
Pharmacophore-Based Virtual Screening, Quantum Mechanics Calculations, and Molecular Dynamics Simulation Approaches Identified Potential Natural Antiviral Drug Candidates against MERS-CoV S1-NTD


 , ,
, ,  , , ,
, , ,  ,
, 
Abstract
:1. Introduction
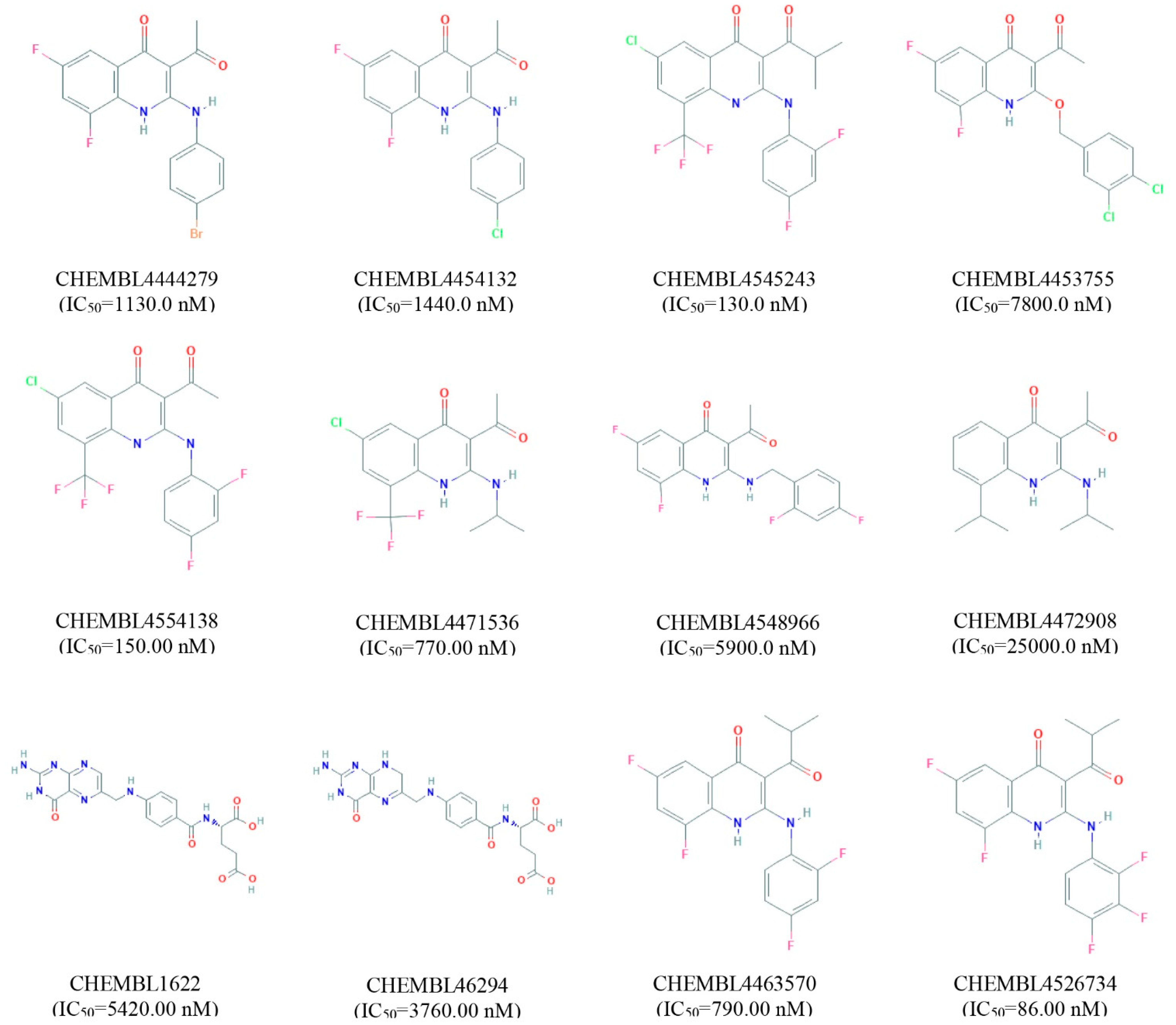
2. Results
2.1. Results of Pharmacophore Modeling
2.2. Molecule Library Preparation
2.3. Active Compounds Identification and Decoy Set Generation
2.4. Pharmacophore-Based Virtual Screening
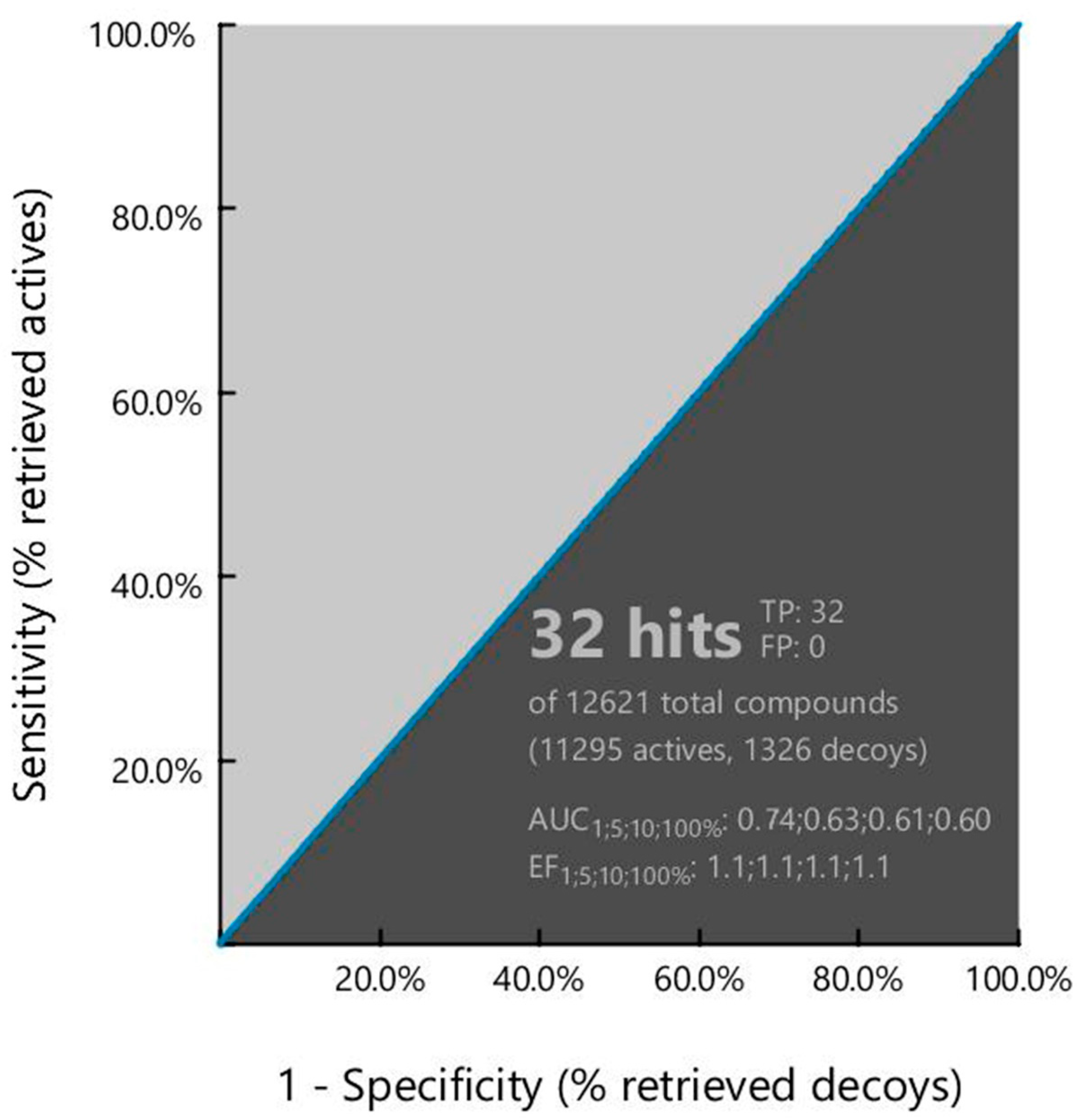
2.5. Pharmacophore Model Performance Analysis
2.6. Binding Site Identification and Receptor Grid Generation
2.7. Molecular Docking Simulation
2.8. ADME Analysis
2.9. Toxicity Test
2.10. Theoretical Calculation
Geometry Optimization
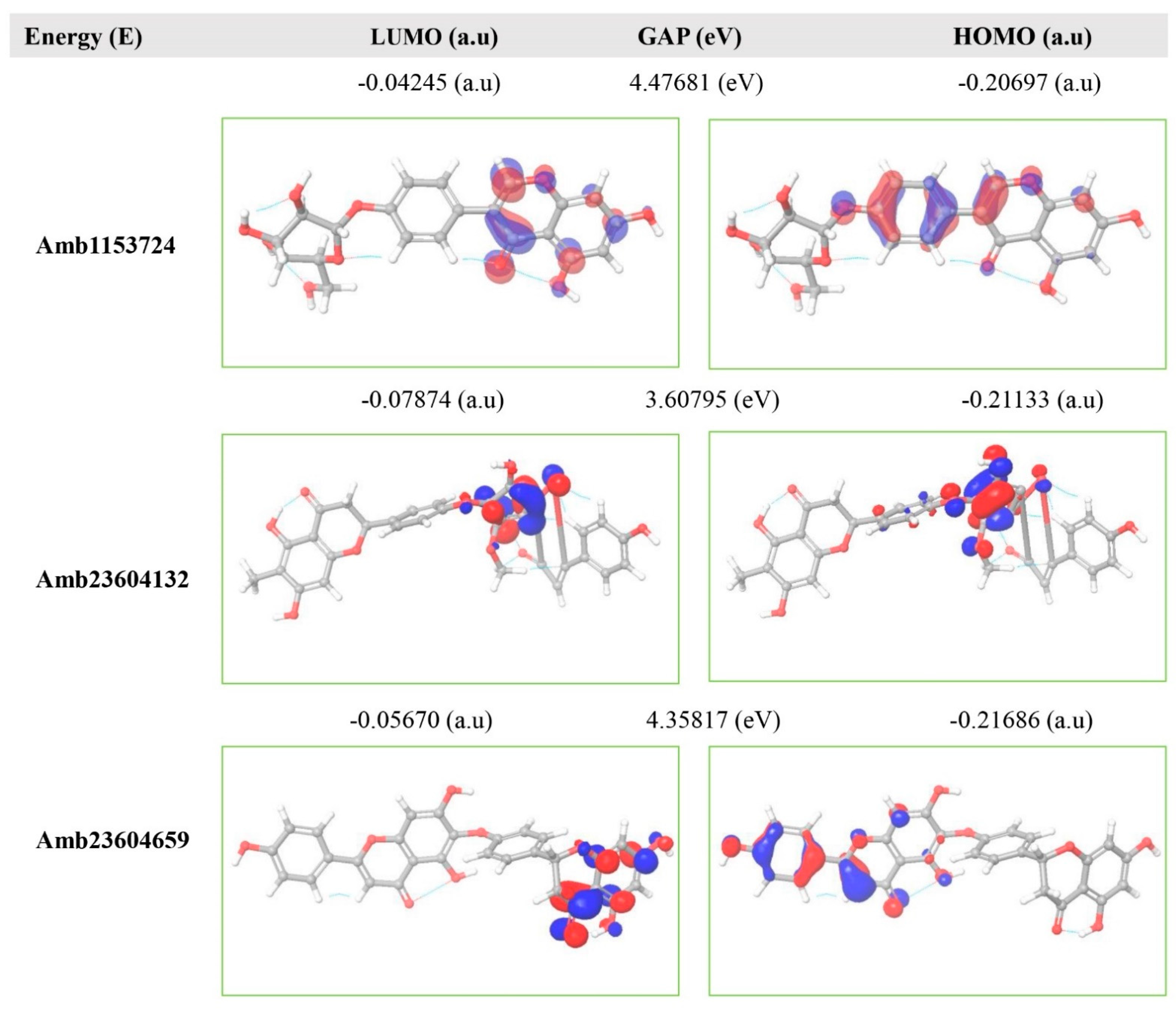
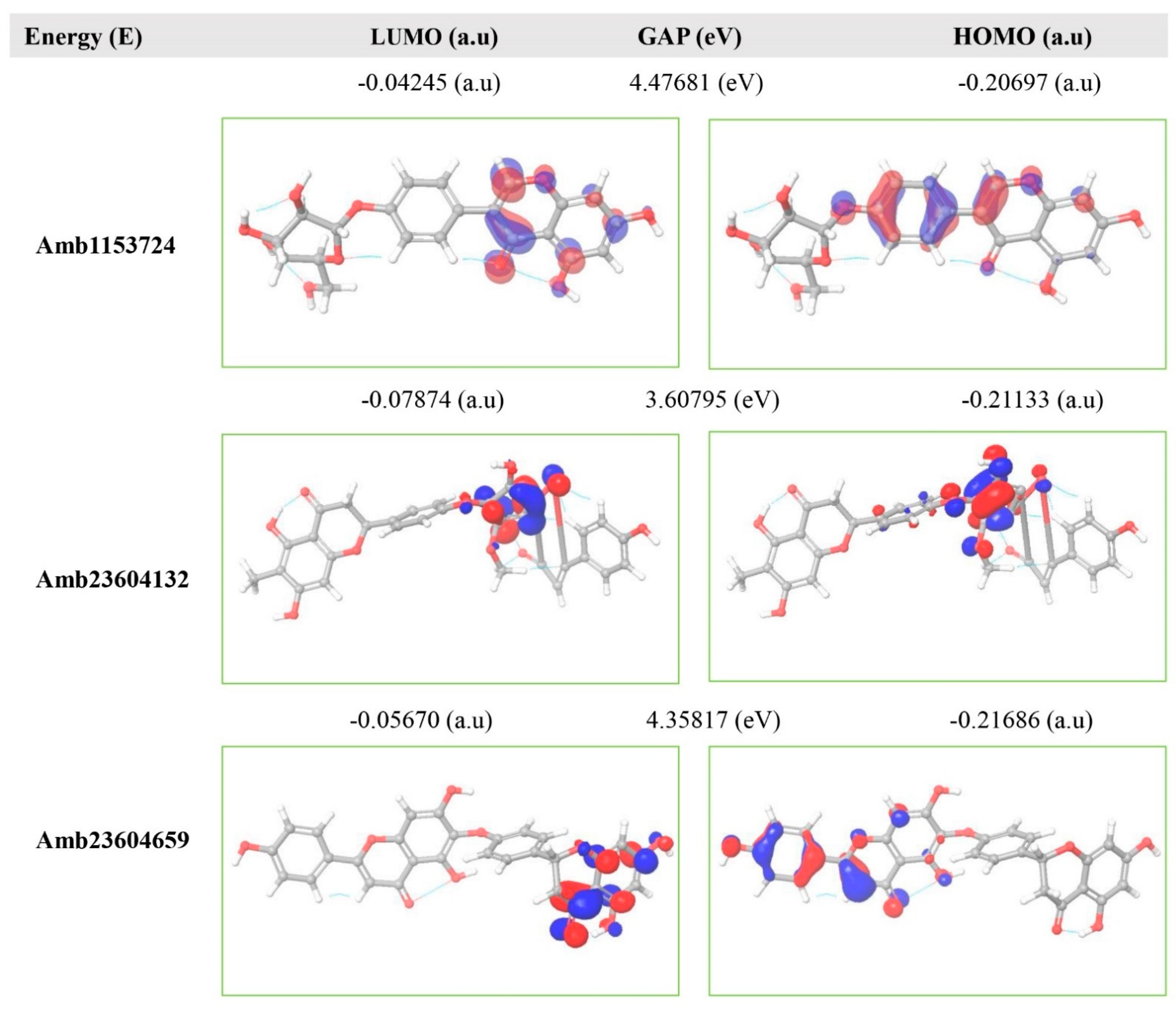
2.11. Frontier Molecular Orbital HOMO/LUMO Calculation
2.12. Re-Docking, Interaction, and Pharmacophore Analysis
2.12.1. Redocking Score
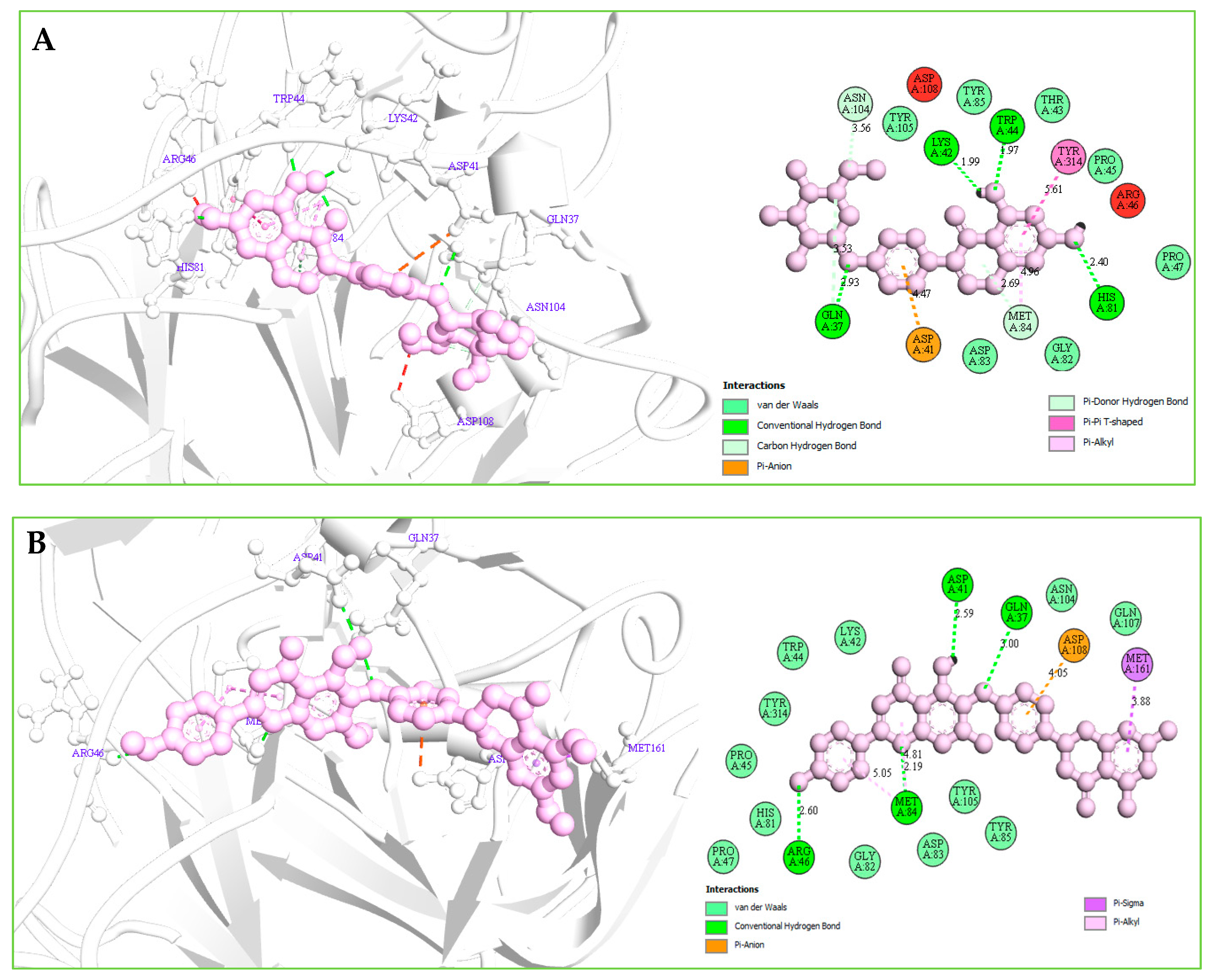
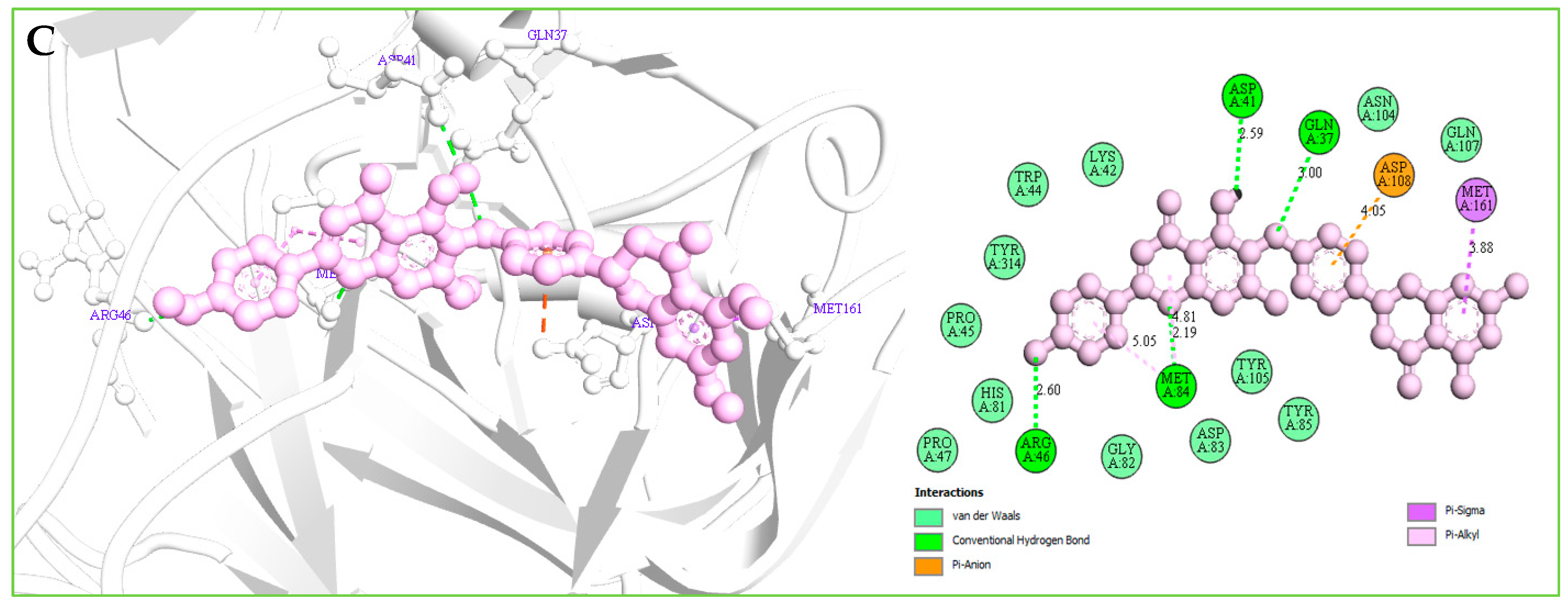
2.12.2. Protein–Ligands Interaction Interpretation
2.12.3. Pharmacophore Features Analysis
2.13. MD Simulations Analysis
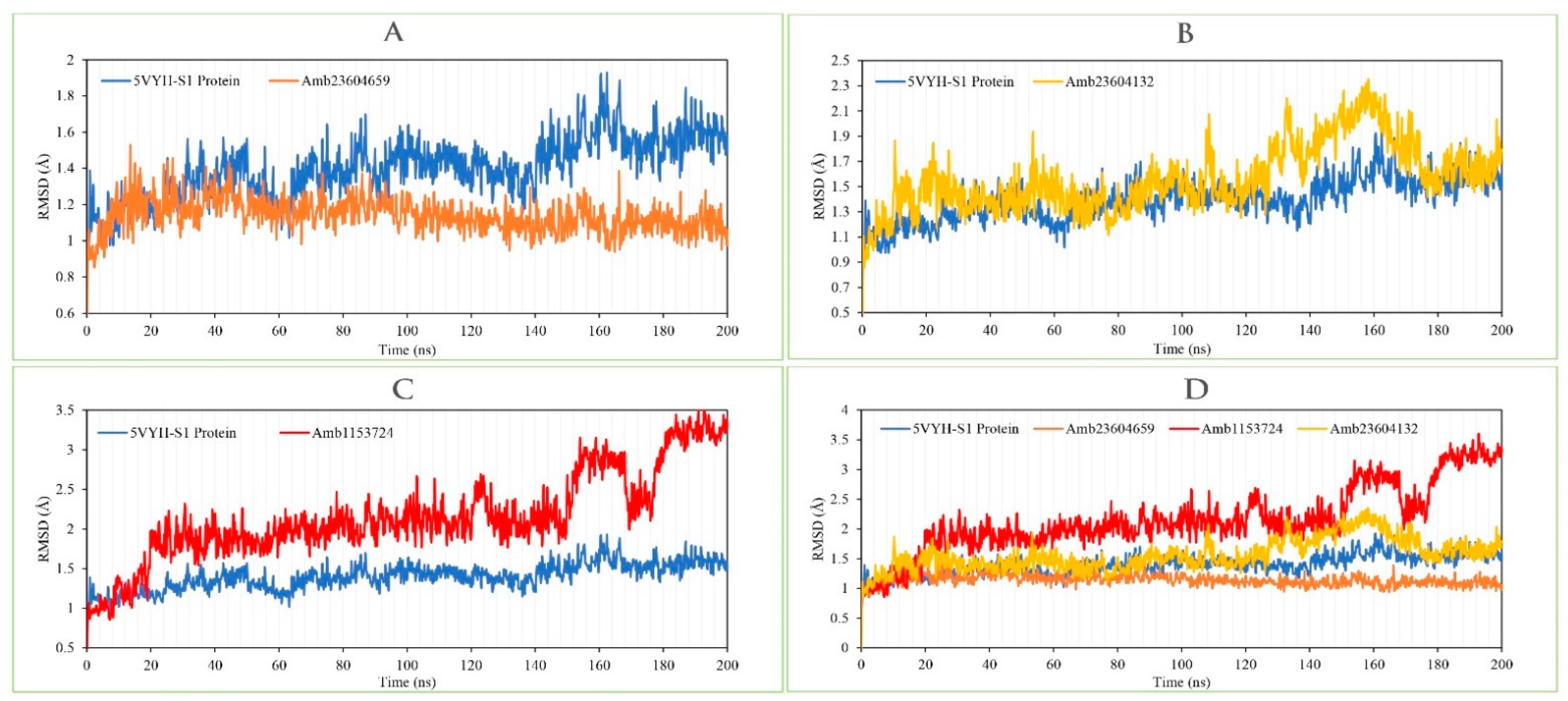
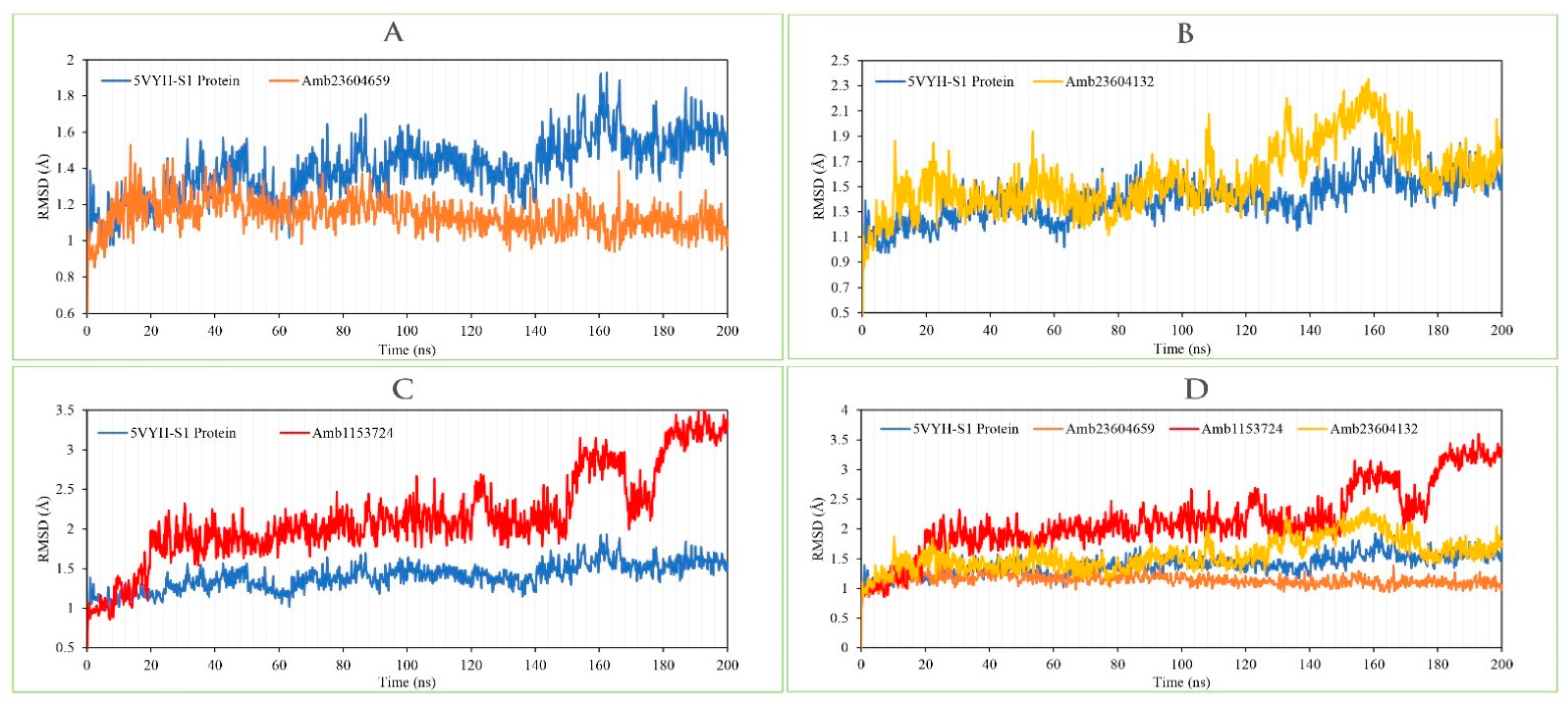
2.13.1. RMSD Analysis
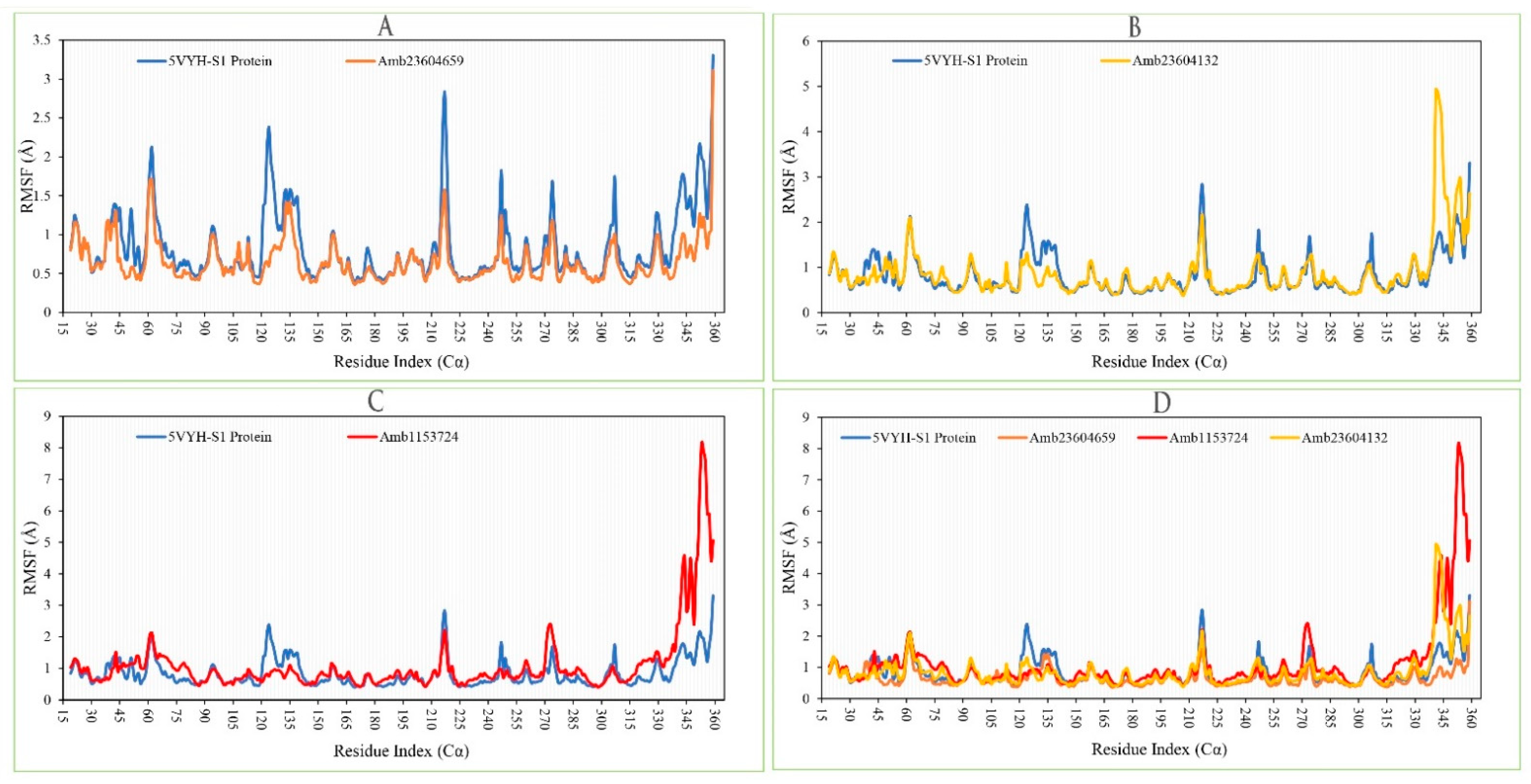
2.13.2. RMSF Analysis
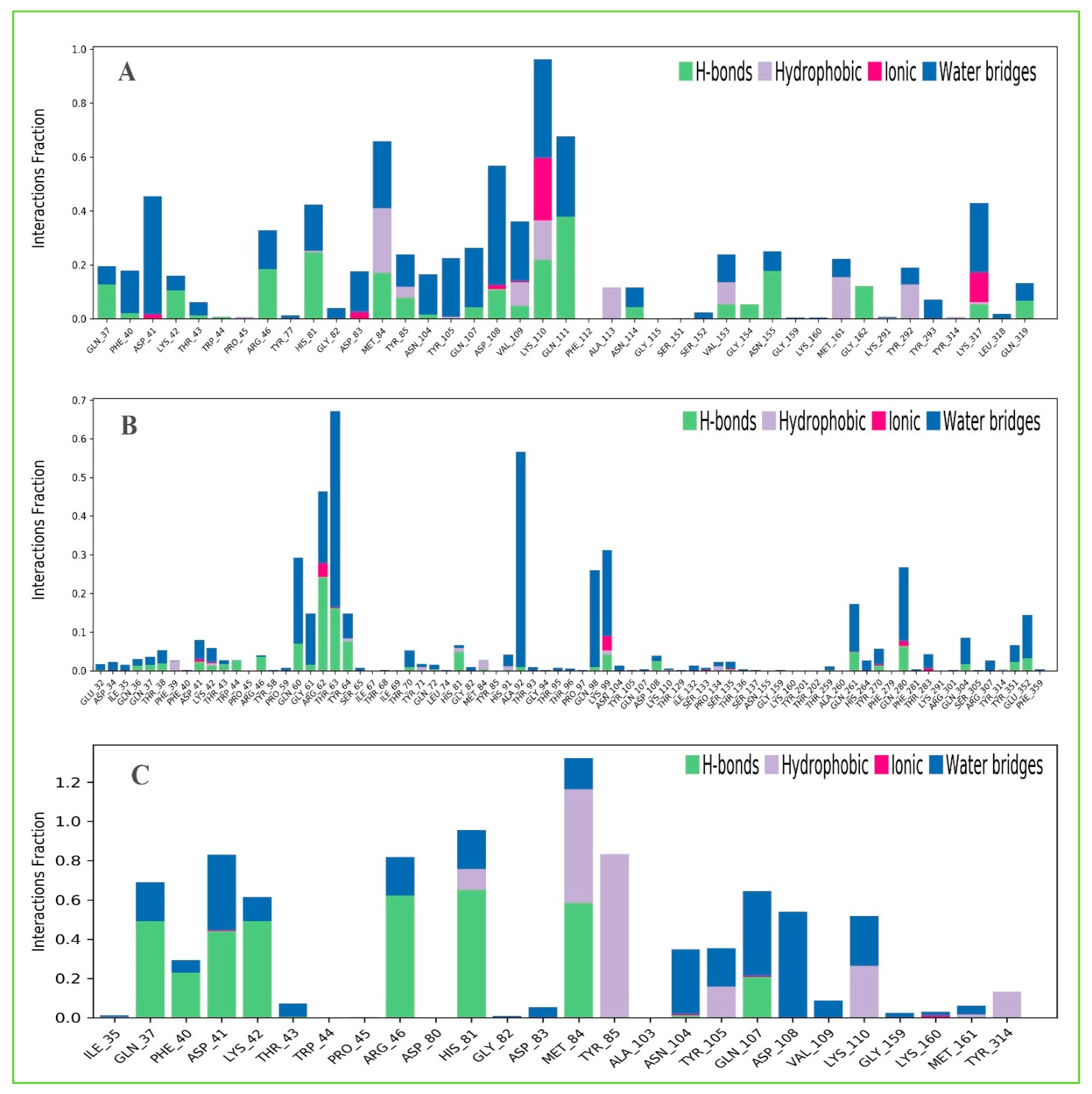
2.13.3. Protein–Ligands Contact Analysis
2.13.4. Ligand Properties Analysis
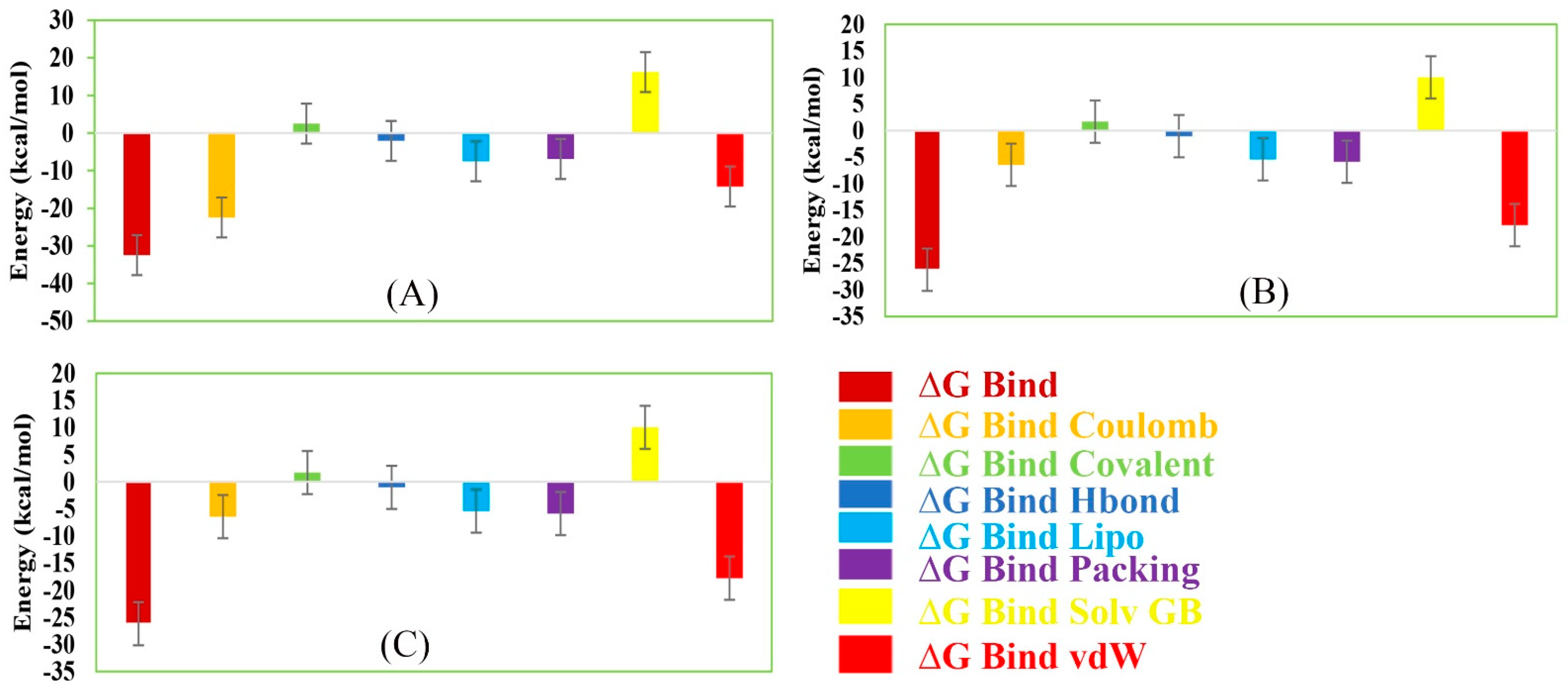
2.14. MM/GBSA Analysis
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Pharmacophore Modeling
5.2. Molecule Library Preparation
5.3. Active Compounds Identification and Decoy Set Generation
5.4. Model Performance Analysis
5.5. Virtual Screening
5.6. Protein and Ligands Preparation
5.7. Binding Site Identification and Grid Box Generation
5.8. Molecular Docking Simulation
5.9. ADME Analysis
5.10. Toxicity Test
5.11. Quantum Mechanics (QM)-Based Calculation
5.12. Frontier Molecular Orbital HOMO/LUMO Calculation
5.13. Re-Docking and Interaction Analysis
5.14. MD Simulation
Analysis of MD Trajectory
5.15. End-Point Binding Free Energy Calculation with MM/GBSA
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. In Coronaviruses: Methods and Protocols; Springer: New York, NY, USA, 2015; Volume 1282, pp. 1–23. ISBN 9781493924387. [Google Scholar]
- Cui, J.; Li, F.; Shi, Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Lu, C.; He, J.; Liu, L.; Zou, Y.; Zhang, Z.; Zhu, Z.; Ge, X.; Wu, A.; Jiang, T.; et al. Identification and characterization of circRNAs encoded by MERS-CoV, SARS-CoV-1 and SARS-CoV-2. Brief. Bioinform. 2021, 22, 1297–1308. [Google Scholar] [CrossRef] [PubMed]
- Samad, A.; Ahammad, F.; Nain, Z.; Alam, R.; Imon, R.R.; Hasan, M.; Rahman, M.S. Designing a multi-epitope vaccine against SARS-CoV-2: An immunoinformatics approach. J. Biomol. Struct. Dyn. 2020, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Cascella, M.; Rajnik, M.; Cuomo, A.; Dulebohn, S.C.; Di Napoli, R. Features, Evaluation and Treatment Coronavirus (COVID-19); StatPearls Publishing, 2020. [Google Scholar]
- Wu, J.T.; Leung, K.; Leung, G.M. Nowcasting and forecasting the potential domestic and international spread of the 2019-nCoV outbreak originating in Wuhan, China: A modelling study. Lancet 2020, 395, 689–697. [Google Scholar] [CrossRef] [Green Version]
- O’Driscoll, M.; Ribeiro Dos Santos, G.; Wang, L.; Cummings, D.A.T.; Azman, A.S.; Paireau, J.; Fontanet, A.; Cauchemez, S.; Salje, H. Age-specific mortality and immunity patterns of SARS-CoV-2. Nature 2021, 590, 140–145. [Google Scholar] [CrossRef]
- Park, Y.J.; Walls, A.C.; Wang, Z.; Sauer, M.M.; Li, W.; Tortorici, M.A.; Bosch, B.J.; DiMaio, F.; Veesler, D. Structures of MERS-CoV spike glycoprotein in complex with sialoside attachment receptors. Nat. Struct. Mol. Biol. 2019, 26, 1151–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Y.; Cao, D.; Zhang, Y.; Ma, J.; Qi, J.; Wang, Q.; Lu, G.; Wu, Y.; Yan, J.; Shi, Y.; et al. Cryo-EM structures of MERS-CoV and SARS-CoV spike glycoproteins reveal the dynamic receptor binding domains. Nat. Commun. 2017, 8, 15092. [Google Scholar] [CrossRef]
- Wang, N.; Rosen, O.; Wang, L.; Turner, H.L.; Stevens, L.J.; Corbett, K.S.; Bowman, C.A.; Pallesen, J.; Shi, W.; Zhang, Y.; et al. Structural Definition of a Neutralization-Sensitive Epitope on the MERS-CoV S1-NTD. Cell Rep. 2019, 28, 3395–3405. [Google Scholar] [CrossRef] [Green Version]
- Du, L.; Yang, Y.; Zhou, Y.; Lu, L.; Li, F.; Jiang, S. MERS-CoV spike protein: A key target for antivirals. Expert Opin. Ther. Targets 2017, 21, 131–143. [Google Scholar] [CrossRef] [Green Version]
- Rahman, S.M.M.; Atikullah, M.; Islam, M.N.S.; Mohaimenul, M.; Ahammad, F.; Islam, M.N.S.; Saha, B.; Rahman, M.H. Anti-inflammatory, antinociceptive and antidiarrhoeal activities of methanol and ethyl acetate extract of Hemigraphis alternata leaves in mice. Clin. Phytoscience 2019, 5, 16. [Google Scholar] [CrossRef]
- Pallesen, J.; Wang, N.; Corbett, K.S.; Wrapp, D.; Kirchdoerfer, R.N.; Turner, H.L.; Cottrell, C.A.; Becker, M.M.; Wang, L.; Shi, W.; et al. Immunogenicity and structures of a rationally designed prefusion MERS-CoV spike antigen. Proc. Natl. Acad. Sci. USA 2017, 114, E7348–E7357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleine-Weber, H.; Elzayat, M.T.; Wang, L.; Graham, B.S.; Müller, M.A.; Drosten, C.; Pöhlmann, S.; Hoffmann, M. Mutations in the Spike Protein of Middle East Respiratory Syndrome Coronavirus Transmitted in Korea Increase Resistance to Antibody-Mediated Neutralization. J. Virol. 2018, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trigueiro-Louro, J.; Correia, V.; Figueiredo-Nunes, I.; Gíria, M.; Rebelo-de-Andrade, H. Unlocking COVID therapeutic targets: A structure-based rationale against SARS-CoV-2, SARS-CoV and MERS-CoV Spike. Comput. Struct. Biotechnol. J. 2020, 18, 2117–2131. [Google Scholar] [CrossRef] [PubMed]
- Leelananda, S.P.; Lindert, S. Computational methods in drug discovery. Beilstein J. Org. Chem. 2016, 12, 2694–2718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agamah, F.E.; Mazandu, G.K.; Hassan, R.; Bope, C.D.; Thomford, N.E.; Ghansah, A.; Chimusa, E.R. Computational/in silico methods in drug target and lead prediction. Brief. Bioinform. 2020, 21, 1663–1675. [Google Scholar] [CrossRef] [PubMed]
- Pal, R.; Kumar, A.; Misra, G. Exploring TEAD2 as a drug target for therapeutic intervention of cancer: A multi-computational case study. Brief. Bioinform. 2021, 2021, 1–10. [Google Scholar] [CrossRef]
- Opo, F.A.D.M.; Rahman, M.M.; Ahammad, F.; Ahmed, I.; Bhuiyan, M.A.; Asiri, A.M. Structure based pharmacophore modeling, virtual screening, molecular docking and ADMET approaches for identification of natural anti-cancer agents targeting XIAP protein. Sci. Rep. 2021, 11, 4049. [Google Scholar] [CrossRef]
- Wolber, G.; Langer, T. LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J. Chem. Inf. Model. 2005, 45, 160–169. [Google Scholar] [CrossRef]
- Ahammad, F.; Fuad, F.A.A. The in silico identification of potent natural bioactive anti-dengue agents by targeting the human hexokinase 2 enzyme. In Proceedings of the 5th International Electronic Conference on Medicinal Chemistry, Basel, Switzerland, 30 October 2019; p. 6342. [Google Scholar]
- Ahammad, F.; Alam, R.; Mahmud, R.; Akhter, S.; Talukder, E.K.; Tonmoy, A.M.; Fahim, S.; Al-Ghamdi, K.; Samad, A.; Qadri, I. Pharmacoinformatics and molecular dynamics simulation-based phytochemical screening of neem plant (Azadiractha indica) against human cancer by targeting MCM7 protein. Brief. Bioinform. 2021, 2021, 1–15. [Google Scholar] [CrossRef]
- Adyani, F.; Fuad, A.; Ahammad, F. Virtual Screening and Molecular Docking Approaches for Identification of Natural Lead Compounds for Improved Anti-Dengue Therapeutics. Kerice 2019. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef] [Green Version]
- Bharadwaj, S.; Dubey, A.; Yadava, U.; Mishra, S.K.; Kang, S.G.; Dwivedi, V.D. Exploration of natural compounds with anti-SARS-CoV-2 activity via inhibition of SARS-CoV-2 Mpro. Brief. Bioinform. 2021, 2021, 1–17. [Google Scholar] [CrossRef]
- Bochevarov, A.D.; Harder, E.; Hughes, T.F.; Greenwood, J.R.; Braden, D.A.; Philipp, D.M.; Rinaldo, D.; Halls, M.D.; Zhang, J.; Friesner, R.A. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 2013, 113, 2110–2142. [Google Scholar] [CrossRef]
- Bento, A.P.; Gaulton, A.; Hersey, A.; Bellis, L.J.; Chambers, J.; Davies, M.; Krüger, F.A.; Light, Y.; Mak, L.; McGlinchey, S.; et al. The ChEMBL bioactivity database: An update. Nucleic Acids Res. 2014, 42, D1083–D1090. [Google Scholar] [CrossRef] [Green Version]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250. [Google Scholar] [CrossRef]
- Friesner, R.A.; Guallar, V. Ab initio quantum chemical and mixed quantum mechanics/molecular mechanics (QM/MM) methods for studying enzymatic catalysis. Annu. Rev. Phys. Chem. 2005, 56, 389–427. [Google Scholar] [CrossRef]
- Li, Y.; Evans, J.N.S. The Fukui Function: A Key Concept Linking Frontier Molecular Orbital Theory and the Hard-Soft-Acid-Base Principle. J. Am. Chem. Soc. 1995, 117, 7756–7759. [Google Scholar] [CrossRef]
- Çlnaroǧlu, S.S.; Timuçin, E. Comprehensive evaluation of the MM-GBSA method on bromodomain-inhibitor sets. Brief. Bioinform. 2020, 21, 2112–2125. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ambinter ID | Molecule Name | Formula | Structure | Binding Affinity (kcal/mol) | Pharmacophore Fit Score |
|---|---|---|---|---|---|
| Amb6600135 | Nicotinamide adenine dinucleotide | C21H28N7O14P2+ |  | −9.2 | 66.6 |
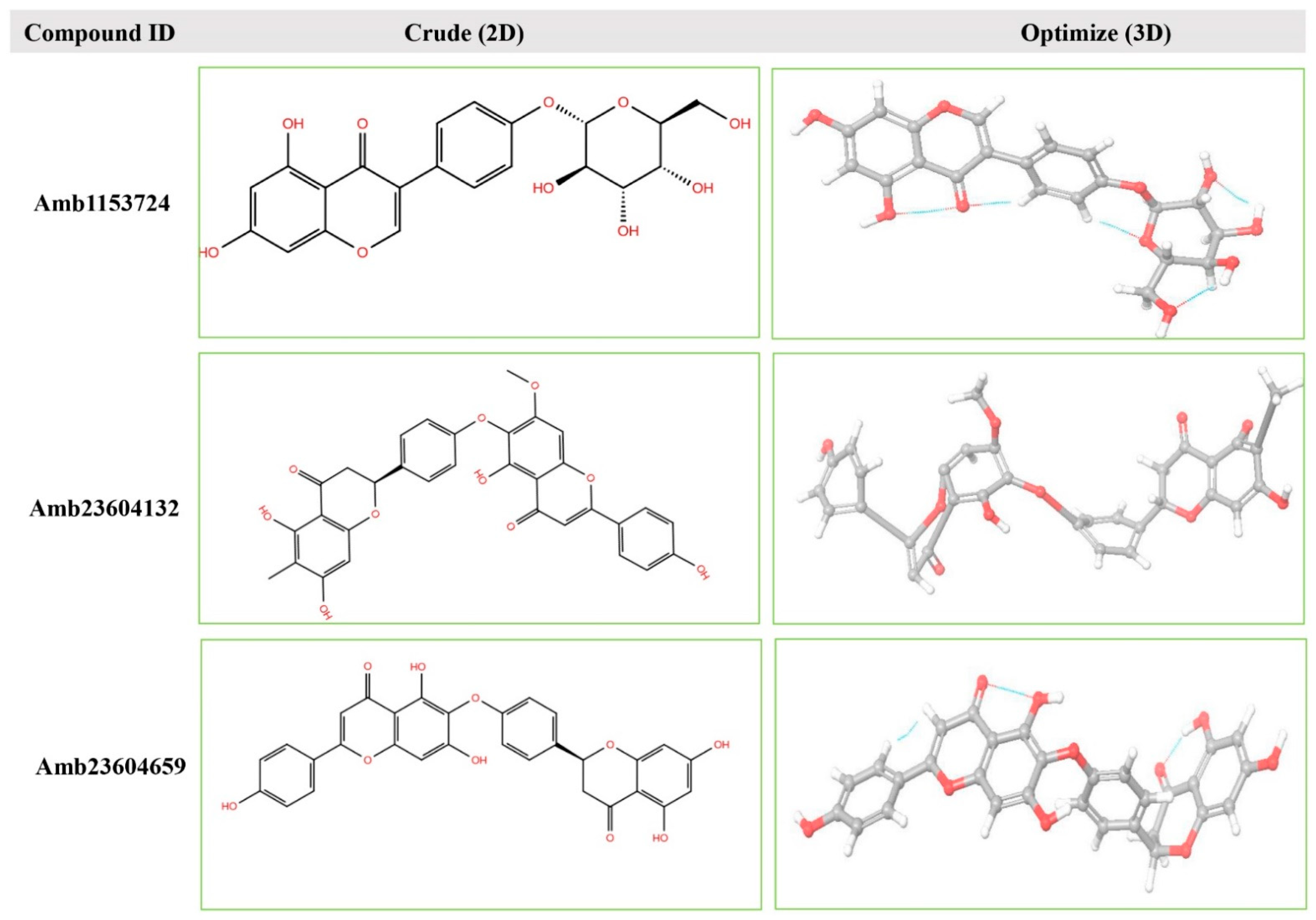
| Amb23604132 | Taiwanhomoflavone B | C32H24O10 | 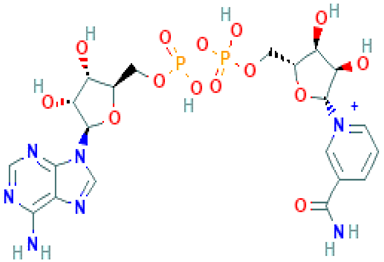 | −9.1 | 65.48 |
| Amb23604659 | 2,3-Dihydrohinokiflavone | C30H20O10 |  | −8.6 | 65.55 |
| Amb1153724 | Sophoricoside | C21H20O10 |  | −8.1 | 66.64 |
| Properties | Amb6600135 | Amb23604132 | Amb23604659 | Amb1153724 | |
|---|---|---|---|---|---|
| Physico-chemical Properties | MW (g/mol) | 664.43 | 568.53 | 540.47 | 432.38 |
| Heavy atoms | 44 | 42 | 40 | 31 | |
| Aro. atoms | 15 | 28 | 28 | 16 | |
| Rotatable bonds | 11 | 5 | 4 | 4 | |
| H-bond acceptors | 17 | 10 | 10 | 10 | |
| H-bond donors | 8 | 4 | 5 | 6 | |
| TPSA (Å2) | 337.88 | 155.89 | 166.89 | 170.05 | |
| Lipophilicity | Log Po/w (Cons) | -5.39 | 4.37 | 3.70 | 0.45 |
| Water Solubility | Log S (ESOL) | High | Soluble | Soluble | Moderate |
| Pharmacokinetics | GI absorption | Low | Moderate | Moderate | Low |
| BBB permeant | No | No | No | No | |
| P-GP substrate | Yes | No | No | No | |
| Drug likeness | Lipinski violations | 3 | 1 | 1 | 1 |
| Medi. Chemistry | Synth. accessibility | Medium | Easy | Easy | Medium |
| Classification | Target | Amb23604132 | Amb23604659 | Amb1153724 |
|---|---|---|---|---|
| Oral toxicity | LD50 (mg/kg) | 5000 | 5000 | 5000 |
| Toxicity Class | 5 | 5 | 5 | |
| Organ toxicity | Hepatotoxicity | Inactive | Inactive | Inactive |
| Toxicity endpoints | Carcinogenicity | Inactive | Inactive | Inactive |
| Mutagenicity | Inactive | Inactive | Inactive | |
| Cytotoxicity | Inactive | Inactive | Inactive |
| Compound | Residues | Bond Distance (Å) | Category | Bond Types |
|---|---|---|---|---|
| Amb1153724 | GLN37 | 2.93083 | Hydrogen Bond | Conventional H-B |
| TRP44 | 1.96777 | Hydrogen Bond | Conventional H-B | |
| HIS81 | 2.39932 | Hydrogen Bond | Conventional H-B | |
| LYS42 | 1.99029 | Hydrogen Bond | Conventional H-B | |
| GLN37 | 3.53458 | Hydrogen Bond | Carbon H-B | |
| ASN104 | 3.56047 | Hydrogen Bond | Carbon H-B | |
| ASP41 | 4.46727 | Electrostatic | Pi-Anion | |
| MET84 | 2.6909 | Hydrogen Bond | Pi-Donor H-B | |
| TYR314 | 5.61379 | Hydrophobic | Pi-Pi T-shaped | |
| MET84 | 4.9701 | Hydrophobic | Pi-Alkyl | |
| MET84 | 4.95866 | Hydrophobic | Pi-Alkyl | |
| Amb23604132 | GLN37 | 2.54483 | Hydrogen Bond | Conventional H-B |
| LYS42 | 2.04393 | Hydrogen Bond | Conventional H-B | |
| MET84 | 2.12397 | Hydrogen Bond | Conventional H-B | |
| PHE40 | 2.49011 | Hydrogen Bond | Conventional H-B | |
| ASP108 | 3.4188 | Hydrogen Bond | Carbon H-B | |
| ASP108 | 4.6773 | Electrostatic | Pi-Anion | |
| TYR314 | 5.74845 | Hydrophobic | Pi-Pi T-shaped | |
| MET161 | 4.84458 | Hydrophobic | Alkyl | |
| MET84 | 4.15196 | Hydrophobic | Pi-Alkyl | |
| MET84 | 5.10244 | Hydrophobic | Pi-Alkyl | |
| Amb23604659 | GLN37 | 2.99559 | Hydrogen Bond | Conventional H-B |
| ARG46 | 2.60436 | Hydrogen Bond | Conventional H-B | |
| MET84 | 2.18527 | Hydrogen Bond | Conventional H-B | |
| ASP41 | 2.59464 | Hydrogen Bond | Conventional H-B | |
| ASP108 | 4.05492 | Electrostatic | Pi-Anion | |
| MET161 | 3.87544 | Hydrophobic | Pi-Sigma | |
| MET84 | 4.80802 | Hydrophobic | Pi-Alkyl | |
| MET84 | 5.05152 | Hydrophobic | Pi-Alkyl |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bouback, T.A.; Pokhrel, S.; Albeshri, A.; Aljohani, A.M.; Samad, A.; Alam, R.; Hossen, M.S.; Al-Ghamdi, K.; Talukder, M.E.K.; Ahammad, F.; et al. Pharmacophore-Based Virtual Screening, Quantum Mechanics Calculations, and Molecular Dynamics Simulation Approaches Identified Potential Natural Antiviral Drug Candidates against MERS-CoV S1-NTD. Molecules 2021, 26, 4961. https://doi.org/10.3390/molecules26164961
Bouback TA, Pokhrel S, Albeshri A, Aljohani AM, Samad A, Alam R, Hossen MS, Al-Ghamdi K, Talukder MEK, Ahammad F, et al. Pharmacophore-Based Virtual Screening, Quantum Mechanics Calculations, and Molecular Dynamics Simulation Approaches Identified Potential Natural Antiviral Drug Candidates against MERS-CoV S1-NTD. Molecules. 2021; 26(16):4961. https://doi.org/10.3390/molecules26164961
Chicago/Turabian StyleBouback, Thamer A., Sushil Pokhrel, Abdulaziz Albeshri, Amal Mohammed Aljohani, Abdus Samad, Rahat Alam, Md Saddam Hossen, Khalid Al-Ghamdi, Md. Enamul Kabir Talukder, Foysal Ahammad, and et al. 2021. "Pharmacophore-Based Virtual Screening, Quantum Mechanics Calculations, and Molecular Dynamics Simulation Approaches Identified Potential Natural Antiviral Drug Candidates against MERS-CoV S1-NTD" Molecules 26, no. 16: 4961. https://doi.org/10.3390/molecules26164961
APA StyleBouback, T. A., Pokhrel, S., Albeshri, A., Aljohani, A. M., Samad, A., Alam, R., Hossen, M. S., Al-Ghamdi, K., Talukder, M. E. K., Ahammad, F., Qadri, I., & Simal-Gandara, J. (2021). Pharmacophore-Based Virtual Screening, Quantum Mechanics Calculations, and Molecular Dynamics Simulation Approaches Identified Potential Natural Antiviral Drug Candidates against MERS-CoV S1-NTD. Molecules, 26(16), 4961. https://doi.org/10.3390/molecules26164961