
The Novel Role of MIF in the Secretion of IL-25, IL-31, and IL-33 from PBMC of Patients with Rheumatoid Arthritis

 , , and
, , and 
Abstract
:1. Introduction
2. Results
2.1. Demographic and Clinical Characteristics of Donors of PBMC
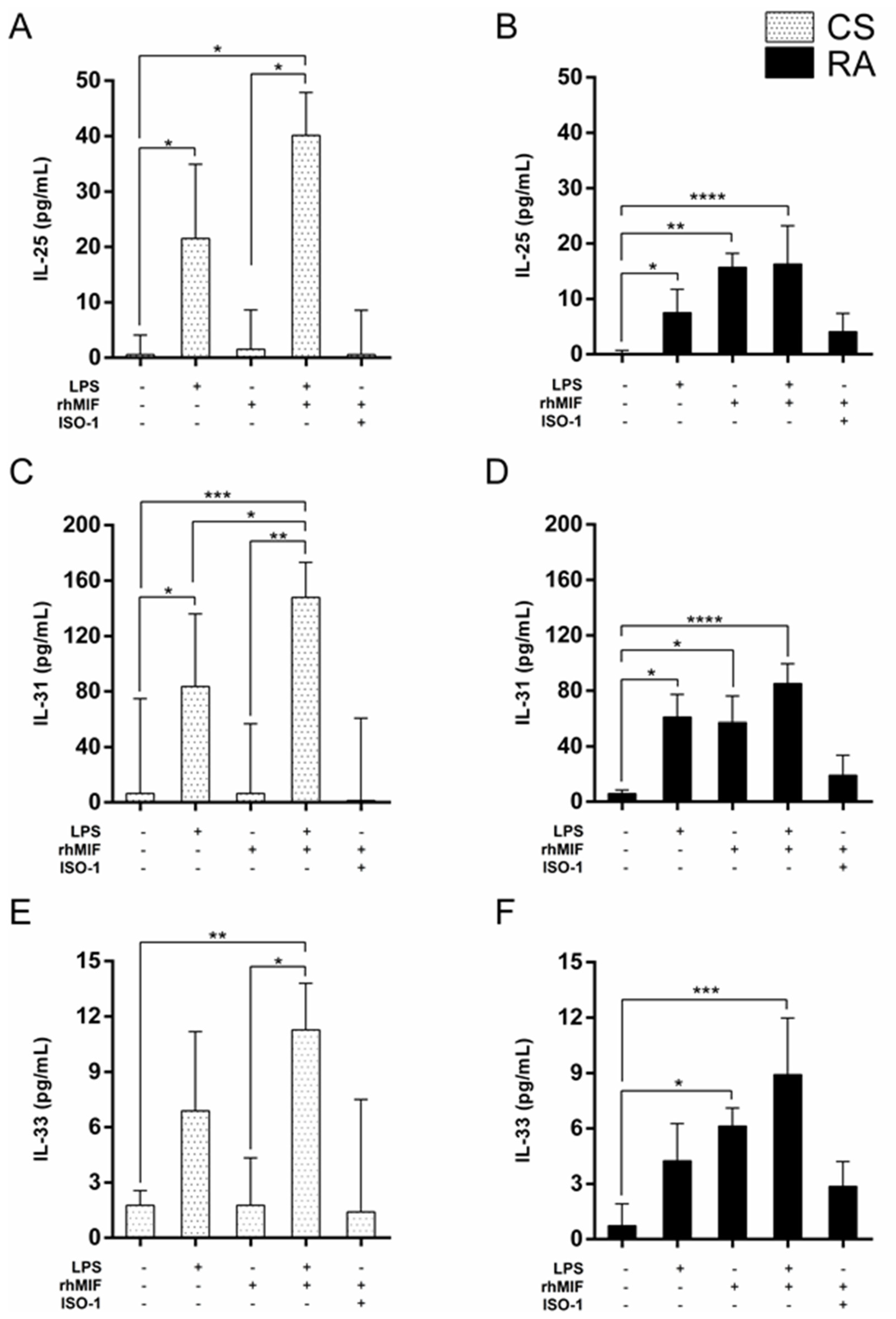
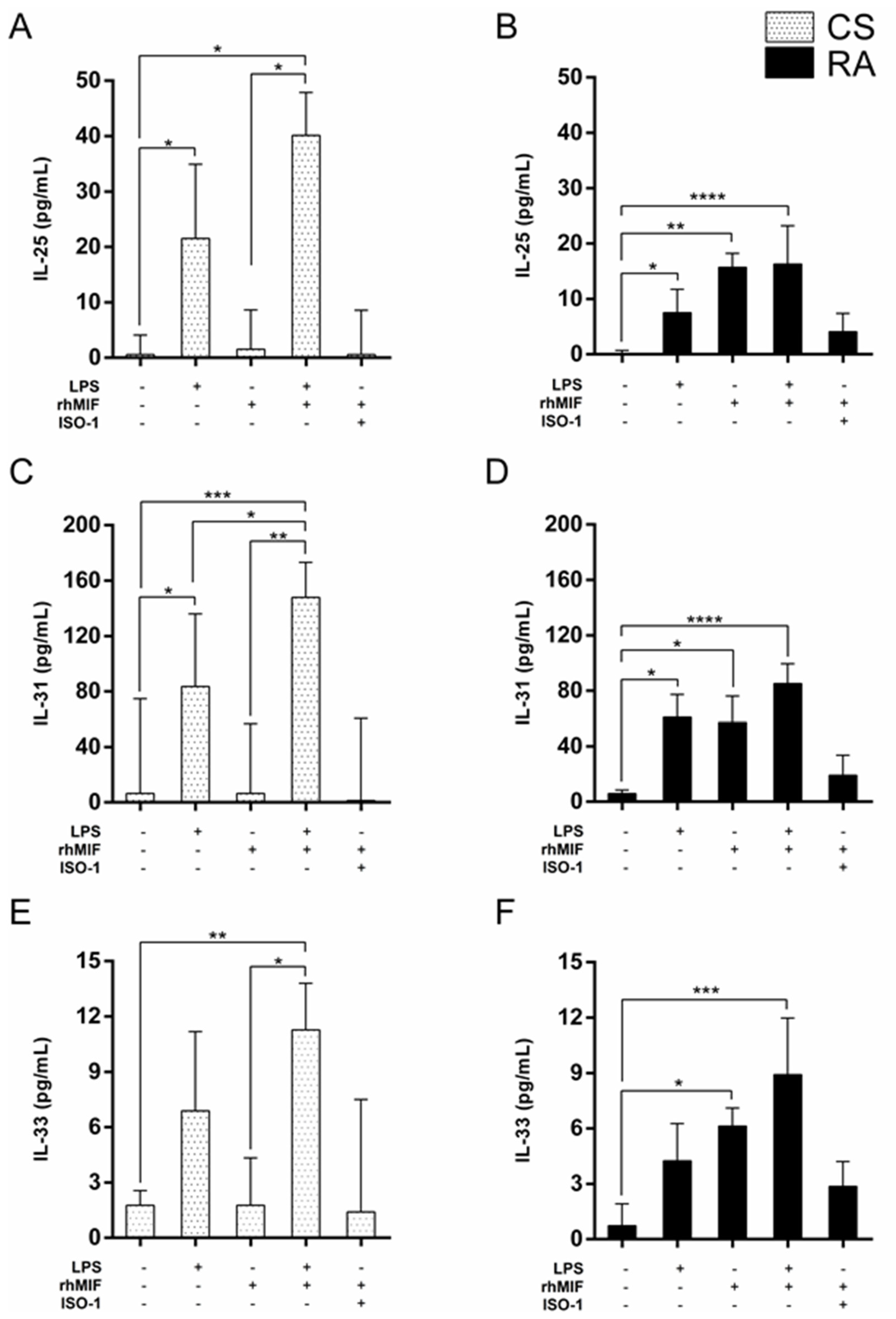
2.2. Effect of LPS and rhMIF on IL-25, IL-31, and IL-33 Secretion from PBMC of CS and RA Patients
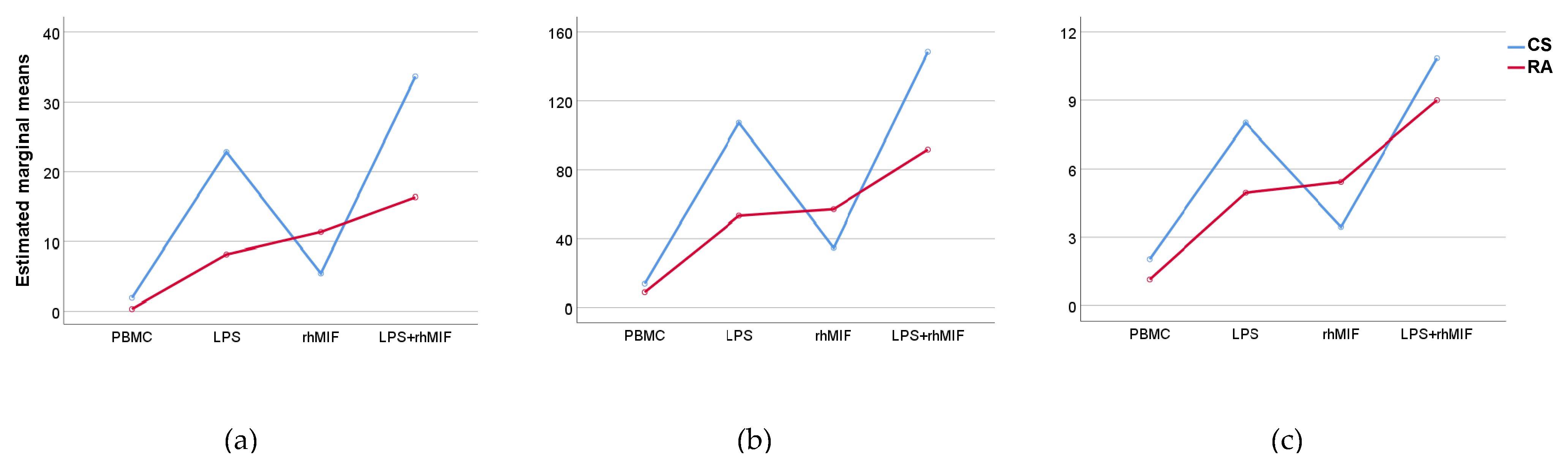
2.3. Effect of Interaction between LPS Plus rhMIF on IL-25, IL-31, and IL-33 Secretion from PBMC of CS and RA Patients
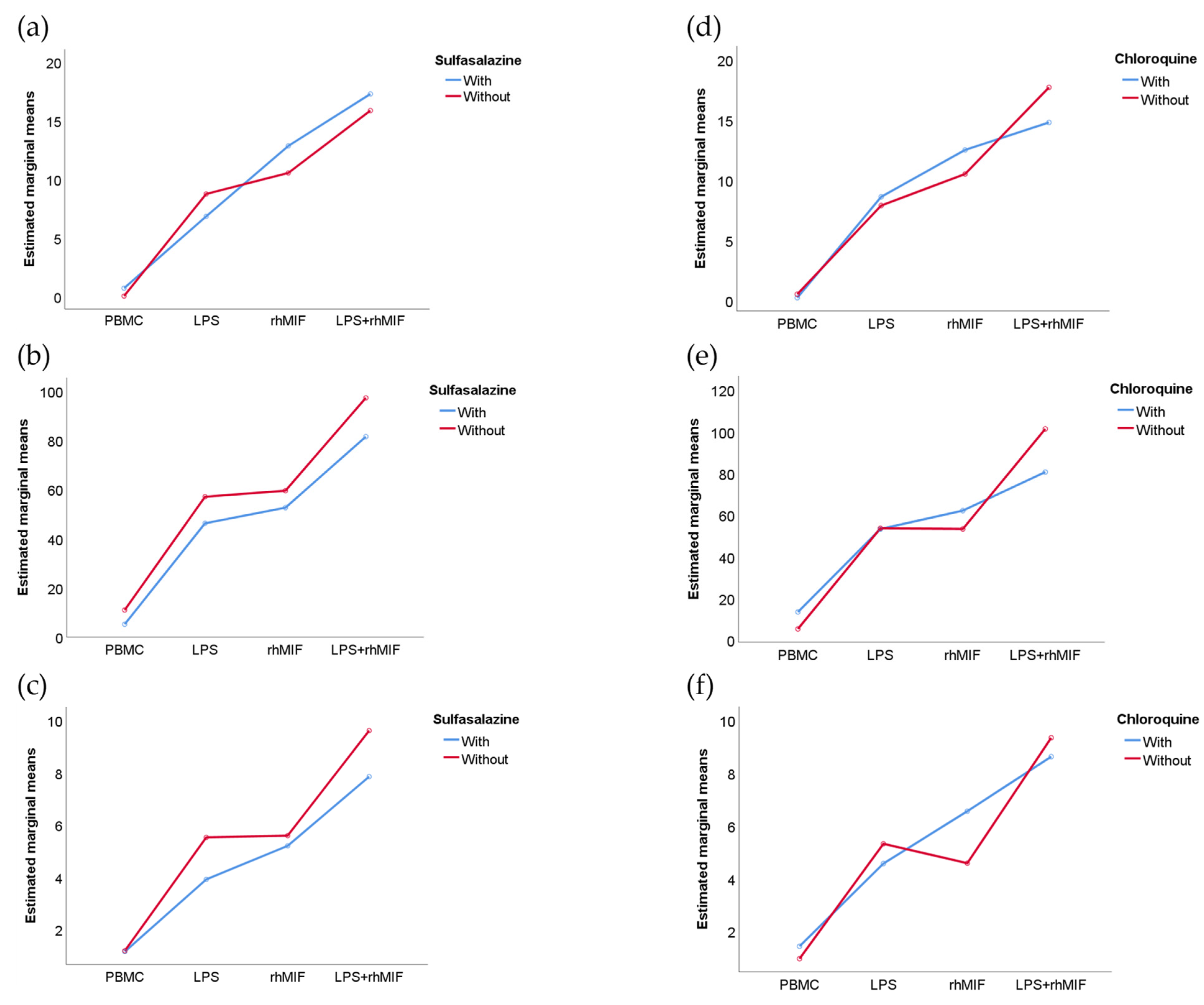
2.4. Analysis of Interactions between Groups of Study or Treatments with Stimuli Effects
3. Discussion
4. Materials and Methods
4.1. Participants
4.2. Isolation of PBMC and Culture Protocol
4.3. Cytokine Assays
4.4. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Smolen, J.S.; Aletaha, D.; Barton, A.; Burmester, G.R.; Emery, P.; Firestein, G.S.; Kavanaugh, A.; McInnes, I.B.; Solomon, D.H.; Strand, V.; et al. Rheumatoid arthritis. Nat. Rev. Dis. Prim. 2018, 4, 18001. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Otsuka, K.; Sato, M.; Takahashi, R.; Odai, T.; Isozaki, T.; Yajima, N.; Miwa, Y.; Kasama, T. Elevated serum levels of macrophage migration inhibitory factor and their significant correlation with rheumatoid vasculitis disease activity. Mod. Rheumatol. 2012, 2, 59–65. [Google Scholar] [CrossRef]
- Kim, K.-W.; Kim, H.-R. Macrophage migration inhibitory factor: A potential therapeutic target for rheumatoid arthritis. Korean J. Intern. Med. 2016, 31, 634–642. [Google Scholar] [CrossRef]
- Stojanović, I.; Cvjetićanin, T.; Lazaroski, S.; Stosic-Grujicic, S.; Miljkovic, D. Macrophage migration inhibitory factor stimulates interleukin-17 expression and production in lymph node cells. Immunology 2009, 126, 74–83. [Google Scholar] [CrossRef]
- Goldberg, M.R.; Nadiv, O.; Luknar-Gabor, N.; Zadik-Mnuhin, G.; Tovbin, J.; Katz, Y. Correlation of Th1-type cytokine ex-pression and induced proliferation to lipopolysaccharide. Am. J. Respir. Cell Mol. Biol. 2008, 38, 733–737. [Google Scholar] [CrossRef]
- Wu, H.-P.; Chung, K.; Lin, C.-Y.; Jiang, B.-Y.; Chuang, D.-Y.; Liu, Y.-C. Associations of T helper 1, 2, 17 and regulatory T lymphocytes with mortality in severe sepsis. Inflamm. Res. 2013, 62, 751–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Shao, Z.; Shangguan, G.; Bie, Q.; Zhang, B. Biological properties and the role of IL-25 in disease pathogenesis. J. Immunol. Res. 2018, 2018, 6519465. [Google Scholar] [CrossRef] [PubMed]
- Saadoun, D.; Terrier, B.; Cacoub, P. Interleukin-25: Key Regulator of Inflammatory and Autoimmune Diseases. Curr. Pharm. Des. 2011, 17, 3781–3785. [Google Scholar] [CrossRef] [PubMed]
- Johnson, G.L.; Lapadat, R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002, 6, 1911–1912. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.S.; Angkasekwinai, P.; Chang, S.H.; Chung, Y.; Dong, C. Protease allergens induce the expression of IL-25 via Erk and p38 MAPK pathway. J. Korean Med. Sci. 2010, 25, 829–834. [Google Scholar] [CrossRef]
- Che, D.N.; Cho, B.O.; Kim, J.-S.; Shin, J.Y.; Kang, H.J.; Jang, S.I. Luteolin and Apigenin Attenuate LPS-Induced Astrocyte Activation and Cytokine Production by Targeting MAPK, STAT3, and NF-κB Signaling Pathways. Inflammation 2020, 43, 1716–1728. [Google Scholar] [CrossRef]
- Li, W.; Tao, W.; Chen, J.; Zhai, Y.; Yin, N.; Wang, Z. Paeoniflorin suppresses IL-33 production by macrophages. Immunopharmacol. Immunotoxicol. 2020, 42, 286–293. [Google Scholar] [CrossRef]
- Qi, F.; Bai, S.; Wang, D.; Xu, L.; Hu, H.; Zeng, S.; Chai, R.; Liu, B. Macrophages produce IL-33 by activating MAPK signaling pathway during RSV infection. Mol. Immunol. 2017, 87, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Caruso, R.; Stolfi, C.; Sarra, M.; Rizzo, A.; Fantini, M.C.; Pallone, F.; Macdonald, T.T.; Monteleone, G. Inhibition of monocyte-derived inflammatory cytokines by IL-25 occurs via p38 Map kinase–dependent induction of Socs-3. Blood 2009, 113, 3512–3519. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xian, Y.-F.; Hu, Z.; Loo, S.K.F.; Ip, S.P.; Chan, W.Y.; Lin, Z.-X.; Wu, J.C.Y. Efficacy and action mechanisms of a Chinese herbal formula on experimental models of atopic dermatitis. J. Ethnopharmacol. 2021, 274, 114021. [Google Scholar] [CrossRef] [PubMed]
- Stott, B.; Lavender, P.; Lehmann, S.; Pennino, D.; Durham, S.; Schmidt-Weber, C.B. Human IL-31 is induced by IL-4 and promotes TH2-driven inflammation. J. Allergy Clin. Immunol. 2013, 132, 446–454. [Google Scholar] [CrossRef]
- Sokol, C.L.; Barton, G.M.; Farr, A.G.; Medzhitov, R. A mechanism for the initiation of allergen-induced T helper type 2 responses. Nat. Immunol. 2008, 9, 310–318. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Chen, L.-Y.; Papadimos, T.J.; Huang, S.; Zuraw, B.L.; Pan, Z.K. Lipopolysaccharide-driven Th2 Cytokine production in macrophages is regulated by both MyD88 and TRAM. J. Biol. Chem. 2009, 284, 29391–29398. [Google Scholar] [CrossRef] [Green Version]
- Nile, C.J.; Barksby, E.; Jitprasertwong, P.; Preshaw, P.M.; Taylor, J.J. Expression and regulation of interleukin-33 in human monocytes. Immunology 2010, 130, 172–180. [Google Scholar] [CrossRef]
- Ali, S.; Mohs, A.; Thomas, M.; Klare, J.; Ross, R.; Schmitz, M.L.; Martin, M.U. The Dual Function Cytokine IL-33 Interacts with the Transcription Factor NF-κB To Dampen NF-κB–Stimulated Gene Transcription. J. Immunol. 2011, 187, 1609–1616. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, C.; Barbour, M.; Fairlie-Clarke, K.J.; Allan, D.; Mu, R.; Jiang, H.R. Emerging role of interleukin-33 in autoimmune dis-eases. Immunology 2014, 141, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Jiang, H.-R.; Kewin, P.; Li, Y.; Mu, R.; Fraser, A.R.; Pitman, N.; Kurowska-Stolarska, M.; McKenzie, A.N.J.; McInnes, I.; et al. IL-33 exacerbates antigen-induced arthritis by activating mast cells. Proc. Natl. Acad. Sci. USA 2008, 105, 10913–10918. [Google Scholar] [CrossRef] [Green Version]
- Yoo, S.-A.; Leng, L.; Kim, B.-J.; Du, X.; Tilstam, P.V.; Kim, K.H.; Kong, J.-S.; Yoon, H.-J.; Liu, A.; Wang, T.; et al. MIF allele-dependent regulation of the MIF coreceptor CD44 and role in rheumatoid arthritis. Proc. Natl. Acad. Sci. USA 2016, 113, 7917–7926. [Google Scholar] [CrossRef] [Green Version]
- Bilsborrow, J.B.; Doherty, E.; Tilstam, P.V.; Bucala, R. Macrophage migration inhibitory factor (MIF) as a therapeutic target for rheumatoid arthritis and systemic lupus erythematosus. Expert Opin. Ther. Targets 2019, 23, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Alampour-Rajabi, S.; El Bounkari, O.; Rot, A.; Müller-Newen, G.; Bachelerie, F.; Gawaz, M.; Weber, C.; Schober, A.; Bernhagen, J. MIF interacts with CXCR7 to promote receptor internalization, ERK1/2 and ZAP-70 signaling, and lymphocyte chemotaxis. FASEB J. 2015, 29, 4497–4511. [Google Scholar] [CrossRef] [Green Version]
- Nanki, T.; Hayashida, K.; El-Gabalawy, H.S.; Suson, S.; Shi, K.; Girschick, H.J.; Yavuz, S.; Lipsky, P.E. Stromal Cell-Derived Factor-1-CXC Chemokine Receptor 4 Interactions Play a Central Role in CD4+T Cell Accumulation in Rheumatoid Arthritis Synovium. J. Immunol. 2000, 165, 6590–6598. [Google Scholar] [CrossRef] [Green Version]
- Angkasekwinai, P.; Park, H.; Wang, Y.-H.; Wang, Y.-H.; Seon, H.C.; Corry, D.B.; Liu, Y.-J.; Zhu, Z.; Dong, C. Interleukin 25 promotes the initiation of proallergic type 2 responses. J. Exp. Med. 2007, 204, 1509–1517. [Google Scholar] [CrossRef] [Green Version]
- Liew, F.Y.; Girard, J.-P.; Turnquist, H.R. Interleukin-33 in health and disease. Nat. Rev. Immunol. 2016, 16, 676–689. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zheng, J.; Wang, L.; Ding, G.; Luo, P.; Lu, Y.; Pan, W.; Wang, M. Chloroquine protects mice from challenge with CpG ODN and LPS by decreasing proinflammatory cytokine release. Int. Immunopharmacol. 2004, 4, 223–234. [Google Scholar]
- Aggarwal, A.; Misra, R. Methotrexate inhibits interleukin-6 production in patients with juvenile rheumatoid arthritis. Rheumatol. Int. 2003, 23, 134–137. [Google Scholar] [CrossRef]
- Park, G.; Lee, S.H.; Han, J.Y.; Oh, D.S. Altered TNF-α response by Aconibal® and methotrexate in a lipopolysaccha-ride-induced setting of inflammatory conditions: Potential on a synergistic combination. J. Ethnopharmacol. 2018, 213, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Crilly, A.; Madhok, R.; Watson, J.; Capell, H.A.; Sturrock, R.D. Production of interleukin-6 by monocytes isolated from rheumatoid arthritis patients receiving second-line drug therapy. Rheumatology 1994, 33, 821–825. [Google Scholar] [CrossRef]
- Hernández-Palma, L.A.; García-Arellano, S.; Bucala, R.; Llamas-Covarrubias, M.A.; De la Cruz-Mosso, U.; Oregon-Romero, E.; Zerpa-Cruz, S.; Parra-Rojas, I.; Plascencia-Hernández, A.; Muñoz-Valle, J.F. Functional MIF promoter haplotypes modulate Th17-related cytokine expression in peripheral blood mononuclear cells from control subjects and rheumatoid arthritis patients. Cytokine 2019, 115, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Arellano, S.; Hernandez-Palma, L.A.; Bucala, R.; Hernandez-Bello, J.; De la Cruz-Mosso, U.; Garcia-Iglesias, T.; Cerpa-Cruz, S.; Padilla-Gutierrez, J.R.; Valle, Y.; Sonanez-Organis, J.G.; et al. Th1/Th17 cytokine profile is induced by macrophage migration inhibitory factor in peripheral blood mononuclear cells from rheumatoid arthritis patients. Curr. Mol. Med. 2018, 18, 679–688. [Google Scholar] [CrossRef] [PubMed]
- Aletaha, D.; Neogi, T.; Silman, A.J.; Funovits, J.; Felson, D.T.; Bingham, C.O., III; Birnbaum, N.S.; Burmester, G.R.; Bykerk, V.P.; Cohen, M.D.; et al. Rheumatoid arthritis classification criteria: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010, 62, 2569–2581. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Investigated Parameters | Sum of Squares (SS) | Mean of Squares (MS) | F | p Value | Sum of Squares (SS) | Mean of Squares (MS) | F | p Value |
|---|---|---|---|---|---|---|---|---|
| CS | RA | |||||||
| IL-25 | ||||||||
| rhMIF | 460 | 460 | 2.163 | 0.151 | 830.2 | 830.2 | 19.025 | <0.001 |
| LPS | 5440.8 | 5440.8 | 25.578 | <0.001 | 370.7 | 370.7 | 8.495 | 0.006 |
| rhMIF + LPS | 117.9 | 117.9 | 0.554 | 0.462 | 17.8 | 17.8 | 0.407 | 0.528 |
| Error | 6806.9 | 212.7 | 1396.4 | 43.637 | ||||
| Total SS | 21,988.9 | 5565.4 | ||||||
| IL-31 | ||||||||
| rhMIF | 8690.6 | 8690.6 | 4.2 | 0.049 | 16,992 | 16,992 | 32.472 | <0.001 |
| LPS | 96,602.9 | 96,602.9 | 46.692 | <0.001 | 14,176 | 14,176 | 27.091 | <0.001 |
| rhMIF + LPS | 905.8 | 905.8 | 0.438 | 0.513 | 208.3 | 208.3 | 0.398 | 0.533 |
| Error | 66,206.7 | 2069 | 16,745 | 523.3 | ||||
| Total SS | 380,120 | 147,769 | ||||||
| IL-33 | ||||||||
| rhMIF | 40.6 | 40.6 | 2.672 | 0.112 | 156.2 | 156.2 | 16.427 | <0.001 |
| LPS | 402.3 | 402.3 | 26.453 | <0.001 | 122.6 | 122.6 | 12.898 | 0.001 |
| rhMIF + LPS | 4.5 | 4.5 | 0.295 | 0.591 | 0.14 | 0.14 | 0.015 | 0.903 |
| Error | 486.7 | 15.209 | 304.2 | 9.5 | ||||
| Total SS | 2266.1 | 1530.8 | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Arellano, S.; Hernández-Palma, L.A.; Cerpa-Cruz, S.; Sánchez-Zuno, G.A.; Herrera-Godina, M.G.; Muñoz-Valle, J.F. The Novel Role of MIF in the Secretion of IL-25, IL-31, and IL-33 from PBMC of Patients with Rheumatoid Arthritis. Molecules 2021, 26, 4968. https://doi.org/10.3390/molecules26164968
García-Arellano S, Hernández-Palma LA, Cerpa-Cruz S, Sánchez-Zuno GA, Herrera-Godina MG, Muñoz-Valle JF. The Novel Role of MIF in the Secretion of IL-25, IL-31, and IL-33 from PBMC of Patients with Rheumatoid Arthritis. Molecules. 2021; 26(16):4968. https://doi.org/10.3390/molecules26164968
Chicago/Turabian StyleGarcía-Arellano, Samuel, Luis Alexis Hernández-Palma, Sergio Cerpa-Cruz, Gabriela Athziri Sánchez-Zuno, Melva Guadalupe Herrera-Godina, and José Francisco Muñoz-Valle. 2021. "The Novel Role of MIF in the Secretion of IL-25, IL-31, and IL-33 from PBMC of Patients with Rheumatoid Arthritis" Molecules 26, no. 16: 4968. https://doi.org/10.3390/molecules26164968