
Palonosetron/Methyllycaconitine Deactivate Hippocampal Microglia 1, Inflammasome Assembly and Pyroptosis to Enhance Cognition in a Novel Model of Neuroinflammation

 , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
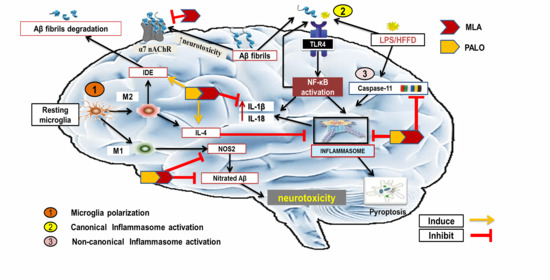
1. Introduction
2. Results
2.1. Verification of the HFFD/LPS-Induced AD Model
2.2. Palonosetron, MLA, or Their Combination Improves Long-Term Spatial Working Memory in AD Rats
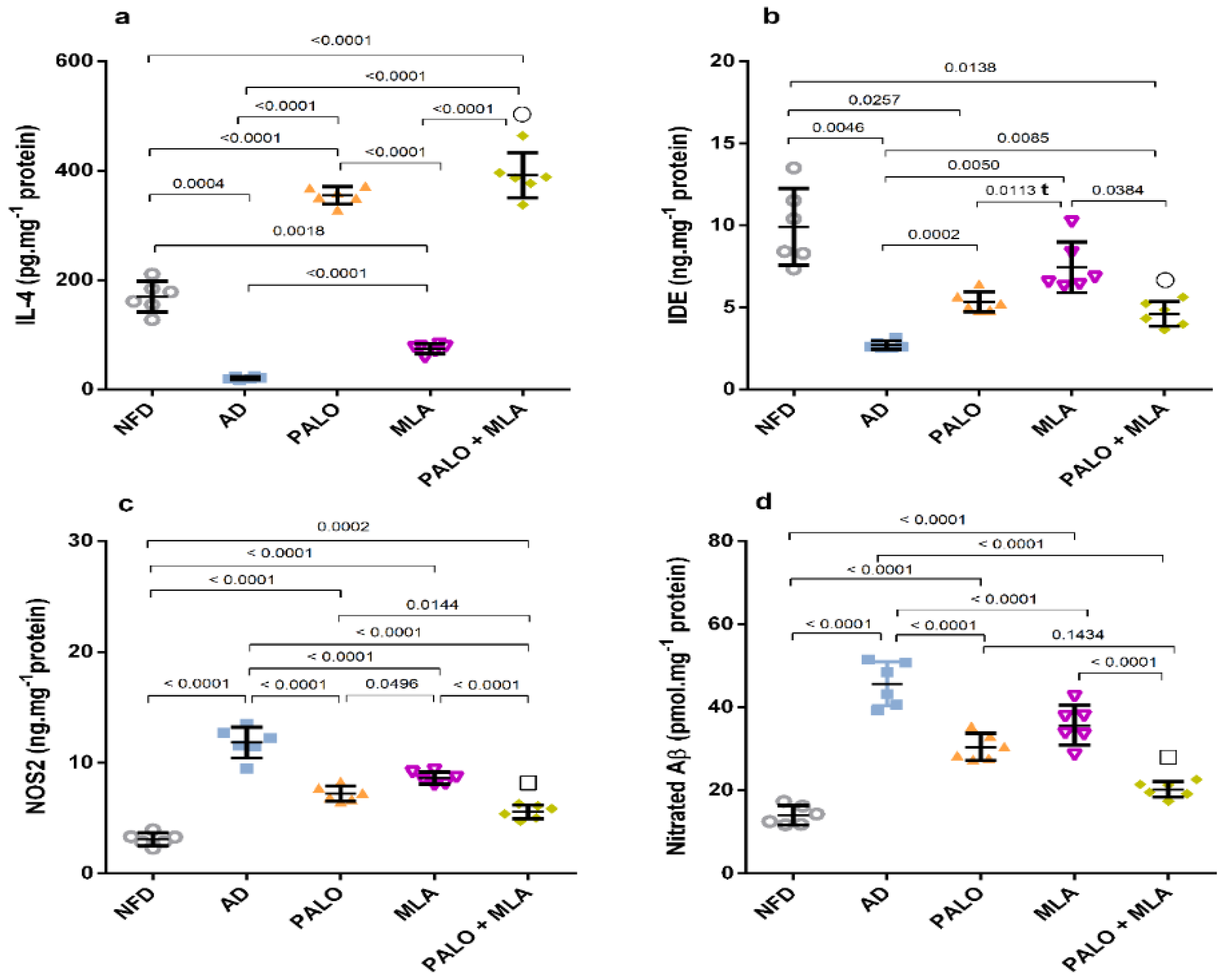
2.3. Palonosetron, MLA, or Their Combination Prevents the Hippocampal Microglial Depolarization in AD Rats
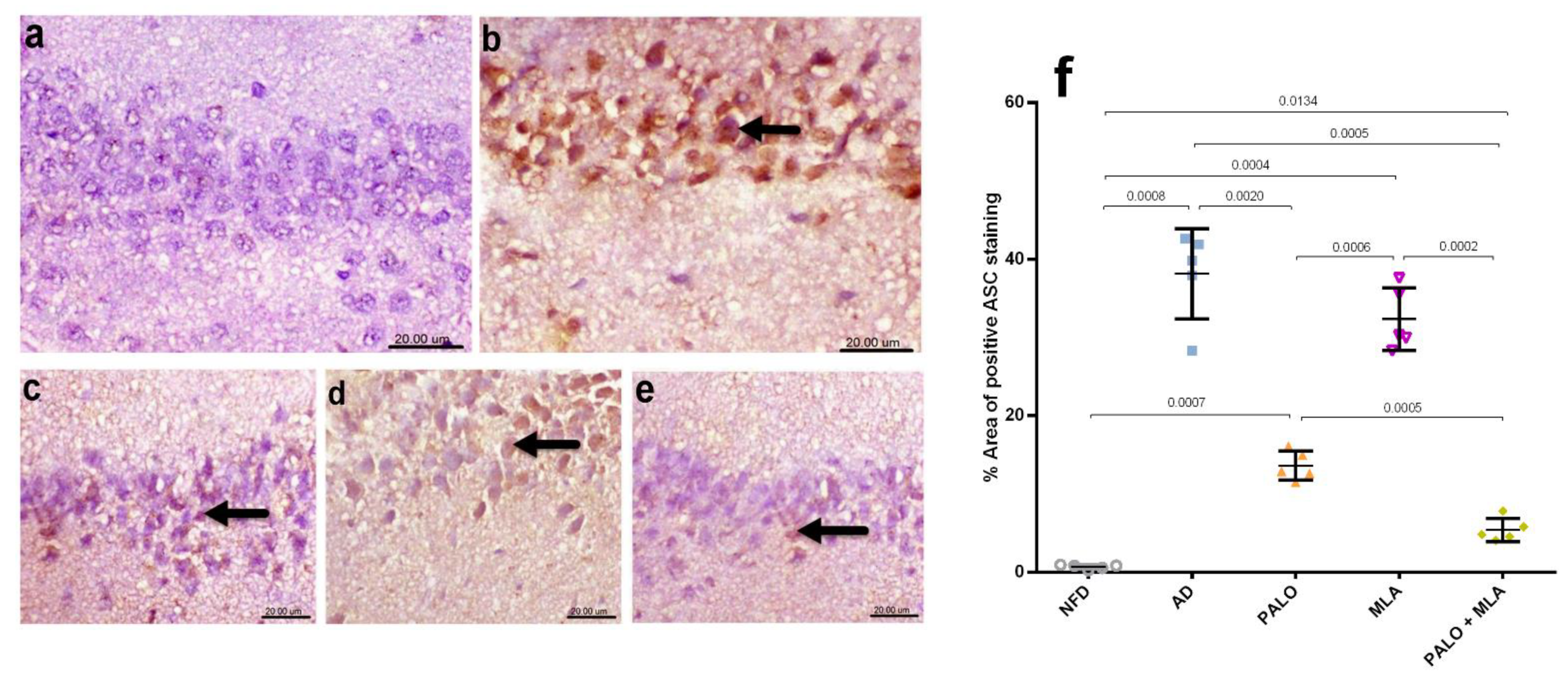
2.4. Palonosetron, MLA or Their Combination Suppresses the Hippocampal Canonical and Non-Canonical Activated Inflammasome Cascades in AD Rats
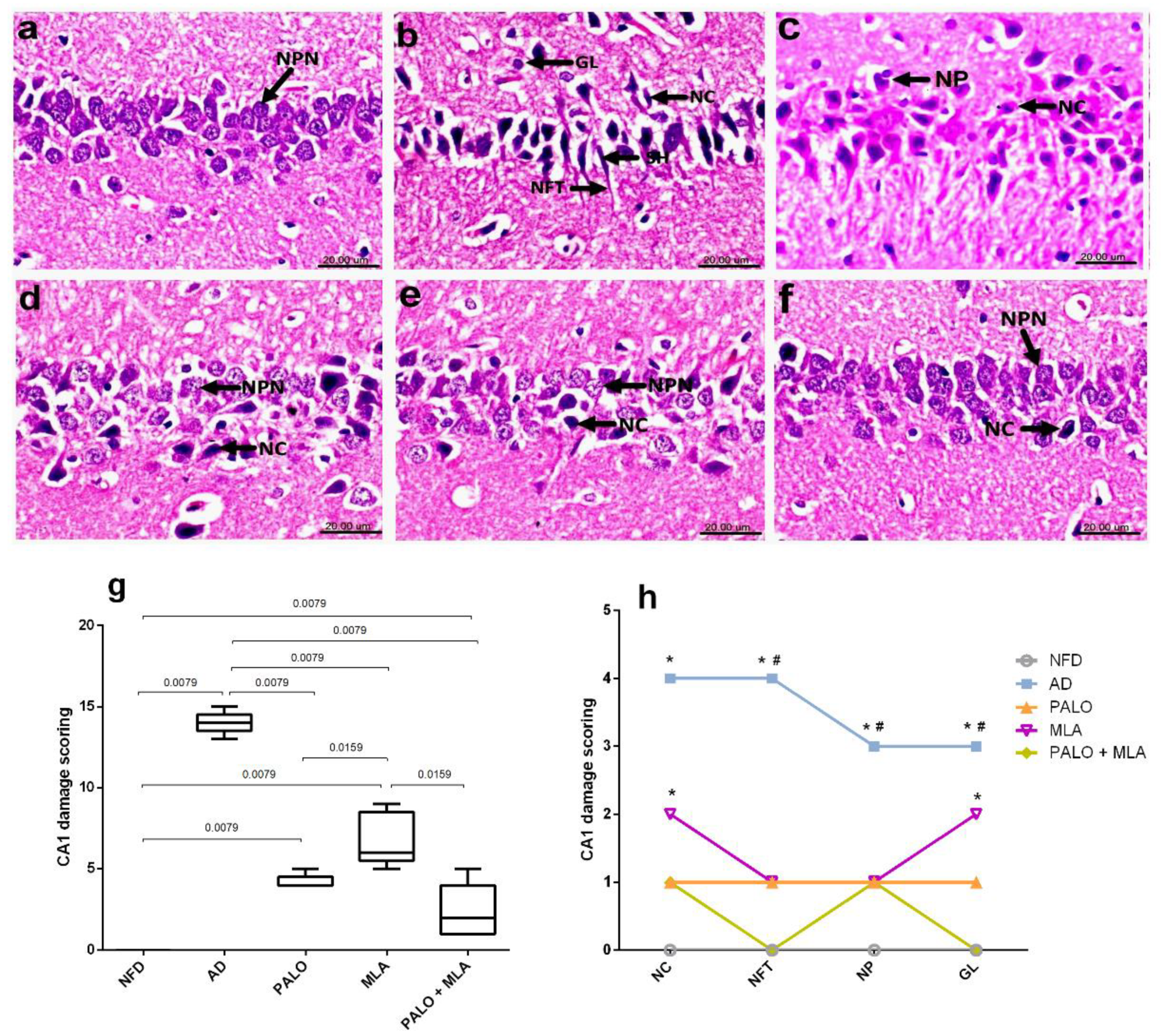
2.5. Palonosetron, MLA, or Their Combination Preserves the Hippocampal Architecture in AD Rats
2.6. Palonosetron, MLA, or Their Combination Reduce Amyloid Plaques-Induced by HFFD/LPS in Rats
2.7. Palonosetron, MLA, or Their Combination Decrease Astrocytes Immunoreactivity in AD Rats
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Drug and Chemicals
4.3. Development of HFFD/LPS Neuroinflammation Model
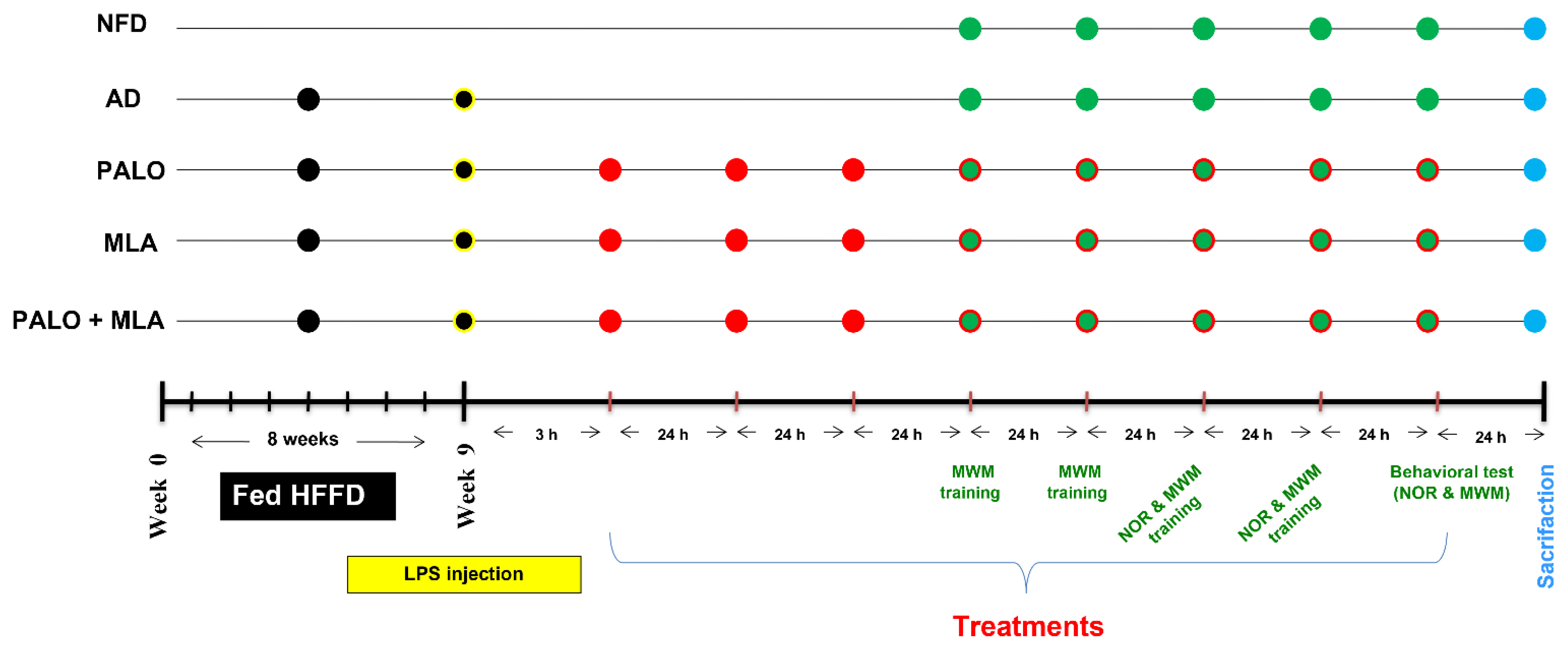
4.4. Experimental Design of the Main Study
4.5. Behavioral Tests
4.5.1. NOR Test
4.5.2. MWM Test
4.6. Biochemical Analysis
4.6.1. Assessment of Serum Insulin, Glucose, and Lipid Profile
4.6.2. Assessment of Hippocampal Aβ1-42, Nitrated Aβ, NOS2, IL-4, and IDE
4.6.3. Assessment of Hippocampal ACh and 5-HT
4.6.4. Determination of Hippocampal Inflammasome Biomarkers
4.7. Histopathological and Immunohistochemical Examinations
Glial Fibrillary Acidic Protein (GFAP)
4.8. Calculation of Combination Effect Using Coefficient Drug Index (CDI)
4.9. Data and Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Piaceri, I.; Nacmias, B.; Sorbi, S. Genetics of familial and sporadic Alzheimer’s disease. Front. Biosci. 2013, 5, 167–177. [Google Scholar] [CrossRef] [Green Version]
- Mendez, M.F. Early-onset Alzheimer disease. Neurol. Clin. 2017, 35, 263–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zubenko, G.S. Molecular neurobiology of Alzheimer’s disease (syndrome?). Harv. Rev. Psychiatry 1997, 5, 177–213. [Google Scholar] [CrossRef] [PubMed]
- Bazzari, F.H.; Abdallah, D.M.; El-Abhar, H.S. Pharmacological Interventions to Attenuate Alzheimer’s Disease Progression: The Story So Far. Curr. Alzheimer Res. 2019, 16, 261–277. [Google Scholar] [CrossRef]
- Zhan, X.; Stamova, B.; Sharp, F.R. Lipopolysaccharide associates with amyloid plaques, neurons and oligodendrocytes in Alzheimer’s disease brain: A review. Front. Aging Neurosci. 2018, 10, 42. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10, 128. [Google Scholar] [CrossRef] [Green Version]
- Shao, B.-Z.; Xu, Z.-Q.; Han, B.-Z.; Su, D.-F.; Liu, C. NLRP3 inflammasome and its inhibitors: A review. Front. Pharm. 2015, 6, 262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katsumoto, A.; Takeuchi, H.; Takahashi, K.; Tanaka, F. Microglia in Alzheimer’s disease: Risk factors and inflammation. Front. Neurol. 2018, 9, 978. [Google Scholar] [CrossRef] [Green Version]
- Cai, H.; Cong, W.-n.; Ji, S.; Rothman, S.; Maudsley, S.; Martin, B. Metabolic dysfunction in Alzheimer’s disease and related neurodegenerative disorders. Curr. Alzheimer Res. 2012, 9, 5–17. [Google Scholar] [CrossRef]
- Walker, J.M.; Harrison, F.E. Shared neuropathological characteristics of obesity, type 2 diabetes and Alzheimer’s disease: Impacts on cognitive decline. Nutrients 2015, 7, 7332–7357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandanmagsar, B.; Youm, Y.-H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179. [Google Scholar] [CrossRef] [PubMed]
- Pistell, P.J.; Morrison, C.D.; Gupta, S.; Knight, A.G.; Keller, J.N.; Ingram, D.K.; Bruce-Keller, A.J. Cognitive impairment following high fat diet consumption is associated with brain inflammation. J. Neuroimmunol. 2010, 219, 25–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowette, K.; Roosen, L.; Tack, J.; Berghe, P.V. Effects of high-fructose diets on central appetite signaling and cognitive function. Front. Nutr. 2015, 2, 5. [Google Scholar] [CrossRef] [Green Version]
- Pratchayasakul, W.; Kerdphoo, S.; Petsophonsakul, P.; Pongchaidecha, A.; Chattipakorn, N.; Chattipakorn, S.C. Effects of high-fat diet on insulin receptor function in rat hippocampus and the level of neuronal corticosterone. Life Sci. 2011, 88, 619–627. [Google Scholar] [CrossRef]
- Chou, L.-M.; Lin, C.-I.; Chen, Y.-H.; Liao, H.; Lin, S.-H. A diet containing grape powder ameliorates the cognitive decline in aged rats with a long-term high-fructose-high-fat dietary pattern. J. Nutr. Biochem. 2016, 34, 52–60. [Google Scholar] [CrossRef]
- Serino, M.; Luche, E.; Gres, S.; Baylac, A.; Bergé, M.; Cenac, C.; Waget, A.; Klopp, P.; Iacovoni, J.; Klopp, C. Metabolic adaptation to a high-fat diet is associated with a change in the gut microbiota. Gut 2012, 61, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Romero, S.; Hereu, M.; Atienza, L.; Casas, J.; Jáuregui, O.; Amézqueta, S.; Dasilva, G.; Medina, I.; Nogués, M.R.; Romeu, M. Mechanistically different effects of fat and sugar on insulin resistance, hypertension, and gut microbiota in rats. Am. J. Physiol. Endocrinol. Metab. 2018, 314, E552–E563. [Google Scholar] [CrossRef]
- Noble, E.E.; Hsu, T.M.; Kanoski, S.E. Gut to brain dysbiosis: Mechanisms linking western diet consumption, the microbiome, and cognitive impairment. Front. Behav. Neurosci. 2017, 11, 9. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, M.J.; Lai, M.K.; Tordera, R.M.; Francis, P.T. Serotonergic therapies for cognitive symptoms in Alzheimer’s disease: Rationale and current status. Drugs 2014, 74, 729–736. [Google Scholar] [CrossRef]
- Mohamed, R.A.; Galal, O.; Mohammed, A.R.; El-Abhar, H.S. Tropisetron modulates peripheral and central serotonin/insulin levels via insulin and nuclear factor kappa B/receptor for advanced glycation end products signalling to regulate type-2 diabetes in rats. RSC Adv. 2018, 8, 11908–11920. [Google Scholar] [CrossRef] [Green Version]
- Fakhfouri, G.; Rahimian, R.; Ghia, J.-E.; Khan, W.I.; Dehpour, A.R. Impact of 5-HT3 receptor antagonists on peripheral and central diseases. Drug Discov. Today 2012, 17, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Rahimian, R.; Fakhfouri, G.; Ejtemaei Mehr, S.; Ghia, J.E.; Genazzani, A.A.; Payandemehr, B.; Dehpour, A.R.; Mousavizadeh, K.; Lim, D. Tropisetron attenuates amyloid-beta-induced inflammatory and apoptotic responses in rats. Eur. J. Clin. Investig. 2013, 43, 1039–1051. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Gautam, S.; Rawat, J.; Singh, M.; Saraf, S.; Kaithwas, G. Effect of palonosetron (5HT-3 antagonist) and pantoprazole (proton pump inhibitor) against surgical esophagitis induced by forestomach and pylorus ligation in albino rats. Hum. Exp. Toxicol. 2016, 35, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.K.; Sammi, S.R.; Rawat, J.K.; Roy, S.; Singh, M.; Gautam, S.; Yadav, R.K.; Devi, U.; Ansari, M.N.; Saeedan, A.S. Palonosetron attenuates 1, 2-dimethyl hydrazine induced preneoplastic colon damage through downregulating acetylcholinesterase expression and up-regulating synaptic acetylcholine concentration. RSC Adv. 2016, 6, 40527–40538. [Google Scholar] [CrossRef]
- Ni, R.; Marutle, A.; Nordberg, A. Modulation of α7 nicotinic acetylcholine receptor and fibrillar amyloid-β interactions in Alzheimer’s disease brain. J. Alzheimers Dis. 2013, 33, 841–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, T.; Xiao, T.; Sun, Q.; Wang, K. The current agonists and positive allosteric modulators of α7 nAChR for CNS indications in clinical trials. Acta Pharm. Sin. B 2017, 7, 611–622. [Google Scholar] [CrossRef]
- Andriambeloson, E.; Huyard, B.; Poiraud, E.; Wagner, S. Methyllycaconitine-and scopolamine-induced cognitive dysfunction: Differential reversal effect by cognition-enhancing drugs. Pharm. Res. Perspect. 2014, 2, e00048. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Lyu, C.; Zhou, J.; Huang, S.; Zhang, Y.; Liu, G.; Liu, K.; Chen, D.; Hu, Y.; Zhou, L. TLR4 signaling pathway mediates the LPS/ischemia-induced expression of monocytechemotactic protein-induced protein 1 in microglia. Neurosci. Lett. 2018, 686, 33–40. [Google Scholar] [CrossRef]
- Nakaoku, Y.; Saito, S.; Yamamoto, Y.; Maki, T.; Takahashi, R.; Ihara, M. The Dipeptidyl Peptidase-4 Inhibitor Linagliptin Ameliorates High-fat Induced Cognitive Decline in Tauopathy Model Mice. Int. J. Mol. Sci. 2019, 20, 2539. [Google Scholar] [CrossRef] [Green Version]
- Pinçon, A.; De Montgolfier, O.; Akkoyunlu, N.; Daneault, C.; Pouliot, P.; Villeneuve, L.; Lesage, F.; Levy, B.I.; Thorin-Trescases, N.; Thorin, É. Non-Alcoholic Fatty Liver Disease, and the Underlying Altered Fatty Acid Metabolism, Reveals Brain Hypoperfusion and Contributes to the Cognitive Decline in APP/PS1 Mice. Metabolites 2019, 9, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selkrig, J.; Wong, P.; Zhang, X.; Pettersson, S. Metabolic tinkering by the gut microbiome: Implications for brain development and function. Gut Microbes 2014, 5, 369–380. [Google Scholar] [CrossRef] [PubMed]
- Erecinska, M.; Cherian, S.; Silver, I.A. Energy metabolism in mammalian brain during development. Prog. Neurobiol. 2004, 73, 397–445. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Stamova, B.; Jin, L.-W.; DeCarli, C.; Phinney, B.; Sharp, F.R. Gram-negative bacterial molecules associate with Alzheimer disease pathology. Neurology 2016, 87, 2324–2332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, H.Q.; Yang, T.; Xiao, W.; Fan, L.; Wu, Y.; Terrando, N.; Wang, T.L. Prolonged neuroinflammation after lipopolysaccharide exposure in aged rats. PLoS ONE 2014, 9, e106331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banks, W.A.; Gray, A.M.; Erickson, M.A.; Salameh, T.S.; Damodarasamy, M.; Sheibani, N.; Meabon, J.S.; Wing, E.E.; Morofuji, Y.; Cook, D.G. Lipopolysaccharide-induced blood-brain barrier disruption: Roles of cyclooxygenase, oxidative stress, neuroinflammation, and elements of the neurovascular unit. Neuroinflammation 2015, 12, 223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosari, S.; Badoer, E.; Nguyen, J.C.; Killcross, A.S.; Jenkins, T.A. Effect of western and high fat diets on memory and cholinergic measures in the rat. Behav. Brain Res. 2012, 235, 98–103. [Google Scholar] [CrossRef]
- Luo, D.; Hou, X.; Hou, L.; Wang, M.; Xu, S.; Dong, C.; Liu, X. Effect of pioglitazone on altered expression of Aβ metabolism-associated molecules in the brain of fructose-drinking rats, a rodent model of insulin resistance. Eur. J. Pharm. 2011, 664, 14–19. [Google Scholar] [CrossRef]
- Kar, S.; Slowikowski, S.P.; Westaway, D.; Mount, H.T. Interactions between β-amyloid and central cholinergic neurons: Implications for Alzheimer’s disease. J. Psychiatry Neurosci. 2004, 29, 427–441. [Google Scholar]
- Fakhfouri, G.; Mousavizadeh, K.; Mehr, S.E.; Dehpour, A.R.; Zirak, M.R.; Ghia, J.-E.; Rahimian, R. From chemotherapy-induced emesis to neuroprotection: Therapeutic opportunities for 5-HT 3 receptor antagonists. Mol. Neurobiol. 2015, 52, 1670–1679. [Google Scholar] [CrossRef]
- Martin, S.E.; de Fiebre, N.E.C.; Christopher, M. The α7 nicotinic acetylcholine receptor-selective antagonist, methyllycaconitine, partially protects against β-amyloid1–42 toxicity in primary neuron-enriched cultures. Brain Res. 2004, 1022, 254–256. [Google Scholar] [CrossRef]
- Escubedo, E.; Chipana, C.; Perez-Sanchez, M.; Camarasa, J.; Pubill, D. Methyllycaconitine prevents methamphetamine-induced effects in mouse striatum: Involvement of α7 nicotinic receptors. J. Pharm. Exp. Ther. 2005, 315, 658–667. [Google Scholar] [CrossRef] [Green Version]
- Thomsen, M.S.; Mikkelsen, J.D. The α7 nicotinic acetylcholine receptor ligands methyllycaconitine, NS6740 and GTS-21 reduce lipopolysaccharide-induced TNF-α release from microglia. J. Neuroimmunol. 2012, 251, 65–72. [Google Scholar] [CrossRef]
- Zheng, X.; Xie, Z.; Zhu, Z.; Liu, Z.; Wang, Y.; Wei, L.; Yang, H.; Yang, H.; Liu, Y.; Bi, J. Methyllycaconitine alleviates amyloid-β peptides-induced cytotoxicity in SH-SY5Y cells. PLoS ONE 2014, 9, e111536. [Google Scholar] [CrossRef] [Green Version]
- Burke, D.A.; Heshmati, P.; Kholdebarin, E.; Levin, E.D. Decreasing nicotinic receptor activity and the spatial learning impairment caused by the NMDA glutamate antagonist dizocilpine in rats. Eur. J. Pharm. 2014, 741, 132–139. [Google Scholar] [CrossRef] [Green Version]
- Van Goethem, N.P.; Paes, D.; Puzzo, D.; Fedele, E.; Rebosio, C.; Gulisano, W.; Palmeri, A.; Wennogle, L.P.; Peng, Y.; Bertrand, D. Antagonizing α7 nicotinic receptors with methyllycaconitine (MLA) potentiates receptor activity and memory acquisition. Cell Signal. 2019, 62, 109338. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C. NLRP3 is activated in Alzheimer´ s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674. [Google Scholar] [CrossRef]
- Tang, Y.; Le, W. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat. Immunol. 2008, 9, 857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baufeld, C.; Osterloh, A.; Prokop, S.; Miller, K.R.; Heppner, F.L. High-fat diet-induced brain region-specific phenotypic spectrum of CNS resident microglia. Acta Neuropathol. 2016, 132, 361–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Xu, Y.; Wang, Y.; Wang, Y.; He, L.; Jiang, Z.; Huang, Z.; Liao, H.; Li, J.; Saavedra, J.M. Telmisartan prevention of LPS-induced microglia activation involves M2 microglia polarization via CaMKKβ-dependent AMPK activation. Brain Behav. Immun. 2015, 50, 298–313. [Google Scholar] [CrossRef]
- Zhao, J.; Bi, W.; Xiao, S.; Lan, X.; Cheng, X.; Zhang, J.; Lu, D.; Wei, W.; Wang, Y.; Li, H. Neuroinflammation induced by lipopolysaccharide causes cognitive impairment in mice. Sci. Rep. 2019, 9, 5790. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Qian, X.; Teng, H.; Ding, Y.; Zhang, L. Association of interleukin-4 genetic polymorphisms with sporadic Alzheimer’s disease in Chinese Han population. Neurosci. Lett. 2014, 563, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, E.; Kawahara, K.; Kajizono, M.; Sawada, M.; Nakayama, H. IL-4-induced selective clearance of oligomeric β-amyloid peptide1–42 by rat primary type 2 microglia. J. Immunol. 2008, 181, 6503–6513. [Google Scholar] [CrossRef] [Green Version]
- Lyons, A.; Griffin, R.J.; Costelloe, C.E.; Clarke, R.M.; Lynch, M.A. IL-4 attenuates the neuroinflammation induced by amyloid-β in vivo and in vitro. J. Neurochem. 2007, 101, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Kiyota, T.; Okuyama, S.; Swan, R.J.; Jacobsen, M.T.; Gendelman, H.E.; Ikezu, T. CNS expression of anti-inflammatory cytokine interleukin-4 attenuates Alzheimer’s disease-like pathogenesis in APP+ PS1 bigenic mice. FASEB J. 2010, 24, 3093–3102. [Google Scholar] [CrossRef] [Green Version]
- Kummer, M.P.; Hermes, M.; Delekarte, A.; Hammerschmidt, T.; Kumar, S.; Terwel, D.; Walter, J.; Pape, H.-C.; König, S.; Roeber, S. Nitration of tyrosine 10 critically enhances amyloid β aggregation and plaque formation. Neuron 2011, 71, 833–844. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Lippi, G.; Carlson, D.M.; Berg, D.K. Activation of α7-containing nicotinic receptors on astrocytes triggers AMPA receptor recruitment to glutamatergic synapses. J. Neurochem. 2013, 127, 632–643. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Holtzman, D.M. Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat. Rev. Immunol. 2018, 18, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Moser, V.A.; Uchoa, M.F.; Pike, C.J. TLR4 inhibitor TAK-242 attenuates the adverse neural effects of diet-induced obesity. J. Neuroinflammation 2018, 15, 306. [Google Scholar] [CrossRef] [PubMed]
- Eren, E.; Özören, N. The NLRP3 inflammasome: A new player in neurological diseases. Turk. J. Biol. 2019, 43, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Thawkar, B.S.; Kaur, G. Inhibitors of NF-κB and P2X7/NLRP3/Caspase 1 pathway in microglia: Novel therapeutic opportunities in neuroinflammation induced early-stage Alzheimer’s disease. J. Neuroimmunol. 2018, 326, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Hwang, I.; Yang, J.; Hong, S.; Ju Lee, E.; Lee, S.H.; Fernandes-Alnemri, T.; Alnemri, E.S.; Yu, J.W. Non-transcriptional regulation of NLRP3 inflammasome signaling by IL-4. Immunol. Cell Biol. 2015, 93, 591–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Walle, L.V.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S. Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479, 117. [Google Scholar] [CrossRef] [PubMed]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef]
- Hagar, J.A.; Powell, D.A.; Aachoui, Y.; Ernst, R.K.; Miao, E.A. Cytoplasmic LPS activates caspase-11: Implications in TLR4-independent endotoxic shock. Science 2013, 341, 1250–1253. [Google Scholar] [CrossRef] [Green Version]
- Wada, N.; Yamada, H.; Motoyama, S.; Saburi, M.; Sugimoto, T.; Kubota, H.; Miyawaki, D.; Wakana, N.; Kami, D.; Ogata, T. Maternal high-fat diet exaggerates diet-induced insulin resistance in adult offspring by enhancing inflammasome activation through noncanonical pathway of caspase-11. Mol. Metab. 2020, 37, 100988. [Google Scholar] [CrossRef]
- Mrak, R.E.; Griffin, W.S.T. Interleukin-1, neuroinflammation, and Alzheimer’s disease. Neurobiol. Aging 2001, 22, 903–908. [Google Scholar] [CrossRef]
- Sciacca, F.; Ferri, C.; Licastro, F.; Veglia, F.; Biunno, I.; Gavazzi, A.; Calabrese, E.; Boneschi, F.M.; Sorbi, S.; Mariani, C. Interleukin-1B polymorphism is associated with age at onset of Alzheimer’s disease. Neurobiol. Aging 2003, 24, 927–931. [Google Scholar] [CrossRef]
- Bossù, P.; Ciaramella, A.; Salani, F.; Vanni, D.; Palladino, I.; Caltagirone, C.; Scapigliati, G. Interleukin-18, from neuroinflammation to Alzheimer’s disease. Curr. Pharm. Des. 2010, 16, 4213–4224. [Google Scholar] [CrossRef]
- Swaroop, S.; Mahadevan, A.; Shankar, S.K.; Adlakha, Y.K.; Basu, A. HSP60 critically regulates endogenous IL-1β production in activated microglia by stimulating NLRP3 inflammasome pathway. J. Neuroinflamm. 2018, 15, 177. [Google Scholar] [CrossRef]
- Sheng, J.G.; Ito, K.; Skinner, R.D.; Mrak, R.E.; Rovnaghi, C.R.; Van Eldik, L.J.; Griffin, W.S.T. In vivo and in vitro evidence supporting a role for the inflammatory cytokine interleukin-1 as a driving force in Alzheimer pathogenesis. Neurobiol. Aging 1996, 17, 761–766. [Google Scholar] [CrossRef]
- Sutinen, E.M.; Pirttiläˆ, T.; Anderson, G.; Salminen, A.; Ojala, J.O. Pro-inflammatory interleukin-18 increases Alzheimer’s disease-associated amyloid-β production in human neuron-like cells. J. Neuroinflamm. 2012, 9, 199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorgensen, I.; Miao, E.A. Pyroptotic cell death defends against intracellular pathogens. Immunol. Rev. 2015, 265, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Zhao, G. Pyroptosis and neurological diseases. Apoptosis 2014, 10, 12. [Google Scholar]
- Council, N.R. Guide for the Care and Use of Laboratory Animals; National Academies Press: Washington, DC, USA, 2011. [Google Scholar]
- Zhang, L.; Chen, C.; Mak, M.S.; Lu, J.; Wu, Z.; Chen, Q.; Han, Y.; Li, Y.; Pi, R. Advance of sporadic Alzheimer’s disease animal models. Med. Res. Rev. 2020, 40, 431–458. [Google Scholar] [CrossRef]
- Schaalan, M.; El-Abhar, H.S.; Barakat, M.; El-Denshary, E.S. Westernized-like-diet-fed rats: Effect on glucose homeostasis, lipid profile, and adipocyte hormones and their modulation by rosiglitazone and glimepiride. J. Diabetes Complicat. 2009, 23, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Matthews, D.; Hosker, J.; Rudenski, A.; Naylor, B.; Treacher, D.; Turner, R. Homeostasis model assessment: Insulin resistance and β-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985, 28, 412–419. [Google Scholar] [CrossRef] [Green Version]
- Wazea, S.A.; Wadie, W.; Bahgat, A.K.; El-Abhar, H.S. Galantamine anti-colitic effect: Role of alpha-7 nicotinic acetylcholine receptor in modulating Jak/STAT3, NF-κB/HMGB1/RAGE and p-AKT/Bcl-2 pathways. Sci. Rep. 2018, 8, 5110. [Google Scholar] [CrossRef] [Green Version]
- Antunes, M.; Biala, G. The novel object recognition memory: Neurobiology, test procedure, and its modifications. Cogn. Process. 2012, 13, 93–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammond, R.S.; Tull, L.E.; Stackman, R.W. On the delay-dependent involvement of the hippocampus in object recognition memory. Neurobiol. Learn. Mem. 2004, 82, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Ennaceur, A.; Delacour, J. A new one-trial test for neurobiological studies of memory in rats. 1: Behavioral data. Behav. Brain Res. 1988, 31, 47–59. [Google Scholar] [CrossRef]
- Nunez, J. Morris water maze experiment. J. Vis. Exp. 2008, 19, e897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suvarna, S.; Layton, C. Bancroft’s Theory and Practice of Histological Techniques; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- El-Shiekh, R.A.; Ashour, R.M.; Abd El-Haleim, E.A.; Ahmed, K.A.; Abdel-Sattar, E. Hibiscus sabdariffa L.: A potent natural neuroprotective agent for the prevention of streptozotocin-induced Alzheimer’s disease in mice. Biomed. Pharmacother. 2020, 128, 110303. [Google Scholar] [CrossRef] [PubMed]
- El-Nasr, N.M.A.; Saleh, D.O.; Mahmoud, S.S.; Nofal, S.M.; Abdelsalam, R.M.; Safar, M.M.; El-Abhar, H.S. Olmesartan attenuates type 2 diabetes-associated liver injury: Cross-talk of AGE/RAGE/JNK, STAT3/SCOS3 and RAS signaling pathways. Eur. J. Pharm. 2020, 874, 173010. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohamed, R.A.; Abdallah, D.M.; El-brairy, A.I.; Ahmed, K.A.; El-Abhar, H.S. Palonosetron/Methyllycaconitine Deactivate Hippocampal Microglia 1, Inflammasome Assembly and Pyroptosis to Enhance Cognition in a Novel Model of Neuroinflammation. Molecules 2021, 26, 5068. https://doi.org/10.3390/molecules26165068
Mohamed RA, Abdallah DM, El-brairy AI, Ahmed KA, El-Abhar HS. Palonosetron/Methyllycaconitine Deactivate Hippocampal Microglia 1, Inflammasome Assembly and Pyroptosis to Enhance Cognition in a Novel Model of Neuroinflammation. Molecules. 2021; 26(16):5068. https://doi.org/10.3390/molecules26165068
Chicago/Turabian StyleMohamed, Reem A., Dalaal M. Abdallah, Amany I. El-brairy, Kawkab A. Ahmed, and Hanan S. El-Abhar. 2021. "Palonosetron/Methyllycaconitine Deactivate Hippocampal Microglia 1, Inflammasome Assembly and Pyroptosis to Enhance Cognition in a Novel Model of Neuroinflammation" Molecules 26, no. 16: 5068. https://doi.org/10.3390/molecules26165068