
Interpretation of Ligand-Based Activity Cliff Prediction Models Using the Matched Molecular Pair Kernel


Abstract
:1. Introduction
2. Result and Discussion
2.1. Performance of the AC Prediction Models
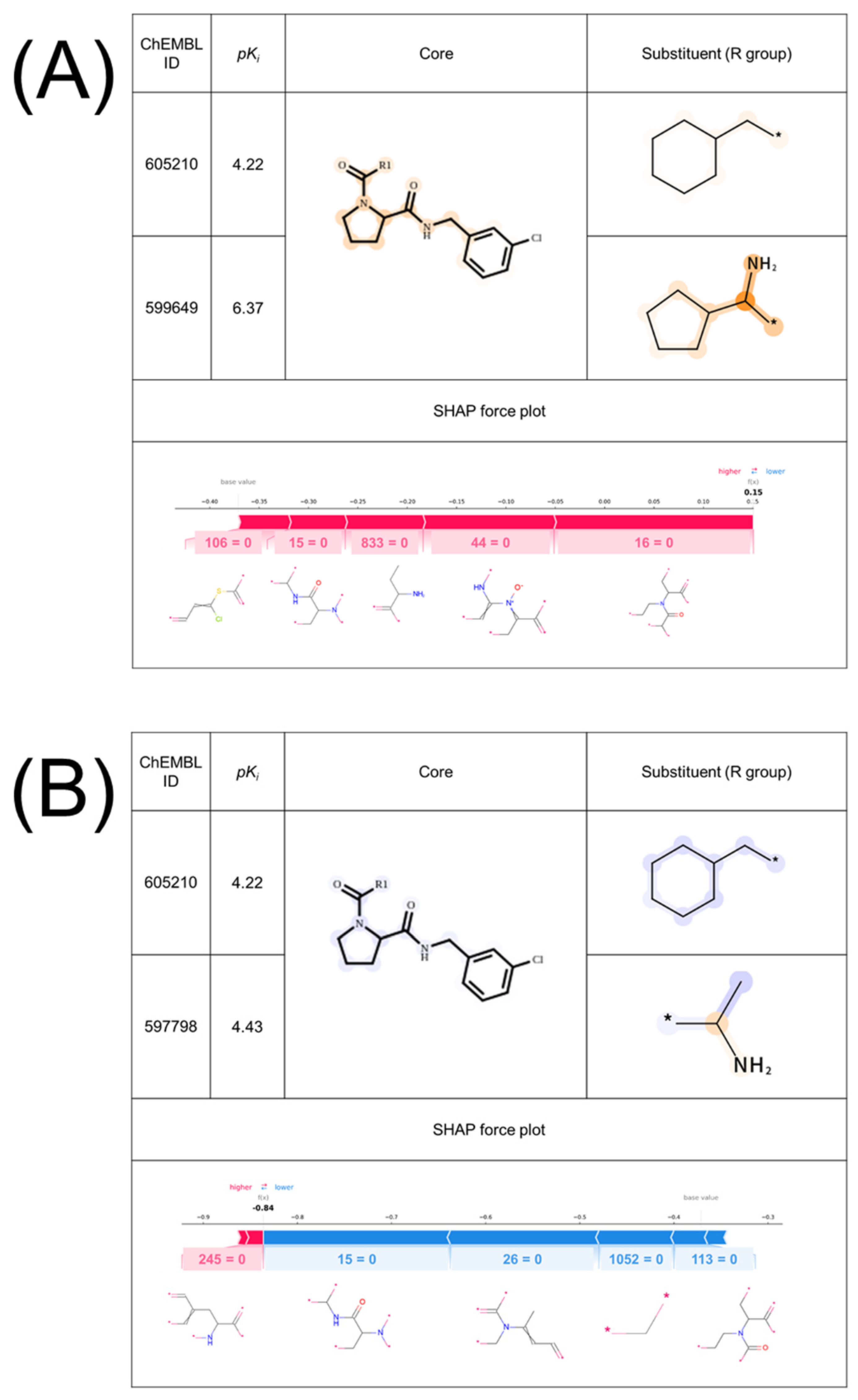
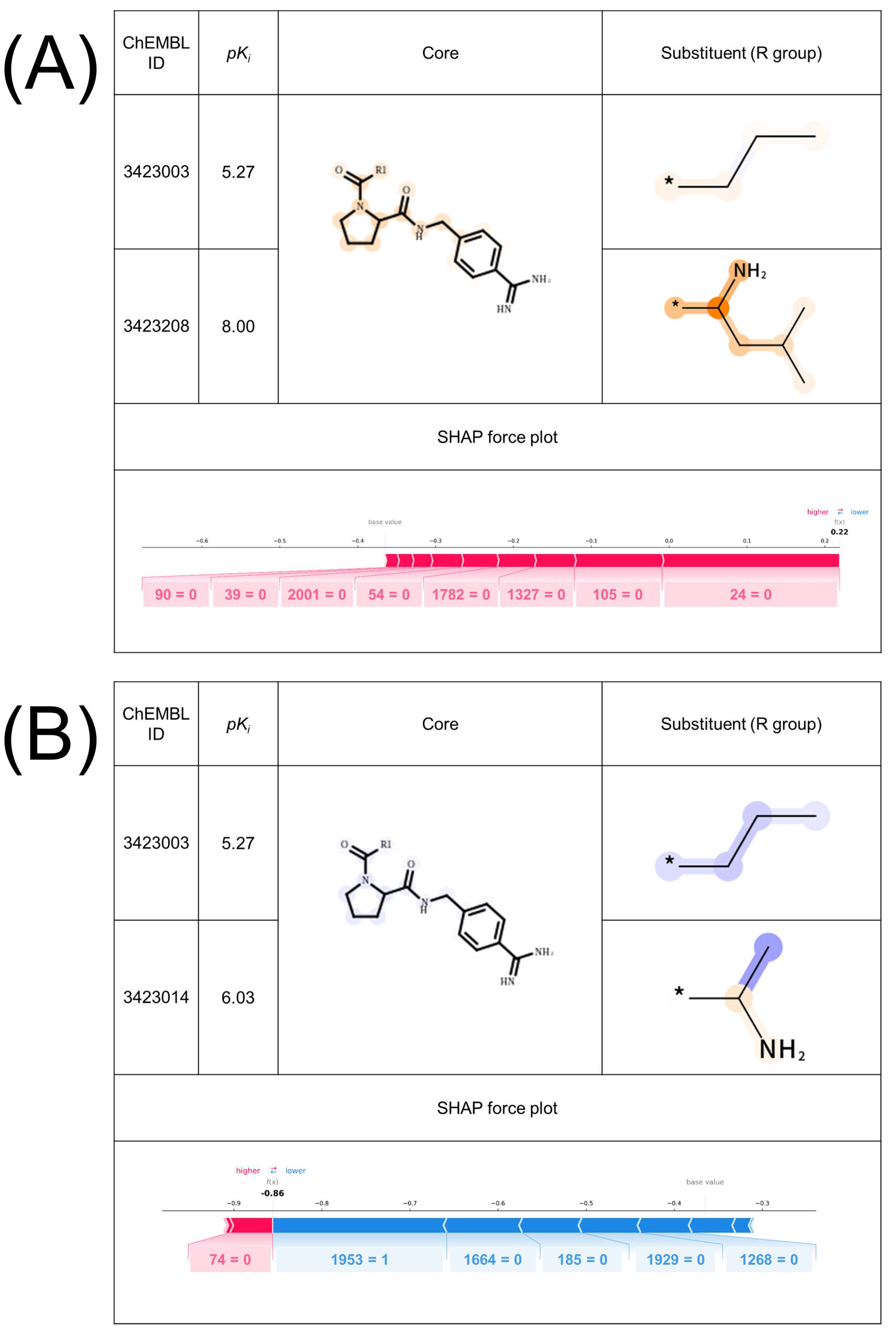
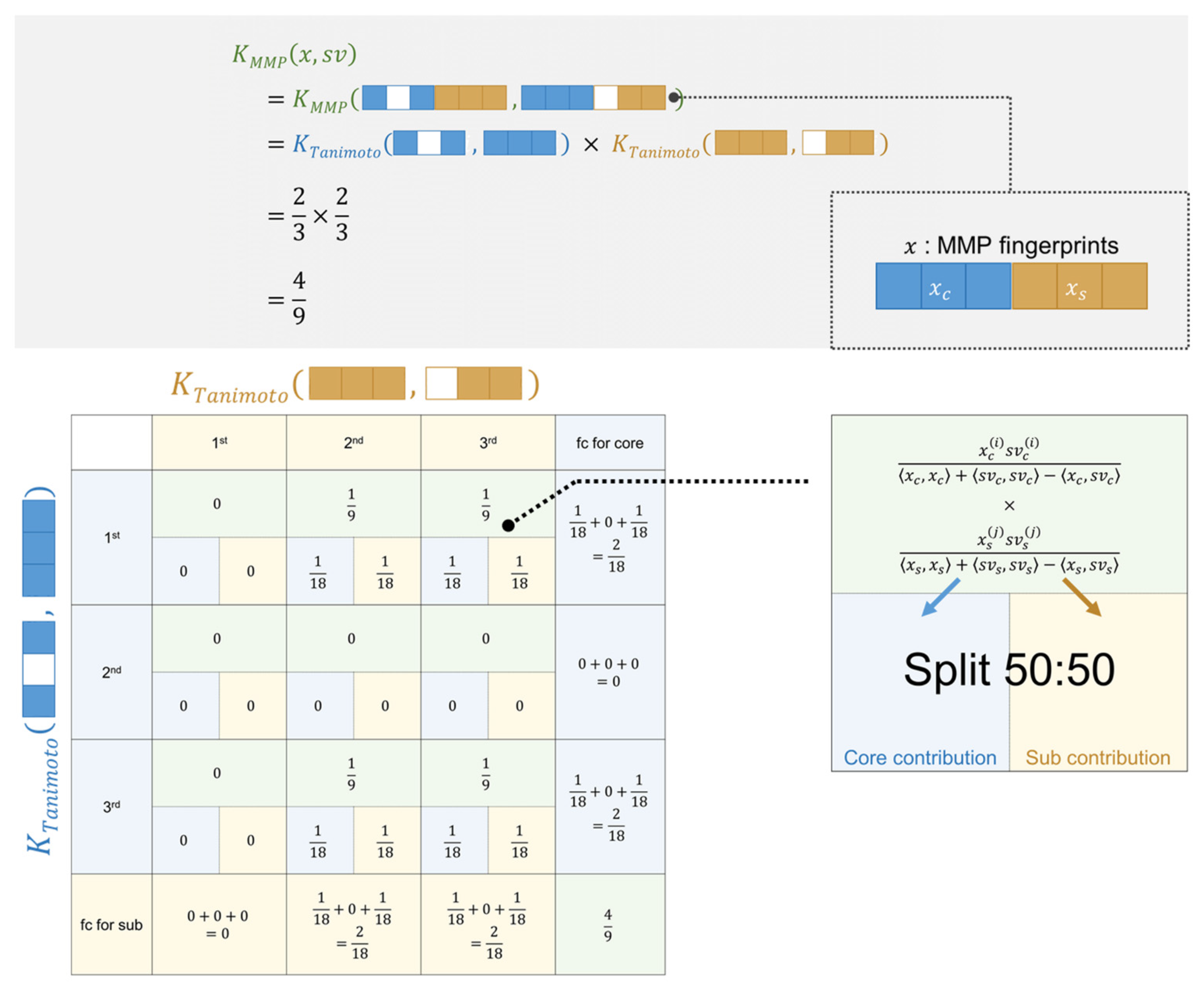
2.2. Interpretability of the Model
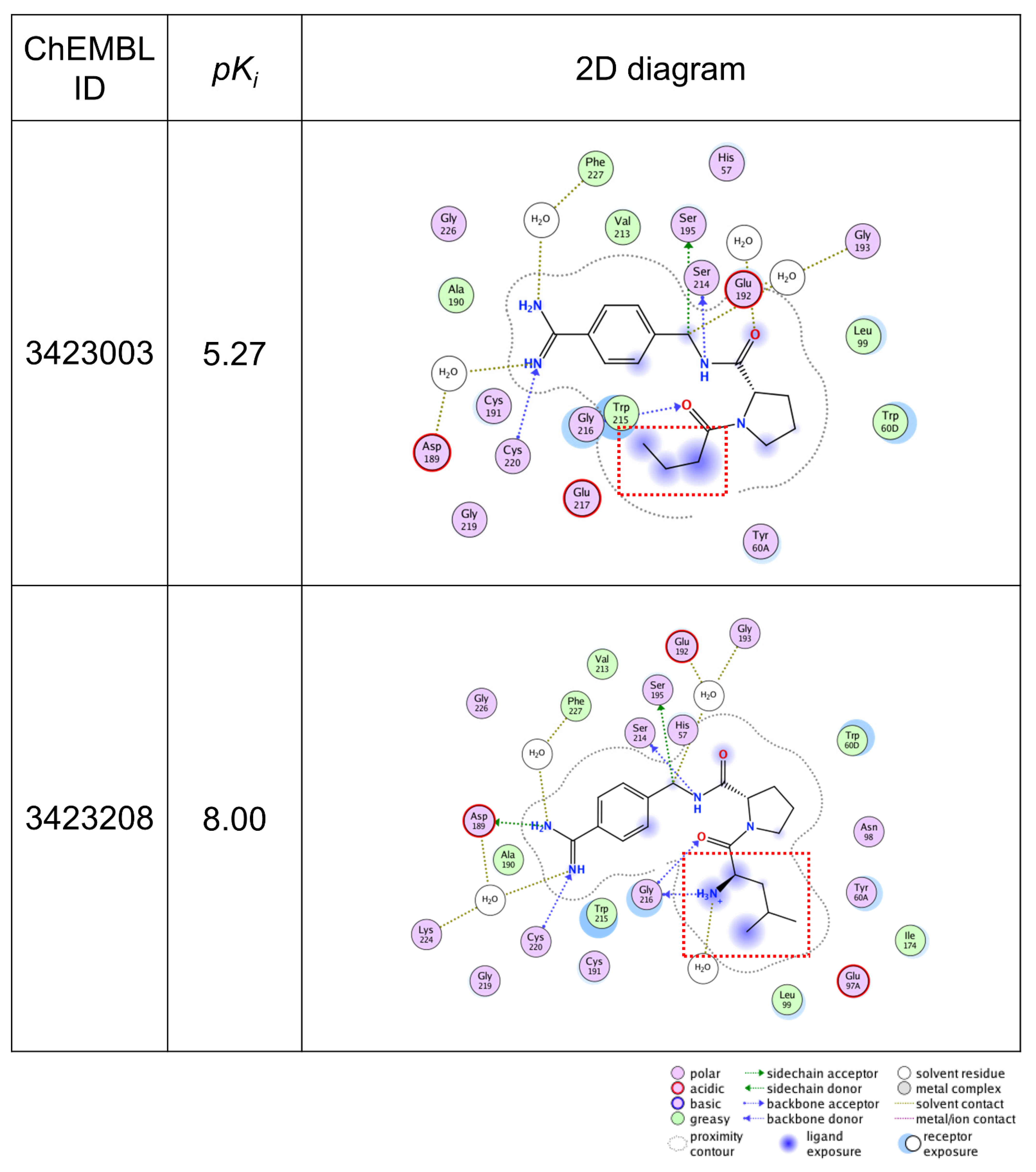
2.3. Validation of Important Features with X-ray Co-Crystal
2.4. Limitations of SHAP for AC Prediction Model Interpretability
3. Materials and Methods
3.1. Data Sets
3.2. MMP Fingerprints
3.3. Construction and Evaluation of the AC Prediction Model
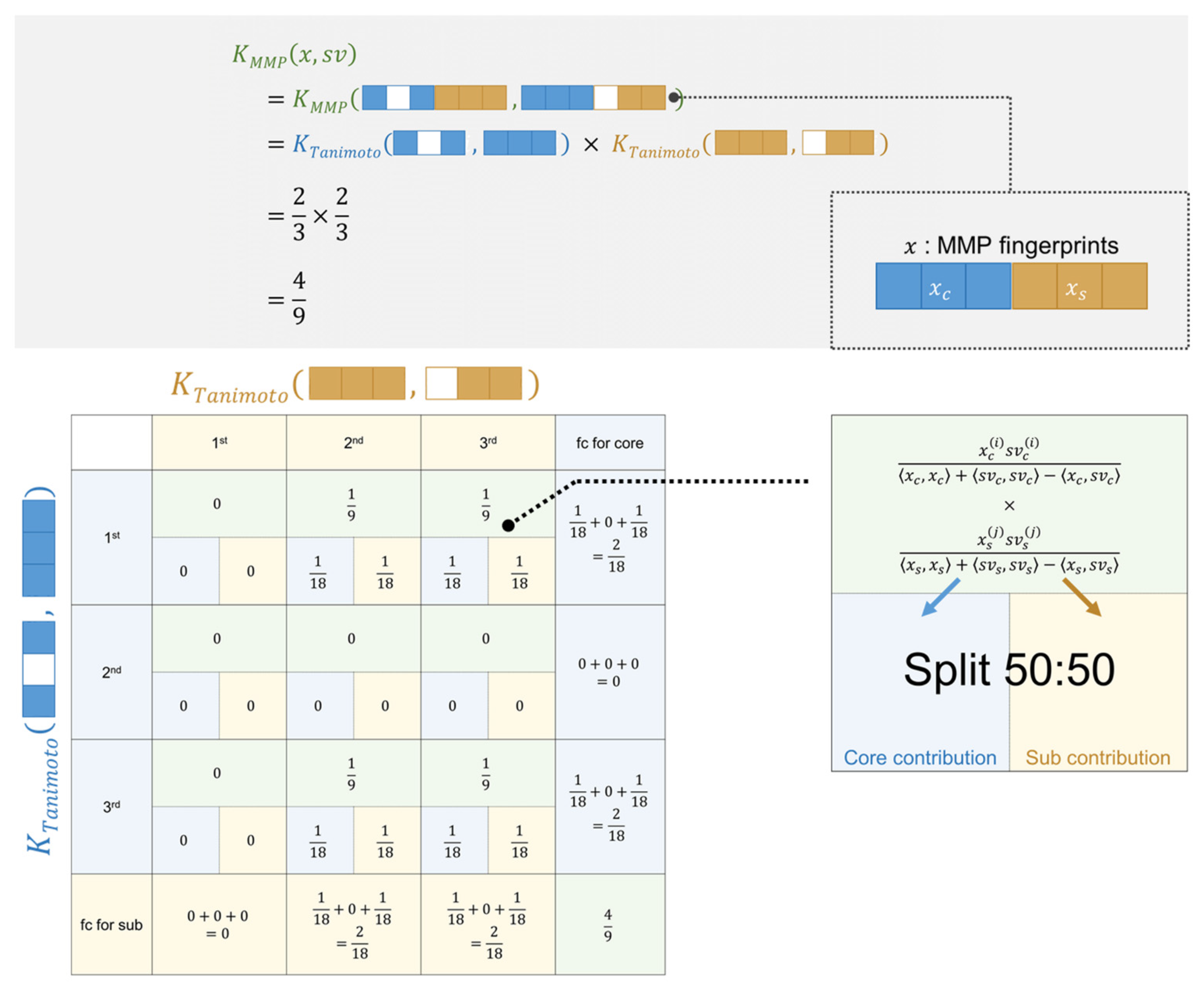
3.4. Feature Contributions for the Tanimoto Kernel in SVM
3.5. Feature Contributions for the MMP Kernel
3.6. SHAP Theory
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Stumpfe, D.; Bajorath, J. Exploring Activity Cliffs in Medicinal Chemistry. J. Med. Chem. 2012, 55, 2932–2942. [Google Scholar] [CrossRef] [PubMed]
- Tyrchan, C.; Evertsson, E. Matched Molecular Pair Analysis in Short: Algorithms, Applications and Limitations. Comput. Struct. Biotechnol. J. 2017, 15, 86–90. [Google Scholar] [CrossRef]
- Hu, X.; Hu, Y.; Vogt, M.; Stumpfe, D.; Bajorath, J. MMP-Cliffs: Systematic Identification of Activity Cliffs on the Basis of Matched Molecular Pairs. J. Chem. Inf. Model 2012, 52, 1138–1145. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Benito, L.; Casajuana-Martin, N.; Jiménez-Rosés, M.; van Vlijmen, H.; Tresadern, G. Predicting Activity Cliffs with Free-Energy Perturbation. J. Chem. Theory Comput. 2019, 15, 1884–1895. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, J.; Vogt, M.; Bajorath, J. Prediction of Activity Cliffs on the Basis of Images Using Convolutional Neural Networks. J. Comput. Aid. Mol. Des. 2021, 1–8. [Google Scholar] [CrossRef]
- Horvath, D.; Marcou, G.; Varnek, A.; Kayastha, S.; León, A.d.l.V.d.; Bajorath, J. Prediction of Activity Cliffs Using Condensed Graphs of Reaction Representations, Descriptor Recombination, Support Vector Machine Classification, and Support Vector Regression. J. Chem. Inf. Model. 2016, 56, 1631–1640. [Google Scholar] [CrossRef]
- Heikamp, K.; Hu, X.; Yan, A.; Bajorath, J. Prediction of Activity Cliffs Using Support Vector Machines. J. Chem. Inf. Model. 2012, 52, 2354–2365. [Google Scholar] [CrossRef]
- Tamura, S.; Miyao, T.; Funatsu, K. Ligand-based Activity Cliff Prediction Models with Applicability Domain. Mol. Inform. 2020, 39, 2000103. [Google Scholar] [CrossRef]
- Maggiora, G.M. On Outliers and Activity CliffsWhy QSAR Often Disappoints. J. Chem. Inf. Model 2006, 46, 1535. [Google Scholar] [CrossRef]
- Vapnik, V.N. The Nature of Statistical Learning Theory, 2nd ed.; Springer: New York, NY, USA, 2000. [Google Scholar] [CrossRef]
- Ralaivola, L.; Swamidass, S.J.; Saigo, H.; Baldi, P. Graph Kernels for Chemical Informatics. Neural Netw. 2005, 18, 1093–1110. [Google Scholar] [CrossRef]
- Tamura, S.; Miyao, T.; Funatsu, K. Development of R-Group Fingerprints Based on the Local Landscape from an Attachment Point of a Molecular Structure. J. Chem. Inf. Model. 2019, 59, 2656–2663. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, S.M.; Lee, S.-I. A Unified Approach to Interpreting Model Predictions. In Proceedings of the 31st International Conference on Neural Information Processing Systems (NIPS ’17), California, CA, USA, 4 December 2017; pp. 4768–4777. [Google Scholar]
- Krishnapuram, B.; Shah, M.; Smola, A.; Aggarwal, C.; Shen, D.; Rastogi, R.; Ribeiro, M.T.; Singh, S.; Guestrin, C. “Why Should I Trust You?”: Explaining the Predictions of Any Classifier. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining (KDD ’16), California, CA, USA, 13 August 2016; pp. 1135–1144. [Google Scholar] [CrossRef]
- Leidner, F.; Yilmaz, N.K.; Schiffer, C.A. Target-Specific Prediction of Ligand Affinity with Structure–Based Interaction Fingerprints. J. Chem. Inf. Model. 2019, 59, 3679–3691. [Google Scholar] [CrossRef]
- Ding, Y.; Chen, M.; Guo, C.; Zhang, P.; Wang, J. Molecular Fingerprint-Based Machine Learning Assisted QSAR Model Development for Prediction of Ionic Liquid Properties. J. Mol. Liq. 2021, 326, 115212. [Google Scholar] [CrossRef]
- Rodríguez-Pérez, R.; Bajorath, J. Interpretation of Machine Learning Models Using Shapley Values: Application to Compound Potency and Multi-Target Activity Predictions. J. Comput. Aid. Mol. Des. 2020, 34, 1013–1026. [Google Scholar] [CrossRef]
- Rodríguez-Pérez, R.; Bajorath, J. Interpretation of Compound Activity Predictions from Complex Machine Learning Models Using Local Approximations and Shapley Values. J. Med. Chem. 2019, 63, 8761–8777. [Google Scholar] [CrossRef] [PubMed]
- Balfer, J.; Bajorath, J. Visualization and Interpretation of Support Vector Machine Activity Predictions. J. Chem. Inf. Model. 2015, 55, 1136–1147. [Google Scholar] [CrossRef]
- Furtmann, N.; Hu, Y.; Gütschow, M.; Bajorath, J. Identification of Interaction Hot Spots in Structures of Drug Targets on the Basis of Three-Dimensional Activity Cliff Information. Chem. Biol. Drug Des. 2015, 86, 1458–1465. [Google Scholar] [CrossRef] [PubMed]
- Baum, B.; Muley, L.; Smolinski, M.; Heine, A.; Hangauer, D.; Klebe, G. Non-Additivity of Functional Group Contributions in Protein–Ligand Binding: A Comprehensive Study by Crystallography and Isothermal Titration Calorimetry. J. Mol. Biol. 2010, 397, 1042–1054. [Google Scholar] [CrossRef]
- Mendez, D.; Gaulton, A.; Bento, A.P.; Chambers, J.; De Veij, M.; Félix, E.; Magariños, M.P.; Mosquera, J.F.; Mutowo, P.; Nowotka, M.; et al. ChEMBL: Towards Direct Deposition of Bioassay Data. Nucleic Acids Res. 2018, 47, gky1075. [Google Scholar] [CrossRef]
- Hussain, J.; Rea, C. Computationally Efficient Algorithm to Identify Matched Molecular Pairs (MMPs) in Large Data Sets. J. Chem. Inf. Model. 2010, 50, 339–348. [Google Scholar] [CrossRef]
- Wawer, M.; Bajorath, J. Local Structural Changes, Global Data Views: Graphical Substructure–Activity Relationship Trailing. J. Med. Chem. 2011, 54, 2944–2951. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Dimova, D.; Bajorath, J. Classification of Matching Molecular Series on the Basis of SAR Phenotypes and Structural Relationships. Medchemcomm 2016, 7, 237–246. [Google Scholar] [CrossRef]
- Rogers, D.; Hahn, M. Extended-Connectivity Fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ChEMBL ID | Target | Abbreviation | #CPDs | #MMPs | #AC | #MMSs | Potency (pKi) | MW | #HA | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Max | Min | Max | Min | Max | Min | |||||||
| 204 | Thrombin | thr | 221 | 839 | 311 | 29 | 10.30 | 2.81 | 693.86 | 280.75 | 50 | 19 |
| 205 | Carbonic anhydrase II | ca2 | 362 | 989 | 248 | 70 | 11.00 | 0.70 | 678.36 | 153.14 | 42 | 11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tamura, S.; Jasial, S.; Miyao, T.; Funatsu, K. Interpretation of Ligand-Based Activity Cliff Prediction Models Using the Matched Molecular Pair Kernel. Molecules 2021, 26, 4916. https://doi.org/10.3390/molecules26164916
Tamura S, Jasial S, Miyao T, Funatsu K. Interpretation of Ligand-Based Activity Cliff Prediction Models Using the Matched Molecular Pair Kernel. Molecules. 2021; 26(16):4916. https://doi.org/10.3390/molecules26164916
Chicago/Turabian StyleTamura, Shunsuke, Swarit Jasial, Tomoyuki Miyao, and Kimito Funatsu. 2021. "Interpretation of Ligand-Based Activity Cliff Prediction Models Using the Matched Molecular Pair Kernel" Molecules 26, no. 16: 4916. https://doi.org/10.3390/molecules26164916
APA StyleTamura, S., Jasial, S., Miyao, T., & Funatsu, K. (2021). Interpretation of Ligand-Based Activity Cliff Prediction Models Using the Matched Molecular Pair Kernel. Molecules, 26(16), 4916. https://doi.org/10.3390/molecules26164916