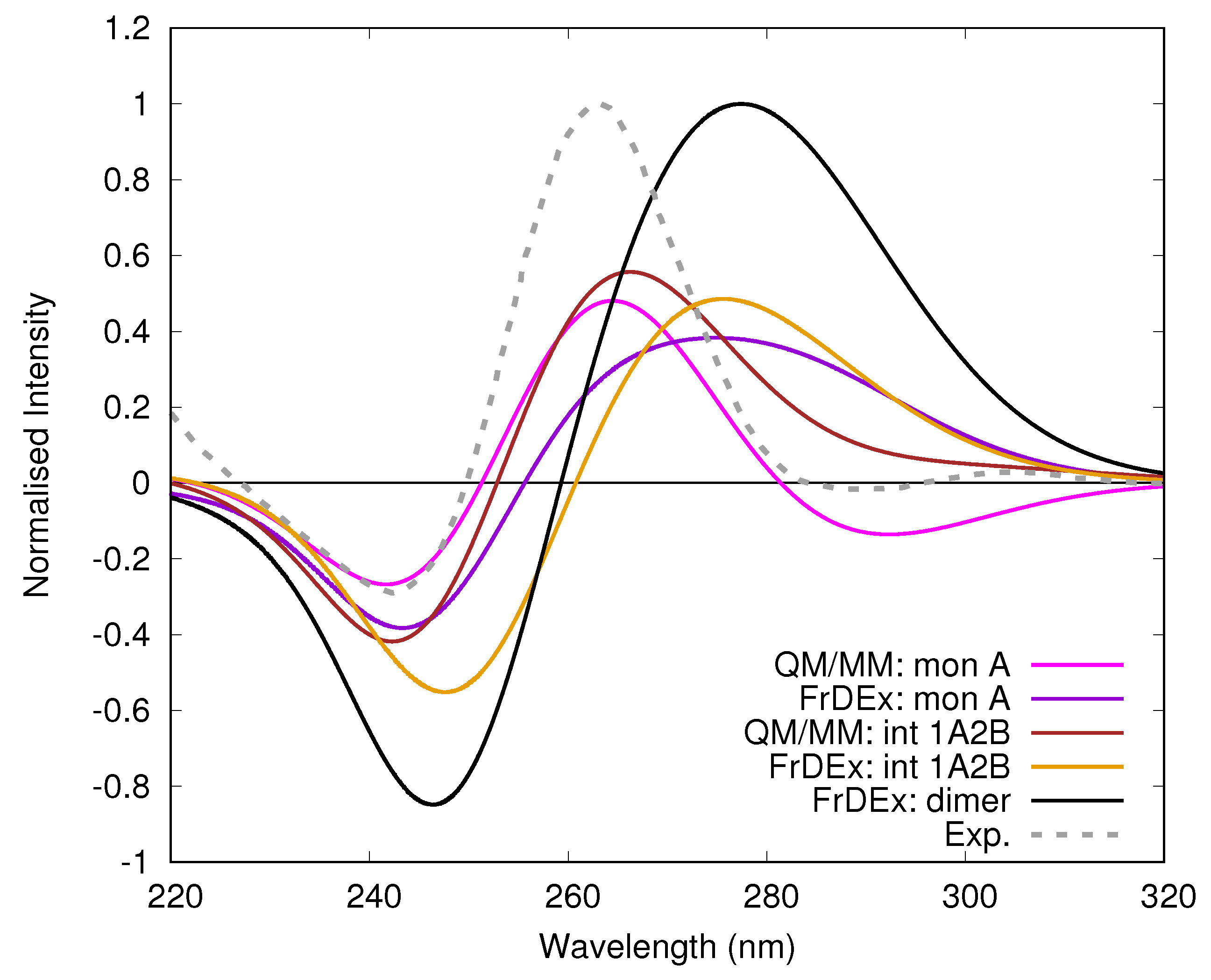
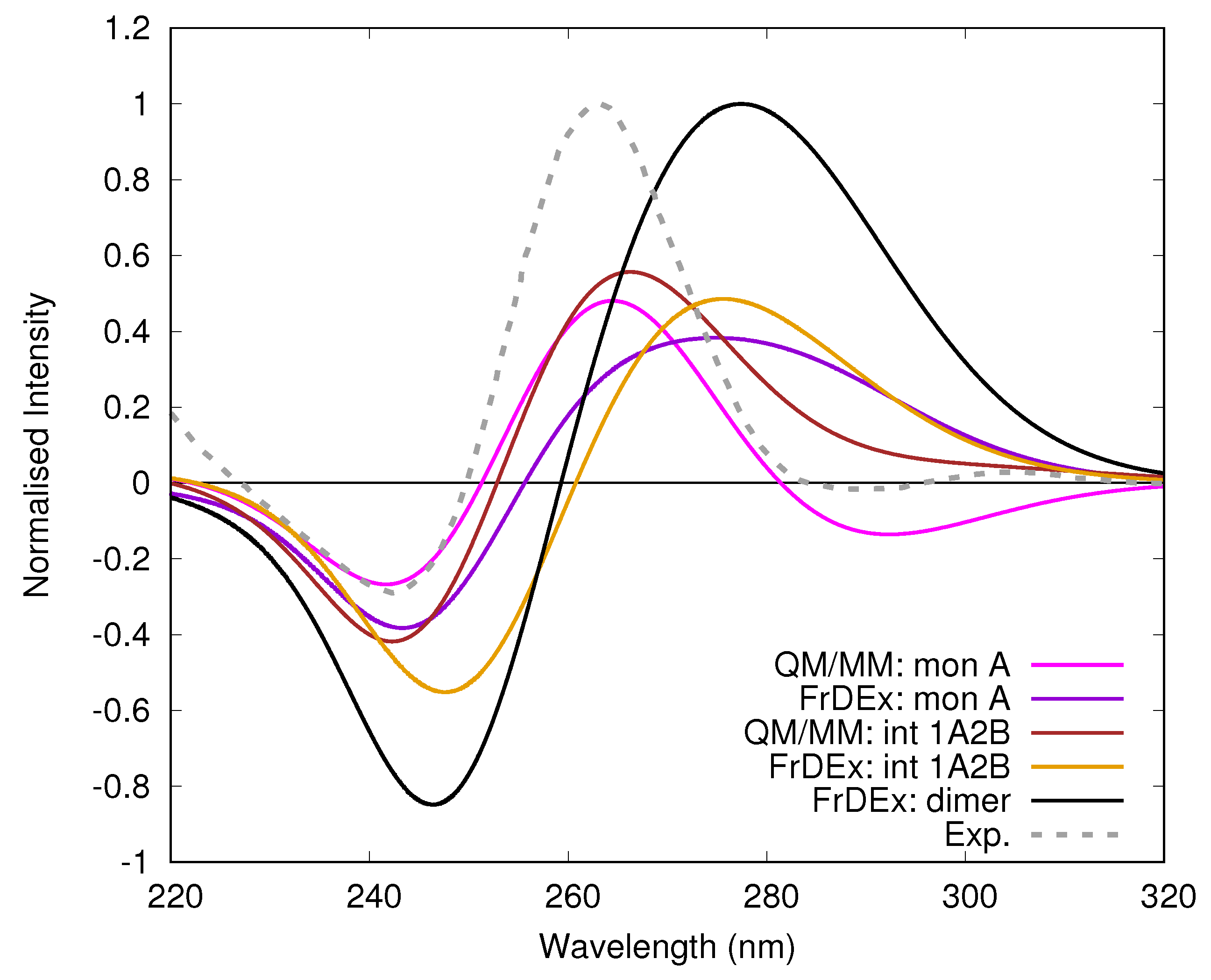
3.2. Monomeric and Dimeric Forms of T30695
4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Neidle, S.; Balasubramanian, S. (Eds.) Quadruplex Nucleic Acids; RSC Biomolecular Sciences; The Royal Society of Chemistry: London, UK, 2006; pp. i–302. [Google Scholar] [CrossRef]
- Rhodes, D.; Lipps, H.J. G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res. 2015, 43, 8627–8637. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.S.; Lin, C. (Eds.) G-Quadruplex Nucleic Acids; Methods in Molecular Biology; Humana: New York, NY, USA, 2019; pp. XIV, 437. [Google Scholar]
- Varshney, D.; Spiegel, J.; Zyner, K.; Tannahill, D.; Balasubramanian, S. The regulation and functions of DNA and RNA G-quadruplexes. Nat. Rev. Mol. Cell Biol. 2020, 21, 459–474. [Google Scholar] [CrossRef]
- Ruggiero, E.; Richter, S.N. G-quadruplexes and G-quadruplex ligands: Targets and tools in antiviral therapy. Nucleic Acids Res. 2018, 46, 3270–3283. [Google Scholar] [CrossRef]
- Shioda, N.; Yabuki, Y.; Yamaguchi, K.; Onozato, M.; Li, Y.; Kurosawa, K.; Tanabe, H.; Okamoto, N.; Era, T.; Sugiyama, H.; et al. Targeting G-quadruplex DNA as cognitive function therapy for ATR-X syndrome. Nat. Med. 2018, 24, 802–813. [Google Scholar] [CrossRef]
- Carvalho, J.; Mergny, J.L.; Salgado, G.F.; Queiroz, J.A.; Cruz, C. G-quadruplex, Friend or Foe: The Role of the G-quartet in Anticancer Strategies. Trends Mol. Med. 2020, 26, 848–861. [Google Scholar] [CrossRef] [PubMed]
- Kosiol, N.; Juranek, S.; Brossart, P.; Heine, A.; Paeschke, K. G-quadruplexes: A promising target for cancer therapy. Mol. Cancer 2021, 20, 40. [Google Scholar] [CrossRef] [PubMed]
- Harada, N.; Nakanishi, K. Optical rotatory power of the benzoate group. J. Am. Chem. Soc. 1968, 90, 7351–7352. [Google Scholar] [CrossRef]
- Harada, N.; Chen, S.M.L.; Nakanishi, K. Quantitative definition of exciton chirality and the distant effect in the exciton chirality method. J. Am. Chem. Soc. 1975, 97, 5345–5352. [Google Scholar] [CrossRef]
- Harada, N.; Nakanishi, K.; Berova, N. Electronic CD Exciton Chirality Method: Principles and Applications. In Comprehensive Chiroptical Spectroscopy; Berova, N., Polavarapu, P.L., Nakanishi, K., Woody, R.W., Eds.; Wiley: Hoboken, NJ, USA, 2012; pp. 115–166. [Google Scholar]
- Pescitelli, G.; Di Bari, L.; Berova, N. Application of electronic circular dichroism in the study of supramolecular systems. Chem. Soc. Rev. 2014, 43, 5211–5233. [Google Scholar] [CrossRef]
- Martínez-Fernández, L.; Esposito, L.; Improta, R. Studying the excited electronic states of guanine rich DNA quadruplexes by quantum mechanical methods: Main achievements and perspectives. Photochem. Photobiol. Sci. 2020, 19, 436–444. [Google Scholar] [CrossRef]
- Albano, G.; Pescitelli, G.; Di Bari, L. Chiroptical Properties in Thin Films of π-Conjugated Systems. Chem. Rev. 2020, 120, 10145–10243. [Google Scholar] [CrossRef]
- Swathi, K.; Sissa, C.; Painelli, A.; George Thomas, K. Supramolecular chirality: A caveat in assigning the handedness of chiral aggregates. Chem. Commun. 2020, 56, 8281–8284. [Google Scholar] [CrossRef] [PubMed]
- Randazzo, A.; Spada, G.P.; da Silva, M.W. Circular Dichroism of Quadruplex Structures. In Quadruplex Nucleic Acids; Chaires, J.B., Graves, D., Eds.; Springer: Berlin/Heidelberg, Germany, 2013; pp. 67–86. [Google Scholar] [CrossRef]
- Loco, D.; Jurinovich, S.; Bari, L.D.; Mennucci, B. A fast but accurate excitonic simulation of the electronic circular dichroism of nucleic acids: How can it be achieved? Phys. Chem. Chem. Phys. 2016, 18, 866–877. [Google Scholar] [CrossRef]
- Jurinovich, S.; Cupellini, L.; Guido, C.A.; Mennucci, B. EXAT: EXcitonic analysis tool. J. Comput. Chem. 2018, 39, 279–286. [Google Scholar] [CrossRef]
- Gattuso, H.; Spinello, A.; Terenzi, A.; Assfeld, X.; Barone, G.; Monari, A. Circular Dichroism of DNA G-Quadruplexes: Combining Modeling and Spectroscopy To Unravel Complex Structures. J. Phys. Chem. B 2016, 120, 3113–3121. [Google Scholar] [CrossRef] [PubMed]
- Krueger, B.P.; Scholes, G.D.; Fleming, G.R. Calculation of Couplings and Energy-Transfer Pathways between the Pigments of LH2 by the ab Initio Transition Density Cube Method. J. Phys. Chem. B 1998, 102, 5378–5386. [Google Scholar] [CrossRef]
- Scholes, G.D.; Gould, I.R.; Cogdell, R.J.; Fleming, G.R. Ab Initio Molecular Orbital Calculations of Electronic Couplings in the LH2 Bacterial Light-Harvesting Complex of Rps-Acidophila. J. Phys. Chem. B 1999, 103, 2543–2553. [Google Scholar] [CrossRef]
- Iozzi, M.F.; Mennucci, B.; Tomasi, J.; Cammi, R. Excitation energy transfer (EET) between molecules in condensed matter: A novel application of the polarizable continuum model (PCM). J. Chem. Phys. 2004, 120, 7029–7040. [Google Scholar] [CrossRef]
- Cupellini, L.; Caprasecca, S.; Guido, C.A.; Müh, F.; Renger, T.; Mennucci, B. Coupling to Charge Transfer States is the Key to Modulate the Optical Bands for Efficient Light Harvesting in Purple Bacteria. J. Phys. Chem. Lett. 2018, 9, 6892–6899. [Google Scholar] [CrossRef] [PubMed]
- Cupellini, L.; Calvani, D.; Jacquemin, D.; Mennucci, B. Charge transfer from the carotenoid can quench chlorophyll excitation in antenna complexes of plants. Nat. Commun. 2020, 11, 662. [Google Scholar] [CrossRef]
- Li, X.; Parrish, R.M.; Liu, F.; Kokkila Schumacher, S.I.L.; Martínez, T.J. An Ab Initio Exciton Model Including Charge-Transfer Excited States. J. Chem. Theory Comput. 2017, 13, 3493–3504. [Google Scholar] [CrossRef]
- Voityuk, A.A. Electronic Couplings for Photoinduced Electron Transfer and Excitation Energy Transfer Computed Using Excited States of Noninteracting Molecules. J. Phys. Chem. A 2017, 121, 5414–5419. [Google Scholar] [CrossRef] [Green Version]
- Tamura, H.; Burghardt, I.; Tsukada, M. Exciton Dissociation at Thiophene/Fullerene Interfaces: The Electronic Structures and Quantum Dynamics. J. Phys. Chem. C 2011, 115, 10205–10210. [Google Scholar] [CrossRef]
- Tamura, H.; Burghardt, I. Potential Barrier and Excess Energy for Electron–Hole Separation from the Charge-Transfer Exciton at Donor–Acceptor Heterojunctions of Organic Solar Cells. J. Phys. Chem. C 2013, 117, 15020–15025. [Google Scholar] [CrossRef]
- Tamura, H. Diabatization for Time-Dependent Density Functional Theory: Exciton Transfers and Related Conical Intersections. J. Phys. Chem. A 2016, 120, 9341–9347. [Google Scholar] [CrossRef] [PubMed]
- Polkehn, M.; Tamura, H.; Burghardt, I. Impact of charge-transfer excitons in regioregular polythiophene on the charge separation at polythiophene-fullerene heterojunctions. J. Phys. B At. Mol. Opt. Phys. 2017, 51, 014003. [Google Scholar] [CrossRef]
- Polkehn, M.; Eisenbrandt, P.; Tamura, H.; Burghardt, I. Quantum dynamical studies of ultrafast charge separation in nanostructured organic polymer materials: Effects of vibronic interactions and molecular packing. Int. J. Quantum Chem. 2018, 118, e25502. [Google Scholar] [CrossRef] [Green Version]
- Popp, W.; Polkehn, M.; Hughes, K.H.; Martinazzo, R.; Burghardt, I. Vibronic coupling models for donor-acceptor aggregates using an effective-mode scheme: Application to mixed Frenkel and charge-transfer excitons in oligothiophene aggregates. J. Chem. Phys. 2019, 150, 244114. [Google Scholar] [CrossRef]
- Popp, W.; Polkehn, M.; Binder, R.; Burghardt, I. Coherent Charge Transfer Exciton Formation in Regioregular P3HT: A Quantum Dynamical Study. J. Phys. Chem. Lett. 2019, 10, 3326–3332. [Google Scholar] [CrossRef]
- Green, J.A.; Asha, H.; Santoro, F.; Improta, R. Excitonic Model for Strongly Coupled Multichromophoric Systems: The Electronic Circular Dichroism Spectra of Guanine Quadruplexes as Test Cases. J. Chem. Theory Comput. 2021, 17, 405–415. [Google Scholar] [CrossRef]
- Jing, N.; Marchand, C.; Liu, J.; Mitra, R.; Hogan, M.E.; Pommier, Y. Mechanism of Inhibition of HIV-1 Integrase by G-tetrad-forming Oligonucleotides in Vitro. J. Biol. Chem. 2000, 275, 21460–21467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Do, N.Q.; Lim, K.W.; Teo, M.H.; Heddi, B.; Phan, A.T. Stacking of G-quadruplexes: NMR structure of a G-rich oligonucleotide with potential anti-HIV and anticancer activity. Nucleic Acids Res. 2011, 39, 9448–9457. [Google Scholar] [CrossRef] [PubMed]
- Imperatore, C.; Varriale, A.; Rivieccio, E.; Pennacchio, A.; Staiano, M.; D’Auria, S.; Casertano, M.; Altucci, C.; Valadan, M.; Singh, M.; et al. Spectroscopic Properties of Two 5′-(4-Dimethylamino)Azobenzene Conjugated G-Quadruplex Forming Oligonucleotides. Int. J. Mol. Sci. 2020, 21, 7103. [Google Scholar] [CrossRef]
- Green, J.A.; Yaghoubi Jouybari, M.; Asha, H.; Santoro, F.; Improta, R. Fragment Diabatization Linear Vibronic Coupling Model for Quantum Dynamics of Multichromophoric Systems: Population of the Charge-Transfer State in the Photoexcited Guanine–Cytosine Pair. J. Chem. Theory Comput. 2021. [Google Scholar] [CrossRef]
- Jurinovich, S.; Pescitelli, G.; Di Bari, L.; Mennucci, B. A TDDFT/MMPol/PCM model for the simulation of exciton-coupled circular dichroism spectra. Phys. Chem. Chem. Phys. 2014, 16, 16407–16418. [Google Scholar] [CrossRef] [PubMed]
- Jurinovich, S.; Guido, C.A.; Bruhn, T.; Pescitelli, G.; Mennucci, B. The role of magnetic–electric coupling in exciton-coupled ECD spectra: The case of bis-phenanthrenes. Chem. Commun. 2015, 51, 10498–10501. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Density Functionals with Broad Applicability in Chemistry. Acc. Chem. Res. 2008, 41, 157–167. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision C.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Improta, R.; Santoro, F.; Blancafort, L. Quantum mechanical studies on the photophysics and the photochemistry of nucleic acids and nucleobases. Chem. Rev. 2016, 116, 3540–3593. [Google Scholar] [CrossRef] [PubMed]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Martinez-Fernandez, L.; Changenet, P.; Banyasz, A.; Gustavsson, T.; Markovitsi, D.; Improta, R. Comprehensive Study of Guanine Excited State Relaxation and Photoreactivity in G-quadruplexes. J. Phys. Chem. Lett. 2019, 10, 6873–6877. [Google Scholar] [CrossRef]
- Martínez-Fernández, L.; Banyasz, A.; Markovitsi, D.; Improta, R. Topology Controls the Electronic Absorption and Delocalization of Electron Holes in Guanine Quadruplexes. Chem. Eur. J. 2018, 24, 15185–15189. [Google Scholar] [CrossRef]
- Improta, R. Quantum Mechanical Calculations Unveil the Structure and Properties of the Absorbing and Emitting Excited Electronic States of Guanine Quadruplex. Chem. Eur. J. 2014, 20, 8106–8115. [Google Scholar] [CrossRef]
- Banyasz, A.; Martínez-Fernández, L.; Balty, C.; Perron, M.; Douki, T.; Improta, R.; Markovitsi, D. Absorption of Low-Energy UV Radiation by Human Telomere G-Quadruplexes Generates Long-Lived Guanine Radical Cations. J. Am. Chem. Soc. 2017, 139, 10561–10568. [Google Scholar] [CrossRef]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Gould, I.R.; Merz, K.M.; Ferguson, D.M.; Spellmeyer, D.C.; Fox, T.; Caldwell, J.W.; Kollman, P.A. A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc. 1995, 117, 5179–5197. [Google Scholar] [CrossRef] [Green Version]
- Dapprich, S.; Komáromi, I.; Byun, K.; Morokuma, K.; Frisch, M.J. A new ONIOM implementation in Gaussian98. Part I. The calculation of energies, gradients, vibrational frequencies and electric field derivatives. J. Mol. Struct. THEOCHEM 1999, 461–462, 1–21. [Google Scholar] [CrossRef]
- Case, D.; Betz, R.; Cerutti, D.; Cheatham, T.R.; Darden, T.; Duke, R.; Giese, T.; Gohlke, H.; Götz, A.; Homeyer, N.; et al. AMBER 2016; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Zgarbová, M.; Šponer, J.; Otyepka, M.; Cheatham, T.E., III; Galindo-Murillo, R.; Jurečka, P. Refinement of the Sugar-Phosphate Backbone Torsion Beta for AMBER Force Fields Improves the Description of Z- and B-DNA. J. Chem. Theory Comput. 2015, 11, 5723–5736. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Grigera, J.R.; Straatsma, T.P. The missing term in effective pair potentials. J. Phys. Chem. 1987, 91, 6269–6271. [Google Scholar] [CrossRef]
- Joung, I.S.; Cheatham, T.E., III. Determination of Alkali and Halide Monovalent Ion Parameters for Use in Explicitly Solvated Biomolecular Simulations. J. Phys. Chem. B 2008, 112, 9020–9041. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Patel, D.J. Solution structure of the human telomeric repeat d[AG3(T2AG3)3] G-tetraplex. Structure 1993, 1, 263–282. [Google Scholar] [CrossRef]
- Lu, X.; Olson, W.K. 3DNA: A software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures. Nucleic Acids Res. 2003, 31, 5108–5121. [Google Scholar] [CrossRef] [Green Version]
- Karunakaran, V.; Kleinermanns, K.; Improta, R.; Kovalenko, S.A. Photoinduced Dynamics of Guanosine Monophosphate in Water from Broad-Band Transient Absorption Spectroscopy and Quantum-Chemical Calculations. J. Am. Chem. Soc. 2009, 131, 5839–5850. [Google Scholar] [CrossRef] [PubMed]
- Avila Ferrer, F.J.; Cerezo, J.; Stendardo, E.; Improta, R.; Santoro, F. Insights for an Accurate Comparison of Computational Data to Experimental Absorption and Emission Spectra: Beyond the Vertical Transition Approximation. J. Chem. Theory Comput. 2013, 9, 2072–2082. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Noguchi, Y.; Sugiyama, H. The new models of the human telomere d[AGGG(TTAGGG)3] in K+ solution. Bioorg. Med. Chem. 2006, 14, 5584–5591. [Google Scholar] [CrossRef]
- del Villar-Guerra, R.; Gray, R.D.; Chaires, J.B. Characterization of Quadruplex DNA Structure by Circular Dichroism. Curr. Protoc. Nucleic Acid Chem. 2017, 68. [Google Scholar] [CrossRef]
- Martinez-Fernandez, L.; Prampolini, G.; Cerezo, J.; Liu, Y.; Santoro, F.; Improta, R. Solvent effect on the energetics of proton coupled electron transfer in guanine-cytosine pair in chloroform by mixed explicit and implicit solvation models. Chem. Phys. 2018, 515, 493–501. [Google Scholar] [CrossRef]
- Islam, B.; Stadlbauer, P.; Krepl, M.; Havrila, M.; Haider, S.; Sponer, J. Structural Dynamics of Lateral and Diagonal Loops of Human Telomeric G-Quadruplexes in Extended MD Simulations. J. Chem. Theory Comput. 2018, 14, 5011–5026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mergny, J.L.; Sen, D. DNA Quadruple Helices in Nanotechnology. Chem. Rev. 2019, 119, 6290–6325. [Google Scholar] [CrossRef]
- Hestand, N.J.; Spano, F.C. Expanded Theory of H- and J-Molecular Aggregates: The Effects of Vibronic Coupling and Intermolecular Charge Transfer. Chem. Rev. 2018, 118, 7069–7163. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asha, H.; Green, J.A.; Martinez-Fernandez, L.; Esposito, L.; Improta, R. Electronic Circular Dichroism Spectra of DNA Quadruple Helices Studied by Molecular Dynamics Simulations and Excitonic Calculations including Charge Transfer States. Molecules 2021, 26, 4789. https://doi.org/10.3390/molecules26164789
Asha H, Green JA, Martinez-Fernandez L, Esposito L, Improta R. Electronic Circular Dichroism Spectra of DNA Quadruple Helices Studied by Molecular Dynamics Simulations and Excitonic Calculations including Charge Transfer States. Molecules. 2021; 26(16):4789. https://doi.org/10.3390/molecules26164789
Chicago/Turabian StyleAsha, Haritha, James A. Green, Lara Martinez-Fernandez, Luciana Esposito, and Roberto Improta. 2021. "Electronic Circular Dichroism Spectra of DNA Quadruple Helices Studied by Molecular Dynamics Simulations and Excitonic Calculations including Charge Transfer States" Molecules 26, no. 16: 4789. https://doi.org/10.3390/molecules26164789