
Solvatomorphism of Moxidectin

 ,
,  and
and
Abstract
:1. Introduction
2. Results and Discussion
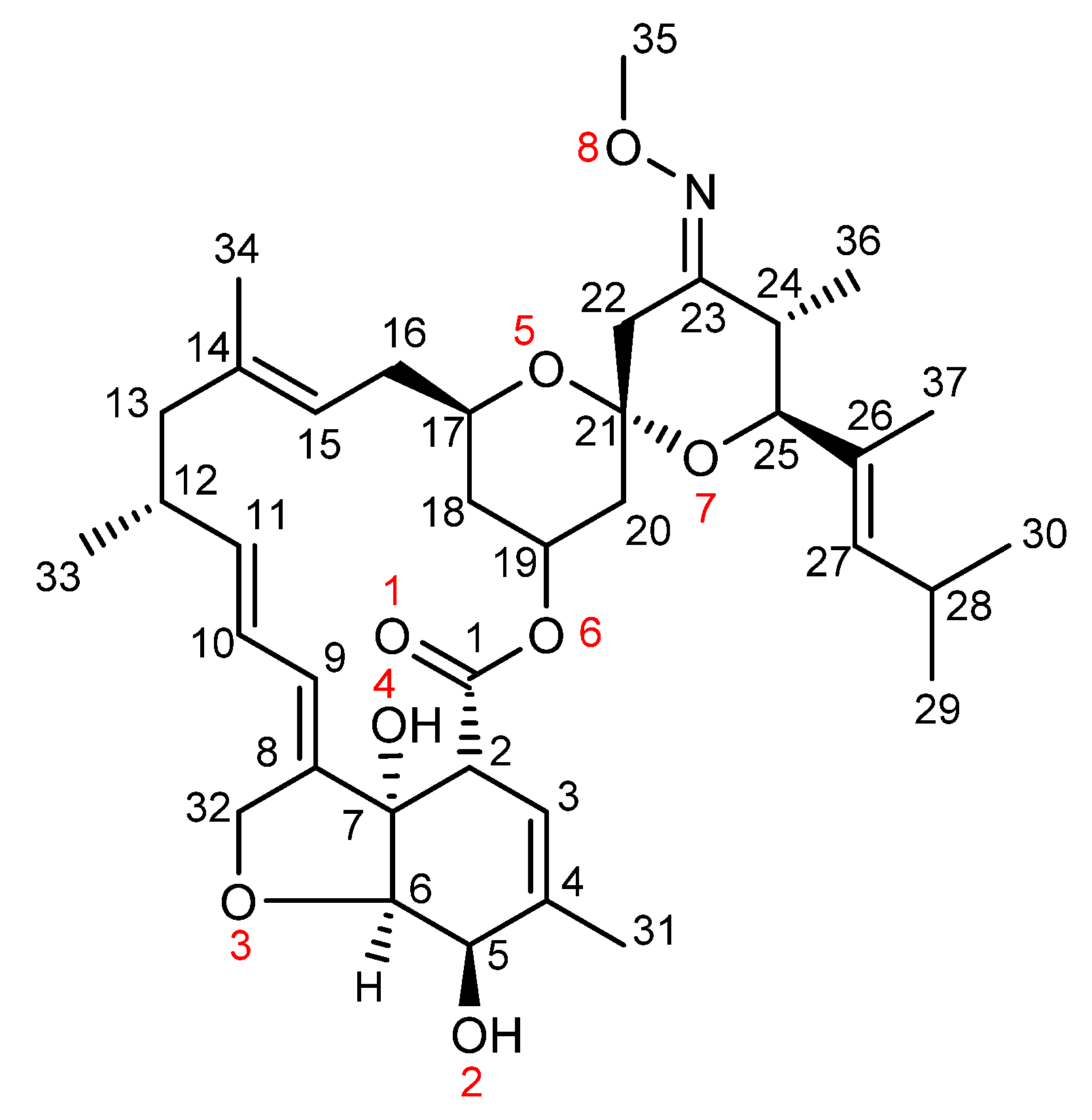
2.1. Preliminary Considerations on Moxidectin Reports
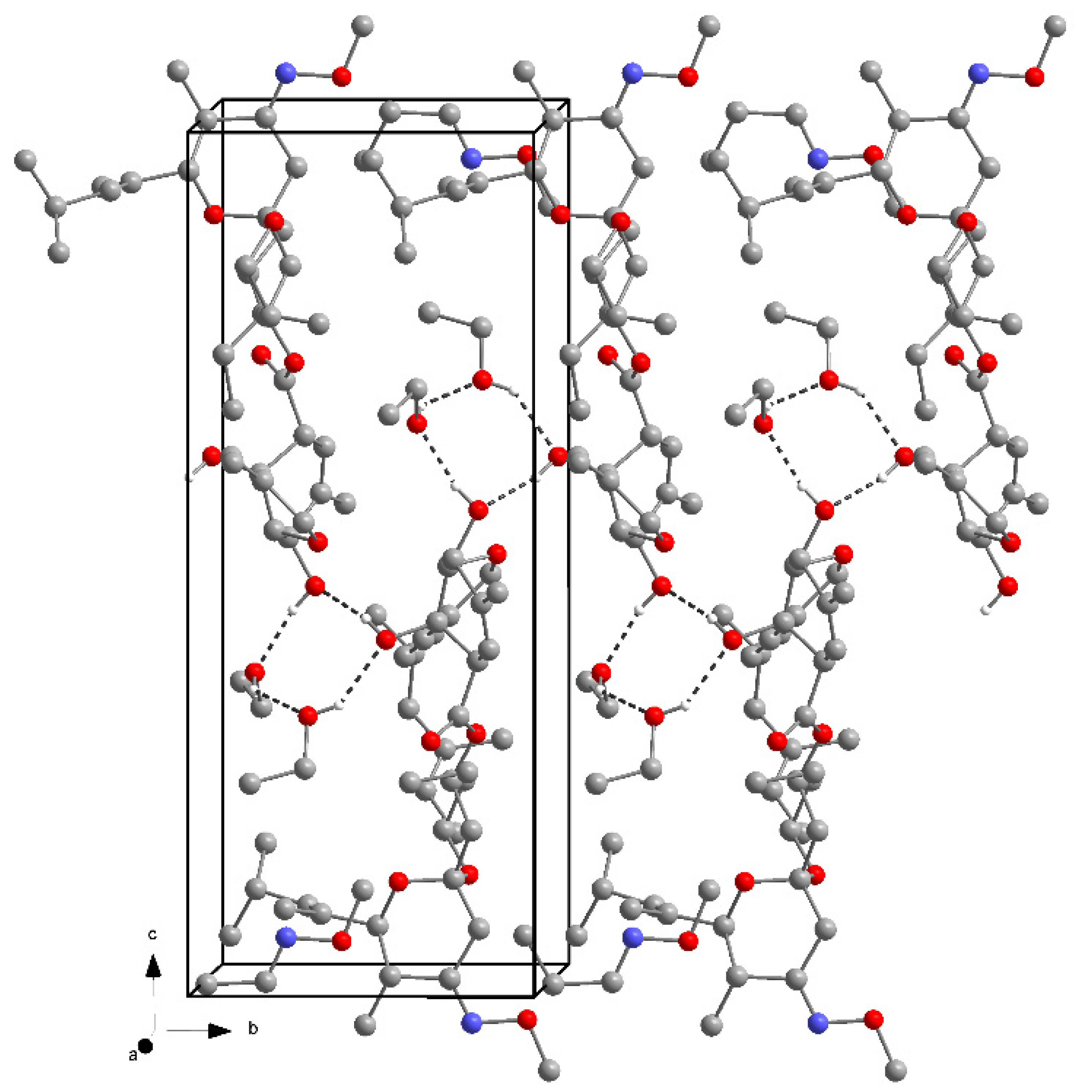
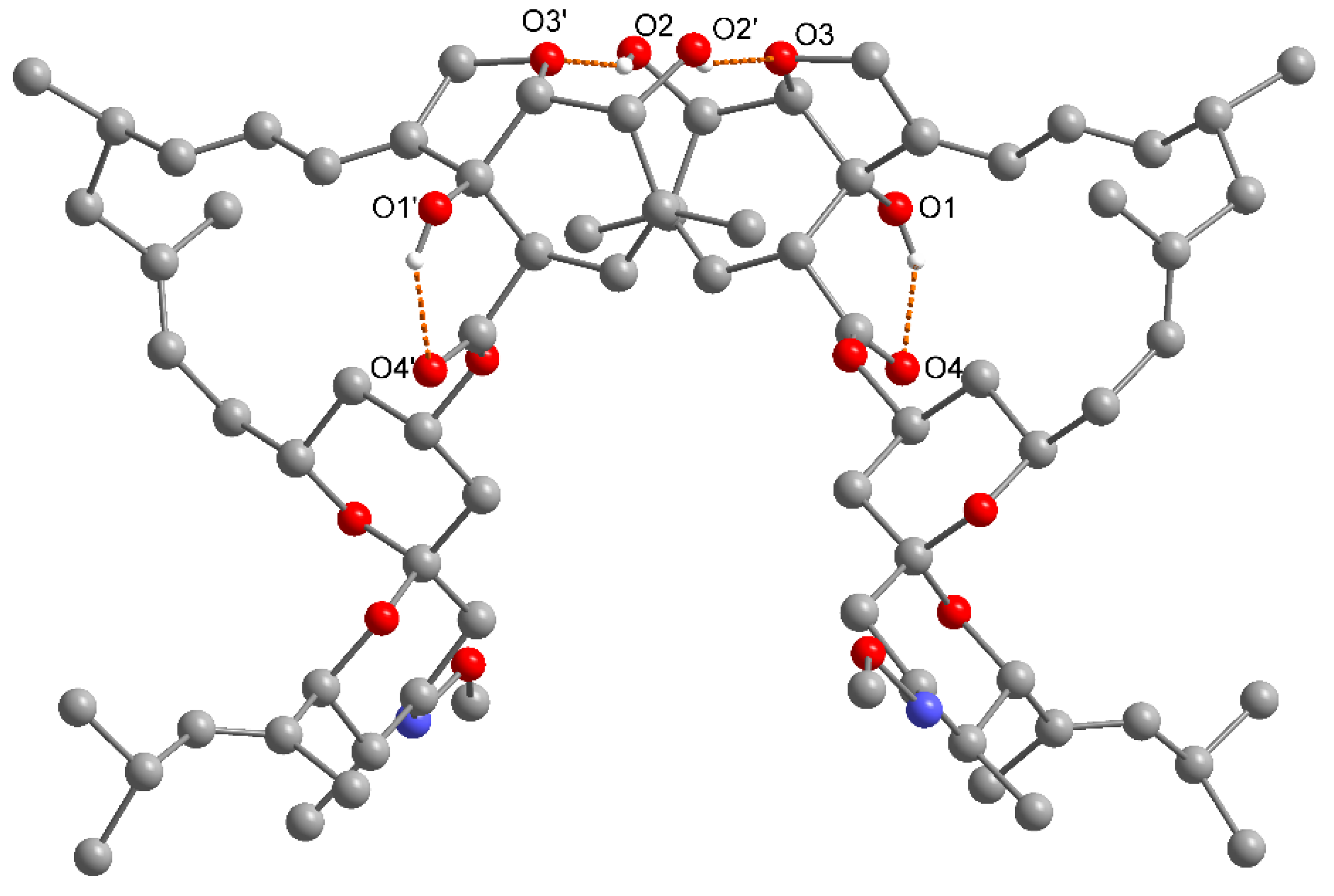
2.2. Ethanol, 2-Propanol, Methanol and 1-Butanol Solvates
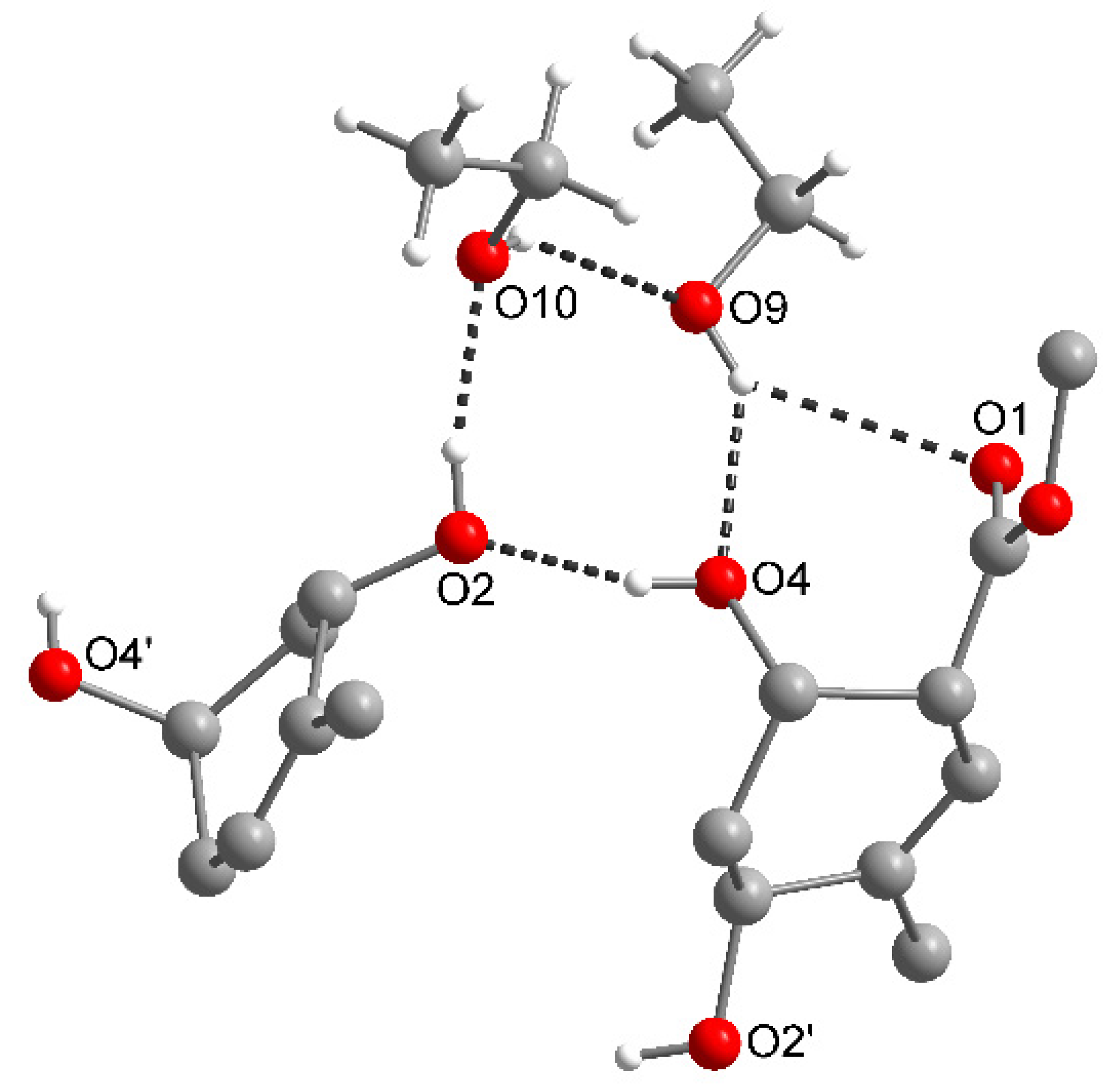
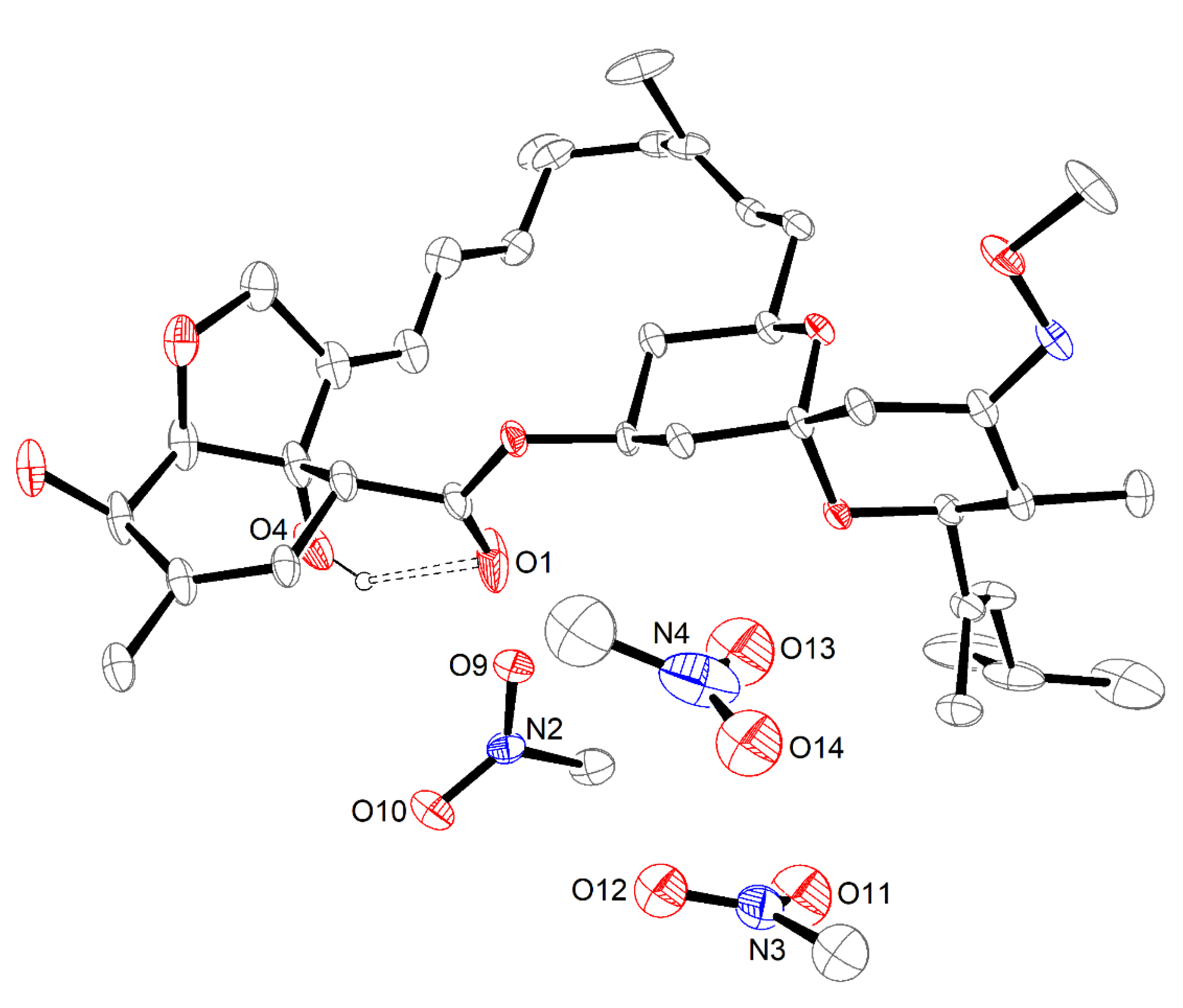
2.3. The Nitromethane Solvatomorph, Moxi·1.5MeNO2
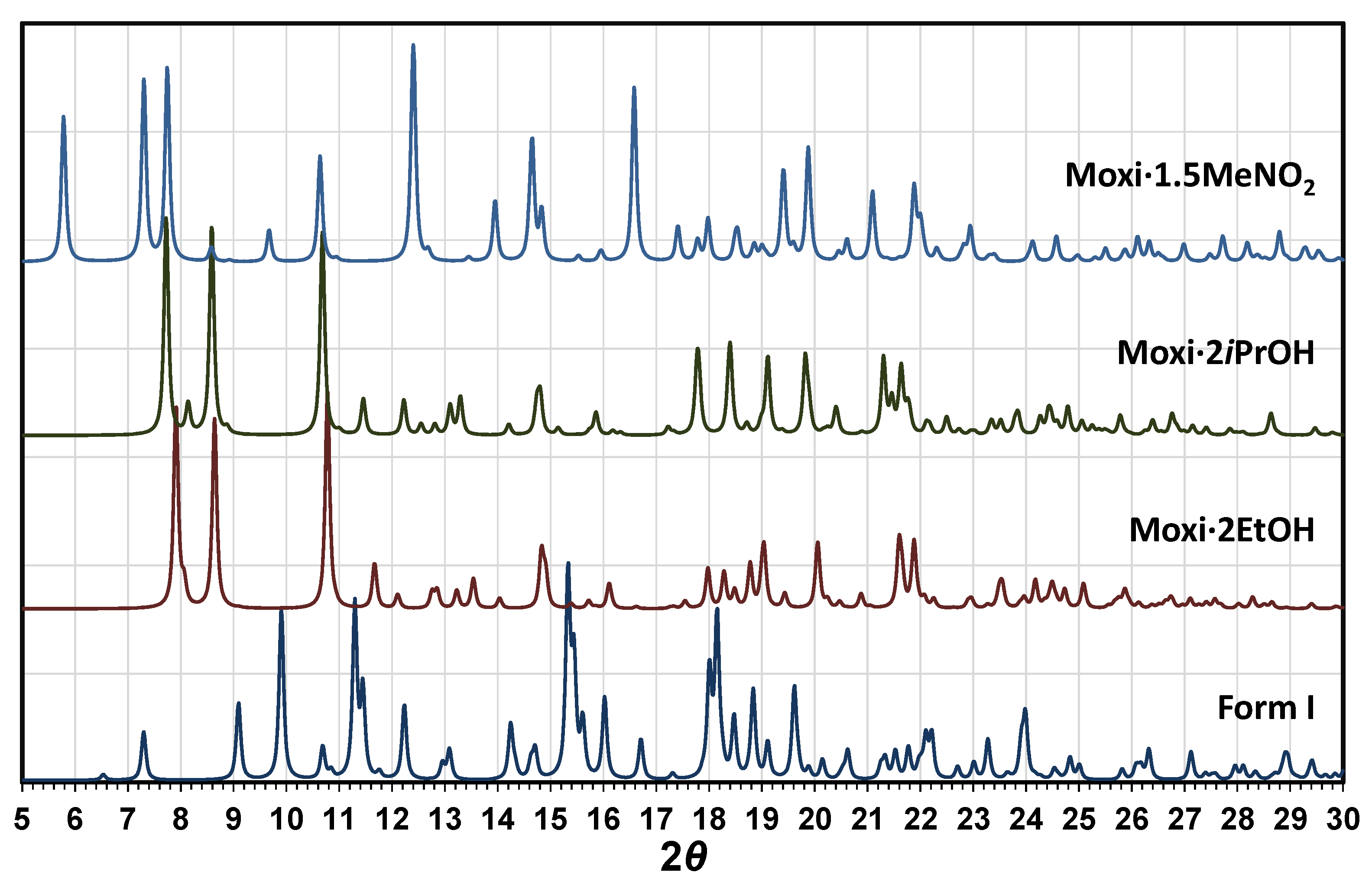
2.4. Comparative Analysis of Moxidectin Crystal Forms
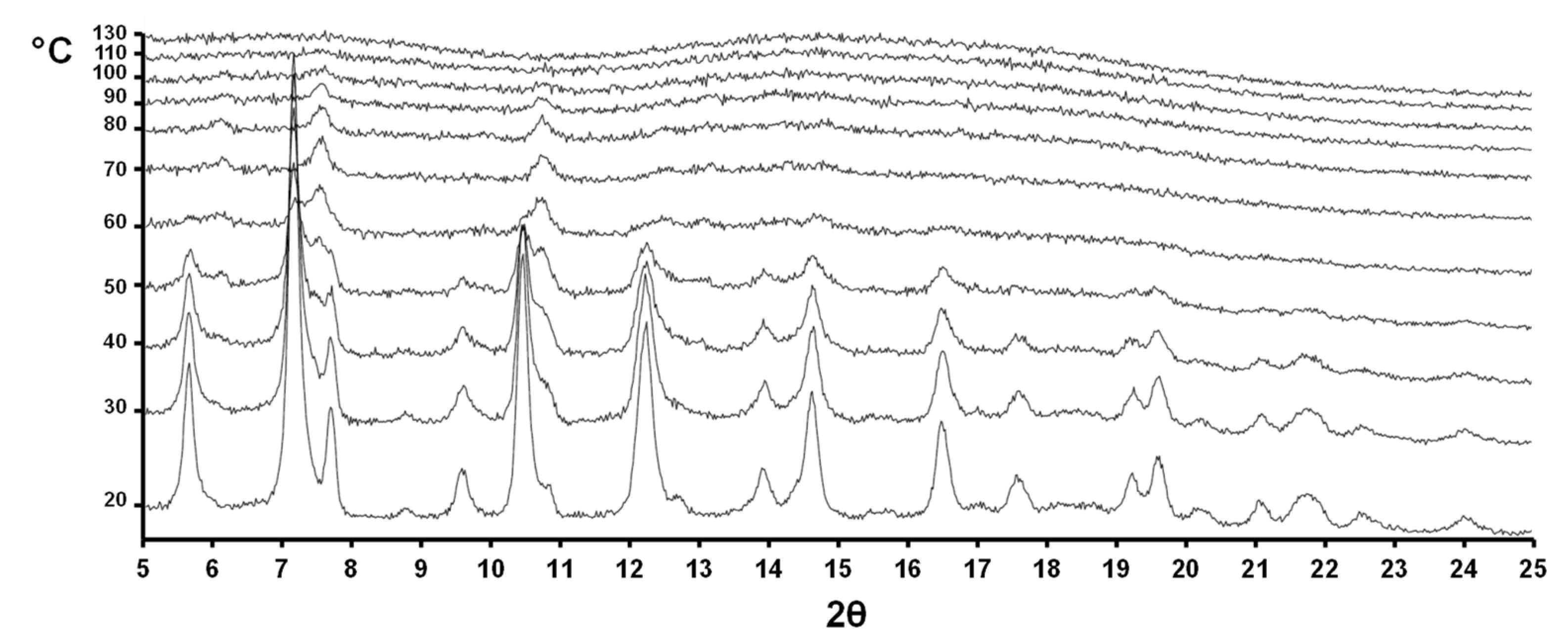
2.5. Variable-Temperature Powder X-ray Diffraction (VT-XRPD) Analysis
3. Experimental Section
3.1. Materials and Methods—Crystallization
3.2. Single Crystal X-ray Diffraction
3.3. Variable-Temperature X-ray Powder Diffraction (VT-XRPD) Analysis
3.4. Quantum Chemical Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Brittain, H.G. Polymorphism in Pharmaceutical Solids; CRC Press: Boca Raton, FL, USA, 2018. [Google Scholar]
- Bernstein, J. Polymorphism in Molecular Crystals; Clarendon Press: Oxford, UK, 2007. [Google Scholar]
- Hilfiker, R.; von Raumer, M. Polymorphism in the Pharmaceutical Industry. In Solid Form and Drug Development; John Wiley & Sons Incorporated: Newark, NJ, USA, 2018. [Google Scholar]
- Bauer, J.; Spanton, S.; Henry, R.; Quick, J.; Dziki, W.; Porter, W.; Morris, J. Ritonavir: An extraordinary example of conformational polymorphism. Pharm. Res. 2001, 18, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Price, S.L. The computational prediction of pharmaceutical crystal structures and polymorphism. Adv. Drug Deliv. Rev. 2004, 56, 301–319. [Google Scholar] [CrossRef]
- Cobb, R.; Boeckh, A. Moxidectin: A review of chemistry, pharmacokinetics and use in horses. Parasites Vectors 2009, 2, S5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prichard, R.; Ménez, C.; Lespine, A. Moxidectin and the avermectins: Consanguinity but not identity. Int. J. Parasitol. Drugs Drug Resist. 2012, 2, 134–153. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.J.; Tranquilli, W.J.; Hutchens, D.E. Safety of moxidectin in avermectin-sensitive collies. Am. J. Vet. Res. 2000, 61, 482–483. [Google Scholar] [CrossRef] [Green Version]
- El-Saber Batiha, G.; Alqahtani, A.; Ilesanmi, O.B.; Saati, A.A.; El-Mleeh, A.; Hetta, H.F.; Magdy Beshbishy, A. Avermectin Derivatives, Pharmacokinetics, Therapeutic and Toxic Dosages, Mechanism of Action, and Their Biological Effects. Pharmaceuticals 2020, 13, 196. [Google Scholar] [CrossRef]
- Boussinesq, M. A new powerful drug to combat river blindness. Lancet 2018, 392, 1170–1172. [Google Scholar] [CrossRef] [Green Version]
- Hood, J.D.; Banks, R.M.; Brewer, M.D.; Fish, J.P.; Manger, B.R.; Poulton, M.E. A novel series of milbemycin antibiotics from Streptomyces strain E225. I. Discovery, fermentation and anthelmintic activity. J. Antibiot. 1989, 42, 1593–1598. [Google Scholar] [CrossRef] [Green Version]
- Beddall, N.E.; Howes, P.D.; Ramsay, M.V.J.; Roberts, S.M.; Slawin, A.M.Z.; Sutherland, D.R.; Tiley, E.P.; Williams, D.J. Chemical transformations of S541 factors (A)-(D): Preparation and reactions of the 23-ketones. Tetrahedron Lett. 1988, 29, 2595–2598. [Google Scholar] [CrossRef]
- Cady, S.M.; Ma, B.; Chapman, R.C.; Yang, C.; Jain, U. Long-Acting Injectable Moxidectin Formulations and Novel Moxidectin Crystal Forms. Patent Number WO2013082373-A1, 6 June 2013. [Google Scholar]
- Barbero, M.; Mossotti, M.; Sironi, A.; Giovenzana, G.B.; Colombo, V. Crystal structure of pirfenidone (5-methyl-1-phenyl-1H-pyridin-2-one): An active pharmaceutical ingredient (API). Acta Crystallogr. Sect. E Crystallogr. Commun. 2019, 75, 984–986. [Google Scholar] [CrossRef] [PubMed]
- Colombo, V.; Masciocchi, N.; Palmisano, G. Crystal chemistry of the antibiotic doripenem. J. Pharm. Sci. 2014, 103, 3641–3647. [Google Scholar] [CrossRef]
- Tasso, B.; Pirisino, G.; Novelli, F.; Garzon, D.; Fruttero, R.; Sparatore, F.; Colombo, V.; Sironi, A. On the self-condensation of aminoguanidine leading to 1,1,4,10,10-pentaamino-2,3,5,6,8,9-hexaazadeca-1,3,5,7,9-pentaene (structure elucidation through X-ray powder diffraction). Tetrahedron 2014, 70, 8056–8061. [Google Scholar] [CrossRef]
- Shi, Z.; Hao, L.; Zhang, M.; Dang, L.; Wei, H. Gel formation and transformation of Moxidectin during the anti-solvent crystallization. J. Cryst. Growth 2017, 469, 8–12. [Google Scholar] [CrossRef]
- Shi, Z.; Wang, Z.; Zhang, T.; Dang, L.; Wei, H. The in situ monitoring of the transformation of moxidectin ethanol solvate to form I in an ethanol–water mixture. RSC Adv. 2015, 5, 98050–98056. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Bizdena, E.; Belyakov, S.; Jure, M.; Grinsteine, I.; Kumpiņš, V.; Turks, M. X-ray structure analysis of a solid solution of milbemycins A3 and A4. Nat. Prod. Res. 2013, 27, 1936–1939. [Google Scholar] [CrossRef]
- Terruzzi, S.; Bellomi, S.; Marras, G.; Barreca, G.; Ventimiglia, G.; Cervellino, A.; Masciocchi, N. Disclosing the Rich Crystal Chemistry of Lesinurad by ab Initio Laboratory X-ray Powder Diffraction Methods. Cryst. Growth Des. 2018, 18, 6863–6872. [Google Scholar] [CrossRef]
- Aprea, A.; Colombo, V.; Galli, S.; Masciocchi, N.; Maspero, A.; Palmisano, G. Thiazolo[5,4-d]thiazole-2,5-dicarboxylic acid, C6H2N2O4S2, and its coordination polymers. Solid State Sci. 2010, 12, 795–802. [Google Scholar] [CrossRef] [Green Version]
- Chierotti, M.R.; Gobetto, R.; Nervi, C.; Bacchi, A.; Pelagatti, P.; Colombo, V.; Sironi, A. Probing hydrogen bond networks in half-sandwich Ru(II) building blocks by a combined 1H DQ CRAMPS solid-state NMR, XRPD, and DFT approach. Inorg. Chem. 2014, 53, 139–146. [Google Scholar] [CrossRef]
- Awasthi, A.; Razzak, M.; Al-Kassas, R.; Harvey, J.; Garg, S. Analytical profile of moxidectin. Profiles Drug Subst. Excip. Relat. Methodol. 2013, 38, 315–366. [Google Scholar] [PubMed]
- Agilent, CrysAlis PRO; Agilent Technologies Ltd.: Yarnton, Oxfordshire, UK, 2019.
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Crystallogr. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—new features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Brandenburg, K.; Putz, H. DIAMOND; Crystal Impact GbR: Bonn, Germany, 1999. [Google Scholar]
- Bruker AXS Inc. EVA; Bruker AXS Inc.: Madison, WI, USA, 2012. [Google Scholar]
- Dovesi, R.; Erba, A.; Orlando, R.; Zicovich-Wilson, C.M.; Civalleri, B.; Maschio, L.; Rérat, M.; Casassa, S.; Baima, J.; Salustro, S.; et al. Quantum-mechanical condensed matter simulations with CRYSTAL. WIREs Comput. Mol. Sci. 2018, 8, e1360. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.V.R. Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21+G basis set for first-row elements, Li-F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self—Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian—Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystal Form | First Report | Structure Report |
|---|---|---|
| Form I (Moxi, unsolvated) | Ref. [12] | Ref. [12], CSD: GETBOW |
| Amorphous (Moxi, unsolvated) | Ref. [13] | – |
| Moxi·2EtOH | Ref. [13] | this work |
| Mox·2iPrOH | Ref. [13] | this work |
| Moxi·2nBuOH | Ref. [13] (reported as Moxi·1.5nBuOH) | – |
| Moxi·1.5MeNO2 | this work | this work |
| Compound | Moxi·2EtOH | Moxi·2iPrOH | Moxi·1.5MeNO2 |
|---|---|---|---|
| Formula | C40H65NO10 | C43H69NO10 | C38.23H56.70N2.23O10.47 |
| FW | 731.94 | 759.99 | 715.05 |
| Crystal System | Monoclinic | Monoclinic | Orthorhombic |
| Space group | P21 | P21 | P21212 |
| a (Å) | 11.1913(4) | 11.4417(3) | 13.9453(5) |
| b (Å) | 8.8440(3) | 8.9507(2) | 19.8096(6) |
| c (Å) | 21.9594(8) | 21.7210(6) | 15.2680(6) |
| α (°) | 90 | 90 | 90 |
| β (°) | 93.747(3) | 92.301(3) | 90 |
| γ (°) | 90 | 90 | 90 |
| V (Å)3 | 2168.80(13) | 2222.68(10) | 4217.8(3) |
| Z | 2 | 2 | 4 |
| Temperature (K) | 120(2) | 120(2) | 100(2) |
| ρ(calcd) (g·cm–3) | 1.121 | 1.136 | 1.126 |
| μ (mm–1) | 0.079 | 0.079 | 0.081 |
| F(000) | 796 | 828 | 1542 |
| Crystal size (mm3) | 0.150 × 0.120 × 0.100 | 0.200 × 0.100 × 0.100 | 0.120 × 0.120 × 0.030 |
| θ range (°) | 1.824 to 31.571 | 1.781 to 31.719 | 1.684 to 26.372 |
| Limiting indices | −16 ≤ h ≤ 16, −12 ≤ k ≤ 13, −32 ≤ l ≤ 32 | −16 ≤ h ≤ 16, −13 ≤ k ≤ 13, −31 ≤ l ≤ 30 | −17 ≤ h ≤ 17, −24 ≤ k ≤ 24, −19 ≤ l ≤ 19 |
| Reflns collected/unique a | 25503/13011 [Rint = 0.0249] | 26404/13370 [Rint = 0.0180] | 36172/8640 [Rint = 0.0244] |
| Data/restraints/param | 13011/9/485 | 13370/9/538 | 8638/2/508 |
| Completeness to θ = 25.242° (%) | 100.0 | 100.0 | 100.00 |
| Max. and min. transmission | 1.00000 and 0.96333 | 1.00000 and 0.90598 | 1.00000 and 0.61508 |
| Final R indices [I > 2σ(I)] b | R1 = 0.0525, wR2 = 0.1160 | R1 = 0.0448, wR2 = 0.1112 | R1 = 0.0536, wR2 = 0.1326 |
| R indices (all data) | R1 = 0.0720, wR2 = 0.1254 | R1 = 0.0549, wR2 = 0.1169 | R1 = 0.0584, wR2 = 0.1357 |
| GooF c on F2 | 1.013 | 1.021 | 1.100 |
| Largest diff peak and hole (e·Å−3) | 0.652 and −0.426 | 0.355 and −0.344 | 0.601 and −0.435 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grell, T.; Barbero, M.; Pattarino, F.; Giovenzana, G.B.; Colombo, V. Solvatomorphism of Moxidectin. Molecules 2021, 26, 4869. https://doi.org/10.3390/molecules26164869
Grell T, Barbero M, Pattarino F, Giovenzana GB, Colombo V. Solvatomorphism of Moxidectin. Molecules. 2021; 26(16):4869. https://doi.org/10.3390/molecules26164869
Chicago/Turabian StyleGrell, Toni, Mauro Barbero, Franco Pattarino, Giovanni Battista Giovenzana, and Valentina Colombo. 2021. "Solvatomorphism of Moxidectin" Molecules 26, no. 16: 4869. https://doi.org/10.3390/molecules26164869
APA StyleGrell, T., Barbero, M., Pattarino, F., Giovenzana, G. B., & Colombo, V. (2021). Solvatomorphism of Moxidectin. Molecules, 26(16), 4869. https://doi.org/10.3390/molecules26164869