1. Introduction
Soybean (
Glycine max) is one of the main crops produced around the world [
1]. Soybean for human consumption is often transformed to obtain different kinds of protein isolates, such as soybean curd or soy milk. Okara is a yellowish fibrous material consisting of insoluble residue remaining after these preparations. Its massive production, about 1.2 kg of okara from 1 kg of soybeans for a total yearly production of 1.4 billion tons [
2], poses serious issues regarding its disposal. Despite being the leftover of protein isolation processes, okara still represents a rich source of valuable compounds. On average, the dry matter contains 15.2–33.4% of proteins, 3.8–5.3% free carbohydrate, 8.3–10.9% oil and fat, while most of the dry matter is composed of insoluble fiber. The high protein content makes okara a low-cost source of plant proteins and, potentially, of biologically active peptides [
3]. In fact, peptides derived from different sources (food, plants, insects) have been shown to exert several bioactivities in animals and plants [
4]. One potential application of these peptides is their use as antimicrobial and antifungal molecules.
Fungi and bacteria seriously damage the growth and yield of crops [
5] being responsible for the largest number of plant diseases. For example,
Fusarium rots and
Rhizoctonia rots can occur in a wide range of crops [
6,
7]. In particular, fungi of the genus
Fusarium are considered economically relevant because they can infect cereals, potatoes, vegetables,
Fabaceae, ornamental, and forest plants. In addition, some
Fusarium species also produce mycotoxins both in the field and in stored grains, posing serious threats to animal and human health [
7]. Fungi of the genus
Rhizoctonia cause several kind of diseases on
Solanaceae and other cultivated plants such as rice, cereals, and sugarbeet [
8]. Bacteria of the genus
Pseudomonas are saprophytes and parasites on plant tissue and surfaces. They are responsible for diseases such as rot, necrosis, and galls [
9].
P. syringae pv. causes, on different tissues of tomato, olive tree, and other plants, local necrotic lesions of various sizes, from tiny flecks to visible lesions [
10,
11];
P. solanacearum is a bacterium distributed worldwide and it causes systemic wilt on different crops like potato, tobacco, tomato, and banana [
12].
Conventional crop protection, based on chemical pesticides, is being revaluated for integrated pest management (IPM) that aims to protect human and environment health. Traditional crop protection is based on ready-to-use tools, while IPM is a dynamic system focused on user needs. To make IPM a successful system, alternatives to conventional pesticides are needed to effectively manage crop pests [
13]. The features of the ideal pesticide have changed: it must be highly selective for the target species, must have high efficacy at a low application rate, and low environmental persistence that avoids bioconcentration, biomagnification, and development of resistance.
Plants have been sources of natural pesticides for centuries [
14]. In particular, the use of plant extracts is considered a good strategy because they have a minimal environmental impact and are potentially nontoxic for consumers [
5]. As a matter of fact, plant’s mechanisms of protection against pathogens include a class of proteins and peptides, called defense-related proteins that represent an important strategy to survive infections. Antifungal proteins and peptides have been found in several plant tissues, mostly in seeds [
15]. Indeed, during germination, these are vulnerable to pathogen attacks due to the rupture of the seed coat, which allow the invasion of the storage tissue [
16]. Antifungal peptides have been described in many seeds, showing inhibitory action against
Botrytis cinerea, Mycosphaerella arachidicola, Fusarium oxysporum.
Trichoderma reesei, and
Candida albicans [
17,
18]. These antifungal peptides are considered a bioactive molecule group with broad-spectrum activities and multiple mechanisms of action [
18]. Antimicrobial peptides have also been found in soybean [
19]; hence, there might be natural bioactive molecules with fungicide activity in okara as well.
In this context, the aim of this work is to describe a rapid and economic procedure to isolate proteins from okara and to produce peptides active against fungal plant pathogens by applying an enzymatic approach. The characterization of okara-derived antifungal bioactive peptides has the potential to turn this by-product into a paradigmatic example of circular economy, since a field-derived food waste is transformed into a source of valuable compounds to be used in field crops protection.
3. Discussion
Reduction of by-products and food wastage to enable full use of natural resources is one of the main challenges the industrialized world faces nowadays. Okara poses, at present, significant disposal issues. Because of the high protein content, it is a by-product interesting and exploitable from a biotechnological point of view. Our small-scale protein extraction trials provided a glimpse into the complexity of the molecular interactions occurring among okara proteins, highlighting a tricky matrix. The process from soybeans to okara likely has detrimental effects on protein stability and solubility [
23], hampering the use of this by-product as a source of proteins and peptides. The thermic treatment faced by okara during its processing led to inter-protein crosslinks, mediated by disulphide bridges. This can be deduced since in all conditions tested, the addition of a reducing agent doubled the extraction yield in almost all the cases. The simultaneous use of reducing agents, detergents, and chaotropic substances enhanced the solubilization of okara proteins (
Figure 1A). The procedure we finally adopted is a fair compromise between yields and use of large amounts of urea, the presence of which is detrimental for subsequent steps, including the production of peptides by proteolytic enzymes. Our results provide insights into the complex interactions that take place between the different constituents of this raw material, those that can affect the biochemical and enzymatic treatments applied for its transformation. Among the seven proteases we used, pancreatin proved to be the most suitable, as it exhibits high levels of proteolysis that, above all, led to a significant production in antimicrobial activity. On the contrary, the use of the other enzymes resulted in an increase in microbial growth, probably caused by the release of single amino acids that may act as a nutritional supplement.
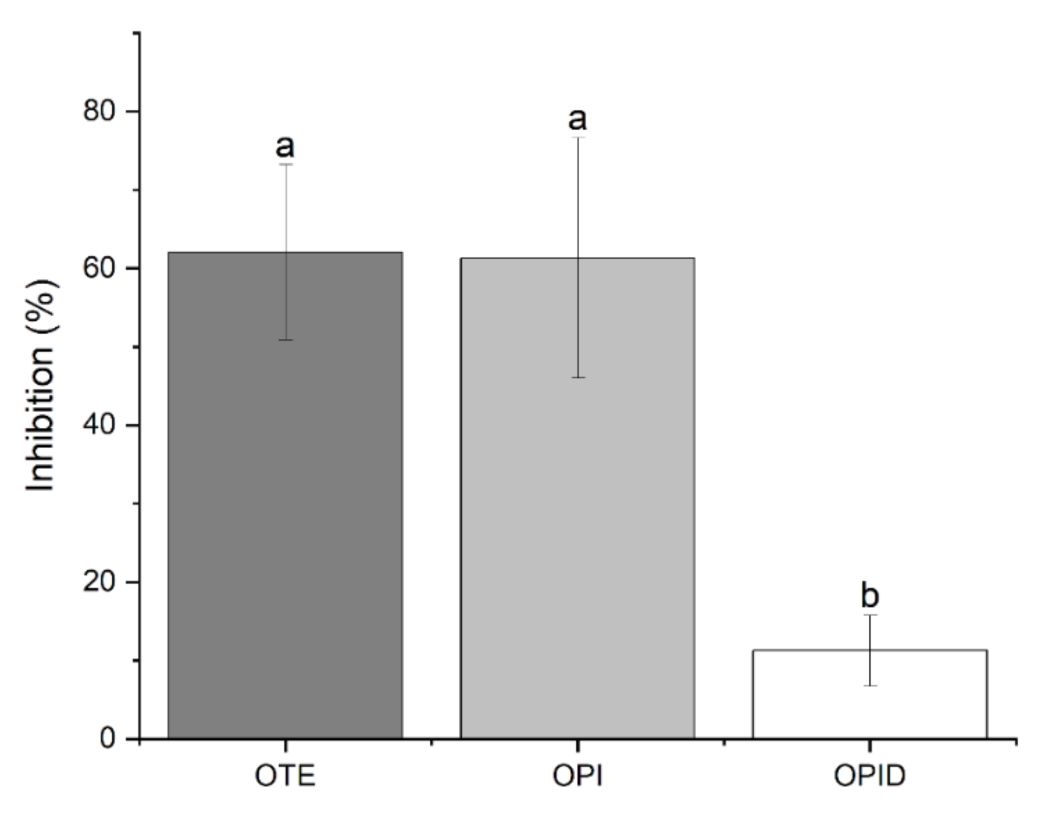
From both the experiments reported in
Figure 6 and
Figure 7, it is evident that OPI itself, at different extents, exerts an inhibitory activity on mycelial growth in dependence either of the fungal strain or the protein concentration. It is reported that trypsin inhibitors may exert antifungal activity as a plant defense response [
24,
25,
26]. Thus, the dose-response dependent inhibition activity of OPI samples is likely attributable to TIA. Meanwhile, it appears unlikely that the activity shown by OPID could be related to the same mechanism since a significant decrease of TIA because of the proteolytic digestion process has been demonstrated. Finally, the selective effect of OPID on the
Fusarium strains, in particular F. graminearum, is noteworthy, while
R. solani growth is partially inhibited by the undigested sample, but this inhibition does not increase following protein digestion.
We developed a rapid, economic, and scalable method of protein extraction and digestion with pancreatin to produce peptides active against pathogens with a relevant economic interest, such as fungi belonging to the genus Fusarium and bacteria like Pseudomonas, which can infect many crops.
We believe that the experimentation we have carried out can be a realistic starting point for the implementation of a production process that transforms okara into a low environmental impact crop protection product. Nowadays, several strategies are used for the control of fungal and bacteria growth by chemical treatments [
27]. New active substances need to be investigated to find compounds that can effectively fight pathogens and have minimal impact on human beings, animals, and the environment [
14].
The production and characterization of okara-derived antifungal bioactive peptides, thus, has the potential to turn this by-product into a paradigmatic example of circular economy, since a field-derived by-product is transformed into a source of valuable compounds to be used in field crops protection.
Finally, these results lay the foundation for further experimentation aimed at testing the peptides produced in vivo using model plants and for final biochemical characterization with the aim of establishing the primary structure of the active peptide(s).
4. Materials and Methods
4.1. Screening of Protein Extraction Methods
Okara pulp (kindly provided frozen by Valsoia, Serravalle Sesia) was milled with liquid nitrogen until obtaining a uniform granulometry and either stored at −80°C (wet okara) or lyophilized (dry okara).
All chemicals used were purchased from Sigma-Aldrich (St. Louis, MO, USA). Different combinations of solvents (dH
2O, 50 mM phosphate Buffer pH 6.0, 50 mM Tris-HCl pH 8.0, 50 mM carbonate Buffer pH 10.0) and additives in different concentrations (500 mM NaCl, 5 mM DTT, SDS, Urea) in a total volume of 10 mL (1:25
w/
v ratio with lyophilized okara) were tested to solubilize proteins at room temperature stirring for 3 h. Supernatants were collected after 15 min centrifugation at 12,000 rpm and protein quantification was performed with Bradford assay [
28]. Extractions were performed in triplicate.
4.2. Screening of Proteolytic Enzymes
Wet okara pulp was suspended in 50 mM Tris-HCl pH 8.0 buffer (1:5 w/v). The enzymes were then tested with the respective ratios: trypsin (Sigma-Aldrich, St. Louis, MO, USA) 50 µg/g wet okara, papain (Sigma-Aldrich, St. Louis, MO, USA), bromelain (Sigma-Aldrich, St. Louis, MO, USA) 1 U/g wet okara, pancreatin (Sigma-Aldrich, St. Louis, MO, USA), Amano A2, S, and N (Amano Enzyme, Inc., Nagoya, Japan) 1 mg/g wet okara. Incubation was performed for 2 h at 25 °C in all the cases, except for trypsin and papain whose incubation lasted for 4 h. Enzymes were inactivated with the addition of NaOH; the obtained proteolyzed okara was centrifuged at 16,000 ×g 90 min at 4 °C, and the supernatant filtered. Okara Total Extract (OTE) was obtained as reference by resuspending wet okara pulp in 50 mM Tris-HCl pH 8.0 buffer (1:5 w/v), without the addition of any enzyme, incubating it for 2 h at 25 °C. The product was finally heated at 65 °C for 30 min to denature enzymes, which were neutralized with HCl and filter sterilized (Millex-GV Filter, 0.22 µM, Millipore, Burlington, MA, USA).
4.3. Okara Protein Extraction Isolation, Digestion, and Haracterization
4.3.1. Okara Protein Isolate Preparation
For large scale protein extraction, milled wet okara pulp was suspended (1:5
w/
v) in 50 mM Tris-HCl pH 8.0 and SDS 1%
w/
v stirring at 25 °C for 3 h. To precipitate free KDS 0.1 vol of 1 M Potassium Phosphate Buffer pH 8.0 was added stirring at 25 °C for 30 min as in Carraro et al. [
29] with slight modifications. The slurry was then centrifuged 16,000×
g 90 min at 4 °C, the supernatant was filtered with Whatman
® quantitative filter paper Grade 42 (Whatman, Maidstone, UK), and the Okara Total Extract (OTE) was collected. To achieve protein precipitation, 5 M HCl was added until reaching pH 4.5 and an incubation overnight at 4°C was performed. After centrifugation at 16,000×
g for 30 min, pelleted Okara Protein Isolate (OPI) was separated from its Supernatant (SN), washed twice with distilled water, and lyophilized. For all the further described procedures, OPI was resuspended in 50 mM Tris-HCl pH 8.0.
4.3.2. Okara Protein Isolate Digestion
Once resuspended in 50 mM Tris-HCl pH 8.0, Okara Protein Isolate (OPI) was autoclaved at 121 °C for 15 min to obtain sterilization. Pancreatin from porcine pancreas (Sigma-Aldrich, St. Louis, MO, USA) was dissolved in 1 mM HCl at 5 mg/mL concentration, filter sterilized (Millex-GV Filter, 0.22 µm, Millipore, Burlington, MA, USA), and added to the resuspended proteins with 1:20 (w/w) proportion. Digestion was performed at 25 °C for 24 h and stopped by heating the solution at 65 °C for 30 min, thus obtaining the Okara Protein Isolate Digested fraction (OPID). Samples after 6 h of digestion were withdrawn and treated with the same procedure in order to control OPI digestion kinetics.
4.3.3. Residual SDS Quantification
Quantification of eventual SDS remaining in Okara Protein Isolate samples was performed with slight modifications of the method described by Žilionis, based on Mukerjee’s photometric method [
30]. Briefly, 100 µL of each sample were incubated with 400 µL of cold acetone (Sigma-Aldrich, St. Louis, MO, USA) for 1 h at −20 °C, then centrifuged 10,000×
g 10 min. 125 µL were then transferred and 1 mL of a solution of 10 µg/mL methylene blue (Carlo Erba, Cornaredo, Italy) in 10 mM HCl, 200 µL of CHCl
3 were added and the samples were thoroughly vortexed. After 10 min incubation at room temperature, a centrifugation step at 2000 g was performed for 10 min. Samples were left 10 min at room temperature, then 800 µL of the upper aqueous layer were transferred to a plastic cuvette and absorbance read at 655 nm against a blank sample. A standard curve was built with the same procedure with SDS standard solutions at different concentrations between 0.0025% and 0.025%. For each sample, the mean absorbance was subtracted to the mean absorbance of the blank solution to obtain a positive correlation curve. Each sample was measured in triplicate.
4.3.4. Spectrophotometric Characterization of Extracts and Protein Quantification
Total okara protein content was determined by the application of the Kjeldahl method [
31] onto three replicates. As for extracted and isolated proteins, common colorimetric quantification methods such as Bradford or Lowry proved to be unreliable due to the presence of SDS, even at negligible concentrations [
32]. Spectra of the different fractions obtained (OTE, OPI, OPID, SN) were collected between 200 and 300 nm on a Lambda 2 spectrophotometer (Perkin Elmer, Waltham, MA, USA). Since high absorption at 260 nm was observed, protein quantification of OTE and OPI fractions was performed using the correction described by Kalb et al. [
33] with the formula:
Digested samples (OPID) were quantified according to the far UV absorbance as described by Scopes [
34] since the previously mentioned method underestimated the protein/peptide content. Lambert-Beer law at 205 nm was applied and molar extinction coefficient ε
0.1% was calculated for each sample using the formula:
4.3.5. SDS–PAGE
SDS–PAGE was carried out according to Laemmli [
35] on 12% polyacrylamide gel. For peptides separations, 15% polyacrylamide gels were used. Gels were stained by Coomassie Blue G-250 (BioRad, Milan, Italy.) Low-range SDS-PAGE Standards (Bio-Rad, Hercules, CA, USA) were used for the Mr calculations.
4.3.6. Trypsin Inhibitory Activity (TIA)
Trypsin Inhibitory Activity (TIA) was measured according to the standard method described in ISO 14902:2001 [
36]. Briefly, trypsin activity was measured by using the synthetic substrate N-Benzoyl-L-arginine 4-nitroanilide hydrochloride (BAPA, Merck, Life Science, Milan, Italy). Trypsin stock solution 0.27 mg/mL was prepared in 1 mM HCl with 5 mM CaCl
2 and then diluted 1:20 to perform the assay. BAPA working solution was prepared by diluting 1:100 the stock solution (1.5 mM in DMSO) in 50 mM Tris-HCl pH 8.2, 5 mM CaCl
2. The assay was performed by mixing 0.1 mL of the protein extract (0.5 mg/mL) with 0.1 mL of BAPA working solution and 0.2 mL of water. After 10 min of incubation at 37 °C, 0.1 mL of trypsin solution was added, and the sample was incubated for 10 min at the same temperature. The reaction was stopped by adding 0.1 mL of 30% acetic acid. The absorbance reading at 410 nm was recorded as a measure of the trypsin activity in the presence of sample inhibitors (As). The reaction was also run in the absence of inhibitors by replacing the sample extract with an equal amount of water to have a reference reading (Ar). Corresponding reagent blanks for the sample readings (Abs) and a reagent blank for the reference readings (Abr) were also prepared by adding the acetic acid solution before the trypsin solution. TIA was calculated as inhibition percent:
Each experiment was performed in triplicate.
4.3.7. Chromatographic Separations
The protein content of OPI and OPID was analyzed by size-exclusion chromatography with a Superdex Peptide Increase 10/300 (GE Healthcare, Chicago, IL, USA) equilibrated in 50 mM Tris-HCl pH 8.0 onto a Waters Delta 600 HPLC system (Waters Corporation, Milford, MA, USA) equipped with a Waters 2487 dual absorbance reader (Waters Corporation, Milford, MA, USA) set at 214 and 280 nm. 100 µL of samples at the same concentrations were loaded and eluted in isocratic mode with a flux of 0.5 mL/min. Calibration was performed with known Mr standards.
4.4. Plant Pathogen Bioassays
4.4.1. Fungal and Bacteria Isolates
To test the inhibitory activity of the proteolyzed samples obtained from okara, three different
Fusarium species were used (
Table 2):
F. graminearum (Fg 3005) collected in Australia in 2001 from barley,
F. oxysporum f. sp. Lycopersici (Fgsc 10446) collected in Italy from tomato, and
F. verticilloides (MUCL43478) collected in California from corn. One strain of
Rhizoctonia solani was also included. As for bacteria tests, two different species of
Pseudomonas were used:
P. solanacearum (Smith) Biotype IV (IPV-2574) and
P. syringeae pv. Tomato (Okabe) (Young et al.).
4.4.2. In Vitro Tests of Fungal Mycelial Growth Inhibition
In vitro tests of fungal mycelial growth inhibition were performed with the same procedure described below to test different conditions, with the proper described controls. Firstly, assays were performed to screen the effect of the different proteolytic enzymes against F. graminearum. Then, digestion kinetics was assayed by testing Okara Protein Isolate (T0) and Okara Protein Isolated digested with pancreatin for 6 h (T6) and 24 h (T24) at a constant concentration of 0.5 mg/mL. Finally, dose-dependent inhibitory activity was assayed by changing the amounts of OPI and OPID digested 24 h (0.05–2 mg/mL) to determine the maximum inhibitory concentration and the concentrations that do not inhibit fungal growth. In these last two cases, inhibitory activity was tested also against F. oxysporum, F. verticilloides and R. solani.
The biological activity of the treatments was determined by incorporating aliquots of the different okara samples into Czapek medium to obtain the desired final protein concentrations. Czapek medium was prepared as follows: 35 g Czapek nutrient broth (Sigma-Aldrich, St. Louis, MO, USA), 2 g yeast extract (Sigma–Aldrich, St. Louis, MO, USA), and 15 g agar (Sigma–Aldrich, St. Louis, MO, USA) dissolved in 750 mL of water. The media were prepared at 1.3× concentration in order to have the same nutrient composition into the final solution, after addition of the proper volumes of okara samples. Okara samples were sterilized and heated at 50 °C to bring them to the same temperature as the medium and then incorporated in it in the proper proportion. pH was corrected after the addition of okara sample to have the same pH of the controls at a final value of 8.0.
Mycelial disks of different fungi were transferred from the edges of a seven-day-old culture to plates filled with the media containing okara samples. Different control experiments were considered: Czapek medium plus undigested OPI (T0), Czapek medium plus equivalent peptone concentration, Czapek medium plus each single enzyme solution at the same concentration used in the digestion procedures, Czapek medium plus SDS 0.04%. Radial mycelial growth was determined after 36 h by calculation of the average of two perpendicular colony diameters for each replicate. Each experiment was performed in triplicate.
4.4.3. Microplate Assay
Bacteria were grown for 48 h in KB liquid medium (Duchefa Biochemie, Haarlem, NL) at 25 °C to reach a concentration of 109 ufc/mL. A flat bottom 96 microplate was prepared with 4 replicates for each okara sample tested. 150 μL of 1.3 × KB medium, either 40 μL of each okara sample or 40 μL of sterile deionized water for the control, and 10 μL of bacteria suspension (107 ufc) were added in each well. Different concentrations (2 mg/mL; 0.5 mg/mL; 0.1 mg/mL; 0.05 mg/mL; 0.005 mg/mL) of OPI and OPID were then assayed to test the dose-dependent inhibitory activity and to determine the maximum inhibitory concentration and the concentrations that do not inhibit growth. Equivalent peptone concentration solutions were set as reference controls. Supernatant (SN) obtained after protein precipitation was also tested by adding 40 μL of the neutralized solution in each well using a solution of 0.004% SDS as control. Controls to determine medium sterility were added for each condition as well. The microplate was closed and incubated at 25 °C. Absorbance readings were performed at 600 nm three times per day over 2 days. All experiments were confirmed twice.
4.5. Statistical Analysis
Statistically significant differences among the different extraction methods tested, in terms of protein extraction yields, and TIA assays were calculated using Tukey’s test for mean comparisons (significance was set at p-value < 0.01). Fungal mycelial growth inhibition expressed as % residual growth of colonies in the presence of OPID, with respect to each proper reference sample, were compared by Student’s t-test (significance was set at p-value < 0.05). Statistical analyses were performed with the software Origin (Pro), Version 2021b. (OriginLab Corporation, Northampton, MA, USA). The same software was used to generate graphs.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}