
Photoregulation of PRMT-1 Using a Photolabile Non-Canonical Amino Acid

 , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. PRMT1 Expression
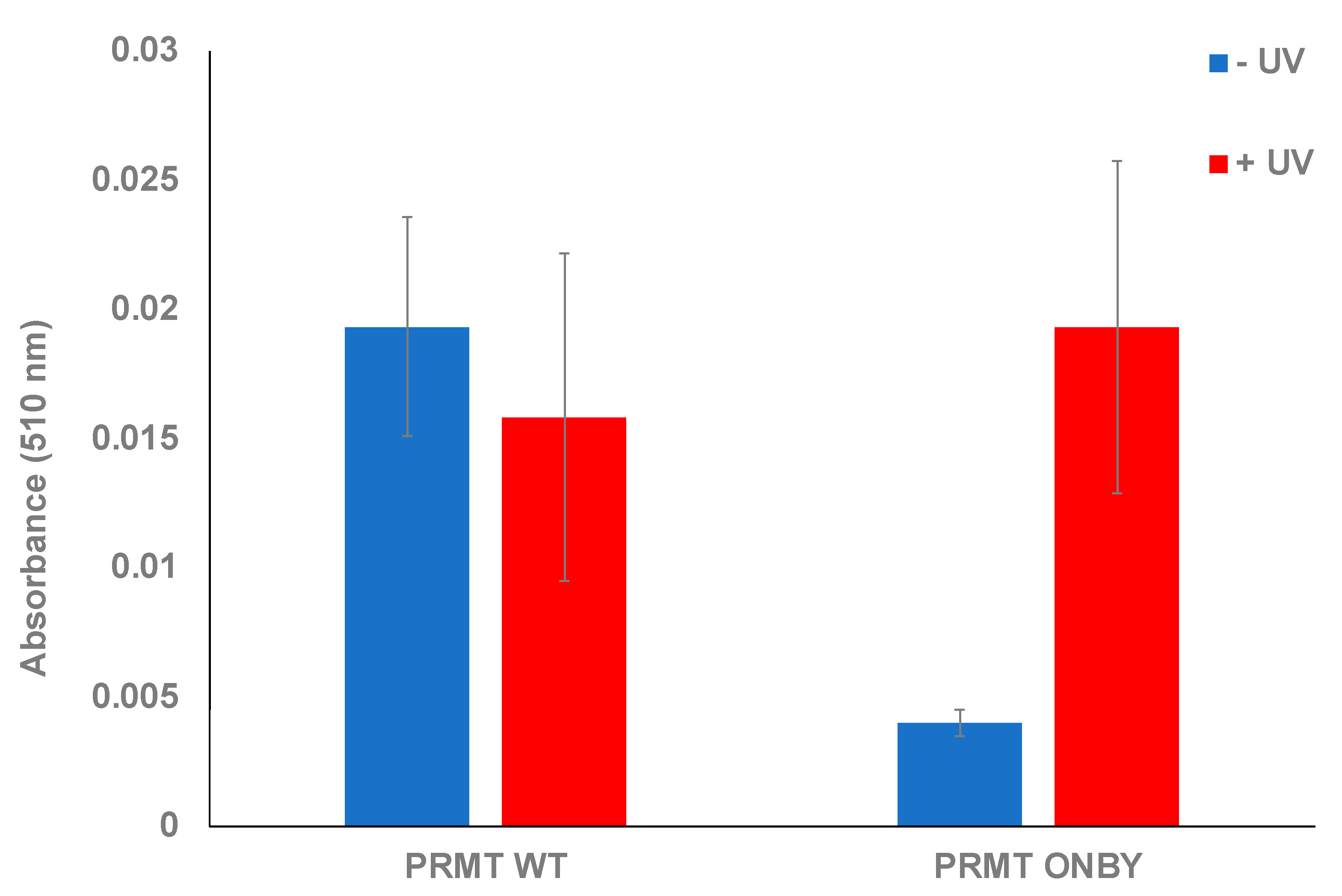
2.2. PRMT1 Activity Assay
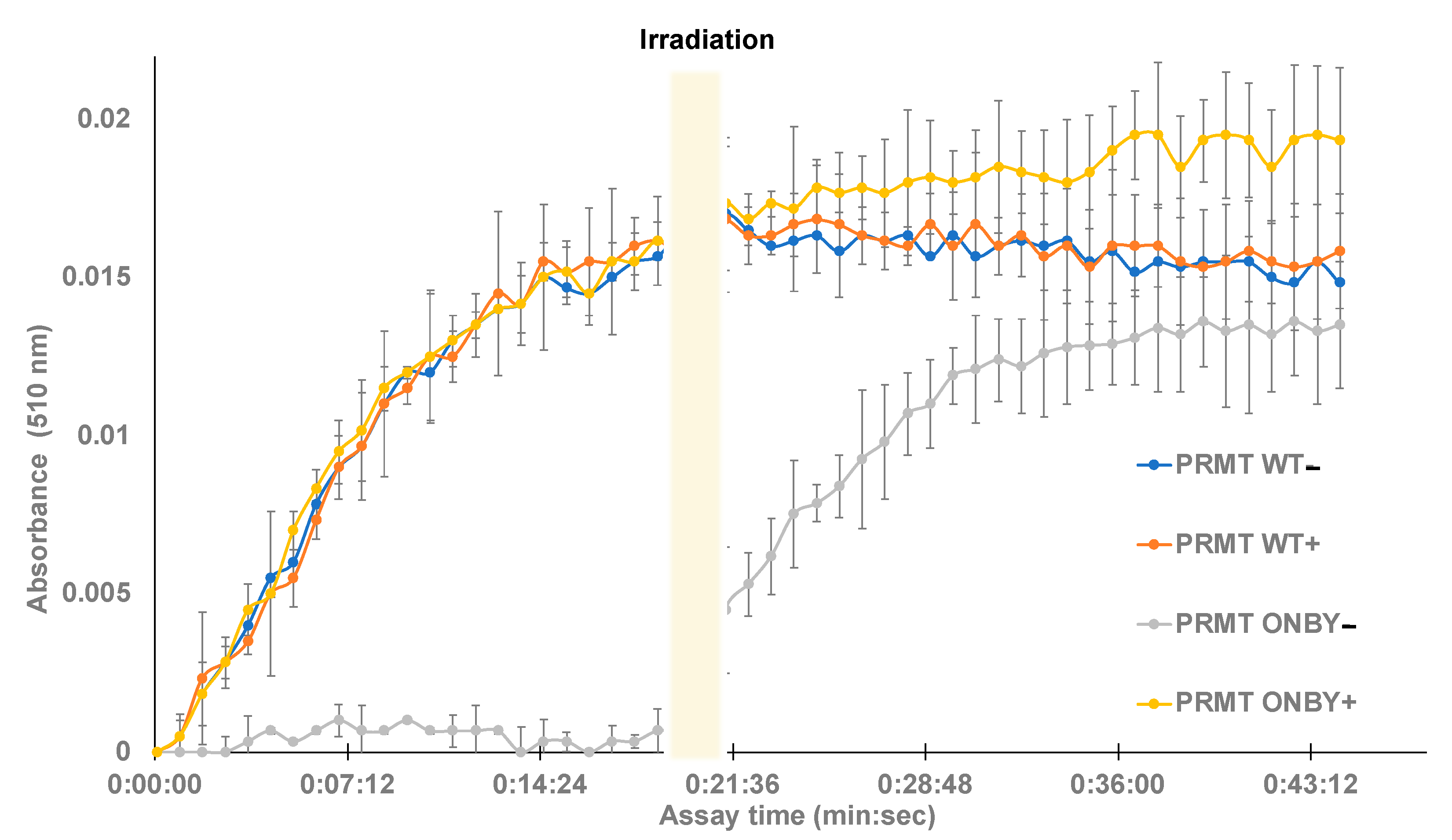
2.3. Temporal Photoregulation of PRMT1 Function
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Kouzarides, T. SnapShot: Histone-Modifying Enzymes. Cell 2007, 131, 822. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Blumenthal, R.M. Mammalian DNA Methyltransferases: A Structural Perspective. Structure 2008, 16, 341–350. [Google Scholar] [CrossRef] [Green Version]
- Schubert, H.L.; Blumenthal, R.M.; Cheng, X. Many Paths to Methyltransfer: A Chronicle of Convergence. Trends Biochem. Sci. 2003, 28, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Schubert, H.L.; Blumenthal, R.M.; Cheng, X. 1 Protein Methyltransferases: Their Distribution among the Five Structural Classes of AdoMet-Dependent Methyltransferases. Enzymes 2006, 24, 3–28. [Google Scholar] [CrossRef] [PubMed]
- Bedford, M.T.; Clarke, S.G. Protein Arginine Methylation in Mammals: Who, What, and Why. Mol. Cell 2009, 33, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuhrmann, J.; Clancy, K.W.; Thompson, P.R. Chemical Biology of Protein Arginine Modifications in Epigenetic Regulation. Chem Rev. 2015, 115, 5413–5461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCabe, M.T.; Mohammad, H.P.; Barbash, O.; Kruger, R.G. Targeting Histone Methylation in Cancer. Cancer J. 2017, 23, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Wu, F.; Wu, J. Targeting Histone Methylation for Cancer Therapy: Enzymes, Inhibitors, Biological Activity and Perspectives. J. Hematol. Oncol. 2016, 9, 49. [Google Scholar] [CrossRef] [Green Version]
- Rust, H.L.; Subramanian, V.; West, G.M.; Young, D.D.; Schultz, P.G.; Thompson, P.R. Using Unnatural Amino Acid Mutagenesis to Probe the Regulation of PRMT1. ACS Chem. Biol. 2014, 9, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Bedford, M.T.; Richard, S. Arginine Methylation an Emerging Regulator of Protein Function. Mol. Cell 2005, 18, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Frankel, A.; Cook, R.J.; Kim, S.; Paik, W.K.; Williams, K.R.; Clarke, S.; Herschman, H.R. PRMT1 Is the Predominant Type I Protein Arginine Methyltransferase in Mammalian Cells. J. Biol. Chem. 2000, 275, 7723–7730. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Cheng, X. Structure of the Predominant Protein Arginine Methyltransferase PRMT1 and Analysis of its Binding to Substrate Peptides. Structure 2003, 11, 509–520. [Google Scholar] [CrossRef] [Green Version]
- Daigle, S.R.; Olhava, E.J.; Therkelsen, C.A.; Majer, C.R.; Sneeringer, C.J.; Song, J.; Johnston, L.D.; Scott, M.P.; Smith, J.J.; Xiao, Y.; et al. Selective Killing of Mixed Lineage Leukemia Cells by a Potent Small-Molecule DOT1L Inhibitor. Cancer Cell 2011, 20, 53–65. [Google Scholar] [CrossRef] [Green Version]
- Copeland, R.A.; Moyer, M.P.; Richon, V.M. Targeting Genetic Alterations in Protein Methyltransferases for Personalized Cancer Therapeutics. Oncogene 2013, 32, 939–946. [Google Scholar] [CrossRef] [Green Version]
- Saloura, V.; Vougiouklakis, T.; Sievers, C.; Burkitt, K.; Nakamura, Y.; Hager, G.; van Waes, C. The Role of Protein Methyltransferases as Potential Novel Therapeutic Targets in Squamous Cell Carcinoma of the Head and Neck. Oral Oncol. 2018, 81, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Yost, J.M.; Korboukh, I.; Liu, F.; Gao, C.; Jin, J. Targets in Epigenetics: Inhibiting the Methyl Writers of the Histone Code. Curr. Chem. Genom. 2011, 5, 72–84. [Google Scholar] [CrossRef] [Green Version]
- Arbely, E.; Torres-Kolbus, J.; Deiters, A.; Chin, J.W. Photocontrol of Tyrosine Phosphorylation in Mammalian Cells via Genetic Encoding of Photocaged Tyrosine. J. Am. Chem. Soc. 2012, 134, 11912–11915. [Google Scholar] [CrossRef]
- Deiters, A. Light Activation as a Method of Regulating and Studying Gene Expression. Curr. Opin. Chem. Biol. 2009, 13, 678–686. [Google Scholar] [CrossRef] [Green Version]
- Edwards, W.; Young, D.; Deiters, A. Light-Activated Cre Recombinase as a Tool for the Spatial and Temporal Control of Gene Function in Mammalian Cells. ACS Chem. Biol. 2009, 4, 441–445. [Google Scholar] [CrossRef]
- Luo, J.; Uprety, R.; Naro, Y.; Chou, C.; Nguyen, D.P.; Chin, J.W.; Deiters, A. Genetically Encoded Optochemical Probes for Simultaneous Fluorescence Reporting and Light Activation of Protein Function with Two-Photon Excitation. J. Am. Chem. Soc. 2014, 136, 15551–15558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, D.; Deiters, A. Photochemical Control of Biological Processes. Org. Biomol. Chem. 2007, 5, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Courtney, T.M.; Deiters, A. Enzyme Allostery: Now Controllable by Light. Cell Chem. Biol. 2019, 26, 1481–1483. [Google Scholar] [CrossRef]
- Zhou, W.; Hankinson, C.P.; Deiters, A. Optical Control of Cellular ATP Levels with a Photocaged Adenylate Kinase. Chembiochem 2020, 21, 1832–1836. [Google Scholar] [CrossRef] [PubMed]
- Riggsbee, C.W.; Deiters, A. Recent Advances in the Photochemical Control of Protein Function. Trends Biotechnol. 2010, 28, 468–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padilla, M.S.; Farley, C.A.; Chatkewitz, L.E.; Young, D.D. Synthesis and Incorporation of a Caged Tyrosine Amino acid Possessing a Bioorthogonal Handle. Tetrahedron Lett. 2016, 57, 4709–4712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceo, L.M.; Koh, J.T. Photocaged DNA Provides New Levels of Transcription Control. Chembiochem 2012, 13, 511–513. [Google Scholar] [CrossRef] [PubMed]
- Koh, M.; Nasertorabi, F.; Han, G.W.; Stevens, R.C.; Schultz, P.G. Generation of an Orthogonal Protein-Protein Interface with a Noncanonical Amino Acid. J. Am. Chem Soc. 2017, 139, 5728–5731. [Google Scholar] [CrossRef]
- Wu, N.; Deiters, A.; Cropp, T.A.; King, D.; Schultz, P.G. A Genetically Encoded Photocaged Amino acid. J. Am. Chem Soc. 2004, 126, 14306–14307. [Google Scholar] [CrossRef]
- Lusic, H.; Young, D.; Lively, M.; Deiters, A. Photochemical DNA Activation. Org. Lett. 2007, 9, 1903–1906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, D.; Deiters, A. Light-Regulated RNA-Small Molecule Interactions. Chembiochem 2008, 9, 1225–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemke, E.A.; Summerer, D.; Geierstanger, B.H.; Brittain, S.M.; Schultz, P.G. Control of Protein Phosphorylation with a Genetically Encoded Photocaged Amino Acid. Nat. Chem. Biol. 2007, 3, 769–772. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.; Mahesh, M.; Elsasser, S.; Hancock, S.; Uttamapinant, C.; Chin, J. Genetic Encoding of Photocaged Cysteine Allows Photoactivation of TEV Protease in Live Mammalian Cells. J. Am. Chem. Soc. 2014, 136, 2240–2243. [Google Scholar] [CrossRef]
- Hauf, M.; Richter, F.; Schneider, T.; Faidt, T.; Martins, B.M.; Baumann, T.; Durkin, P.; Dobbek, H.; Jacobs, K.; Möglich, A.; et al. Photoactivatable Mussel-Based Underwater Adhesive Proteins by an Expanded Genetic Code. Chembiochem 2017. [Google Scholar] [CrossRef] [PubMed]
- Deiters, A.; Groff, D.; Ryu, Y.; Xie, J.; Schultz, P. A Genetically Encoded Photocaged Tyrosine. Angew. Chem. -Int. Ed. 2006, 45, 2728–2731. [Google Scholar] [CrossRef]
- Gautier, A.; Nguyen, D.; Lusic, H.; An, W.; Deiters, A.; Chin, J. Genetically Encoded Photocontrol of Protein Localization in Mammalian Cells. J. Am. Chem. Soc. 2010, 132, 4086. [Google Scholar] [CrossRef]
- Young, D.D.; Schultz, P.G. Playing with the Molecules of Life. ACS Chem. Biol. 2018, 13, 854–870. [Google Scholar] [CrossRef] [PubMed]
- Young, T.S.; Schultz, P.G. Beyond the Canonical 20 Amino Acids: Expanding the Genetic Lexicon. J. Biol. Chem. 2010, 285, 11039–11044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Schultz, P.; Kornberg, R.; Raetz, C.; Rothman, J.; Thorner, J. Adding New Chemistries to the Genetic Code. Annu. Rev. Biochem. 2010, 79, 413–444. [Google Scholar] [CrossRef] [Green Version]
- Niu, W.; Guo, J. Expanding the Chemistry of Fluorescent Protein Biosensors Through Genetic Incorporation of Unnatural Amino Acids. Mol. Biosyst. 2013, 9, 2961–2970. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
King, E.A.; Peairs, E.M.; Uthappa, D.M.; Villa, J.K.; Goff, C.M.; Burrow, N.K.; Deitch, R.T.; Martin, A.K.; Young, D.D. Photoregulation of PRMT-1 Using a Photolabile Non-Canonical Amino Acid. Molecules 2021, 26, 5072. https://doi.org/10.3390/molecules26165072
King EA, Peairs EM, Uthappa DM, Villa JK, Goff CM, Burrow NK, Deitch RT, Martin AK, Young DD. Photoregulation of PRMT-1 Using a Photolabile Non-Canonical Amino Acid. Molecules. 2021; 26(16):5072. https://doi.org/10.3390/molecules26165072
Chicago/Turabian StyleKing, Elizabeth A., Emily M. Peairs, Diya M. Uthappa, Jordan K. Villa, Cameron M. Goff, Naya K. Burrow, Rebecca T. Deitch, Anna K. Martin, and Douglas D. Young. 2021. "Photoregulation of PRMT-1 Using a Photolabile Non-Canonical Amino Acid" Molecules 26, no. 16: 5072. https://doi.org/10.3390/molecules26165072
APA StyleKing, E. A., Peairs, E. M., Uthappa, D. M., Villa, J. K., Goff, C. M., Burrow, N. K., Deitch, R. T., Martin, A. K., & Young, D. D. (2021). Photoregulation of PRMT-1 Using a Photolabile Non-Canonical Amino Acid. Molecules, 26(16), 5072. https://doi.org/10.3390/molecules26165072