
Synthetic Strategies of Pyrimidine-Based Scaffolds as Aurora Kinase and Polo-like Kinase Inhibitors

 ,
,  ,
,  and
and 
Abstract
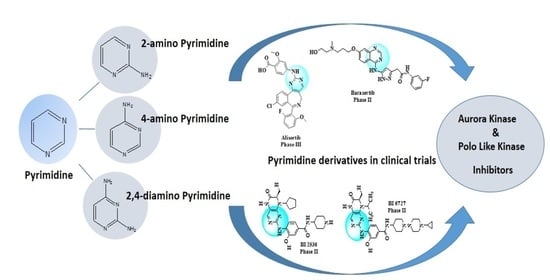
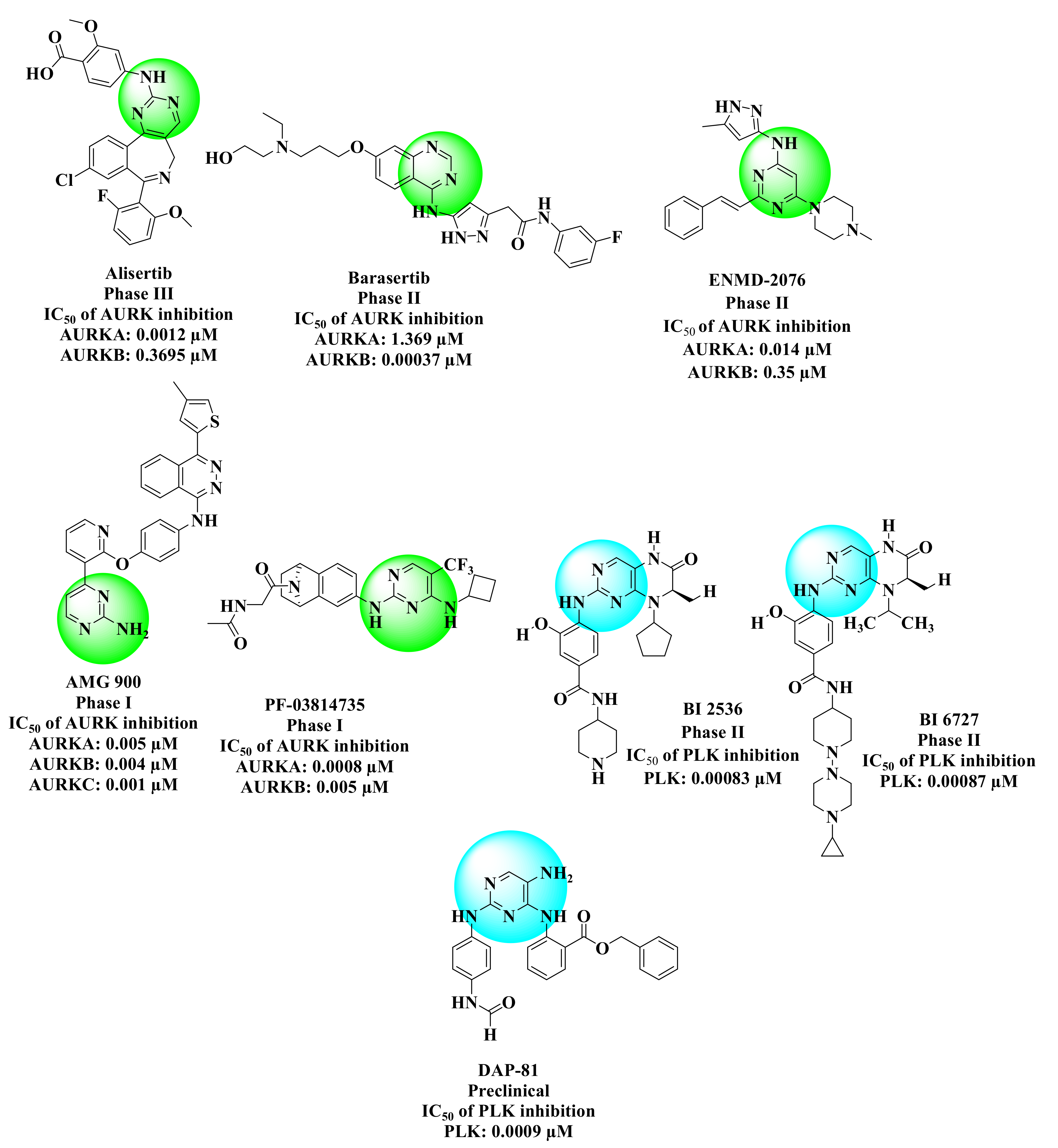
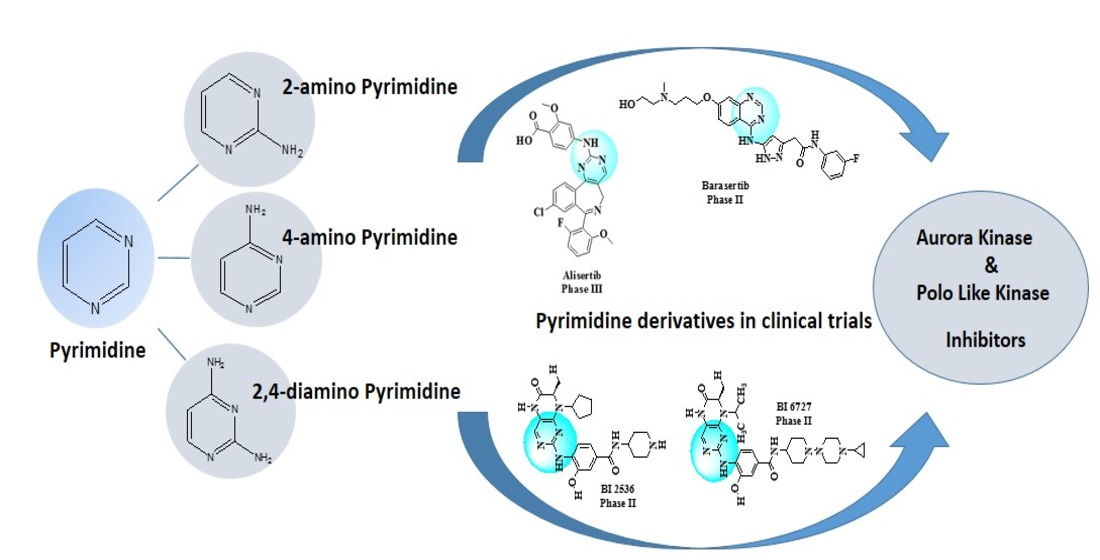
:
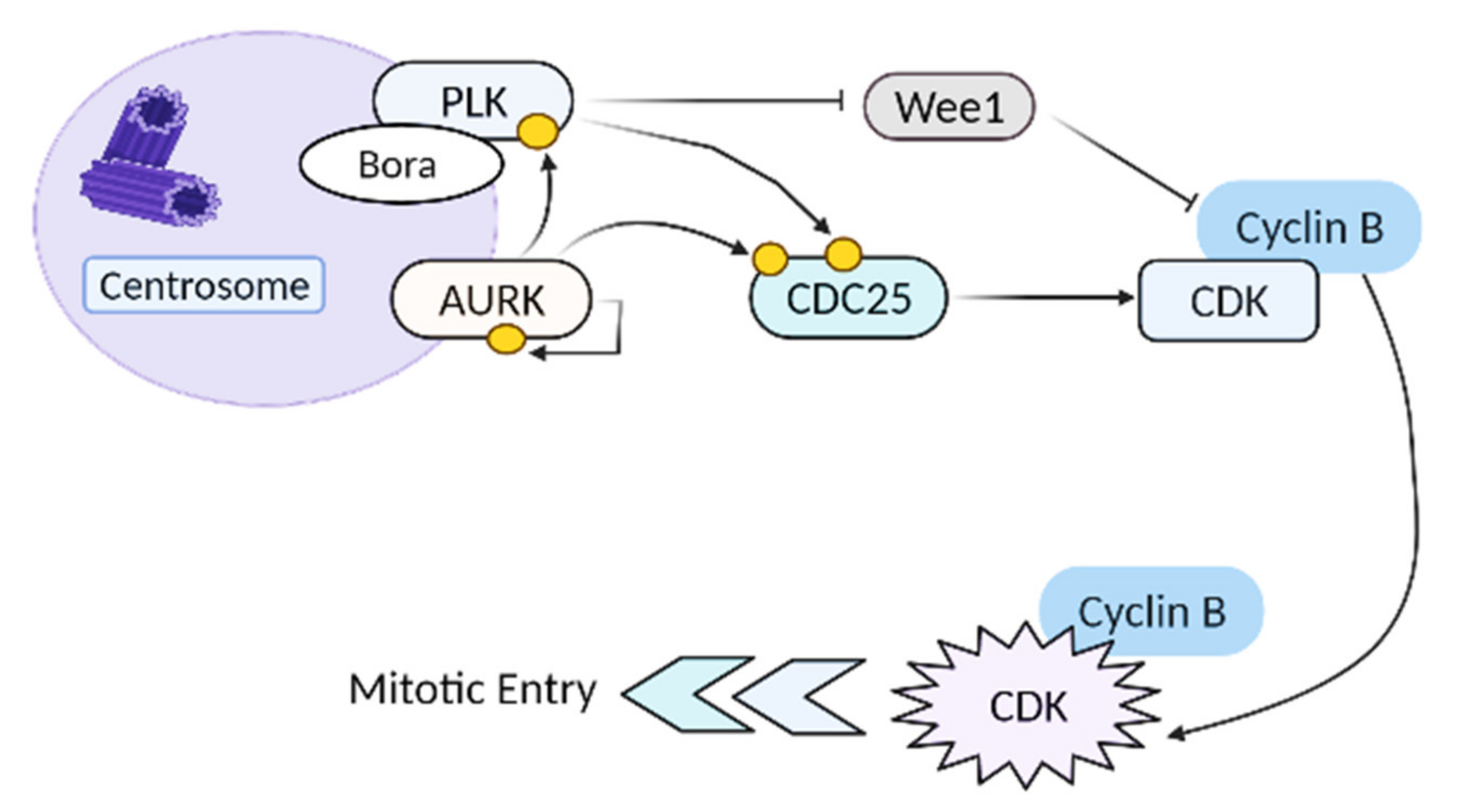
1. Introduction
2. Synthetic Strategies of Pyrimidines
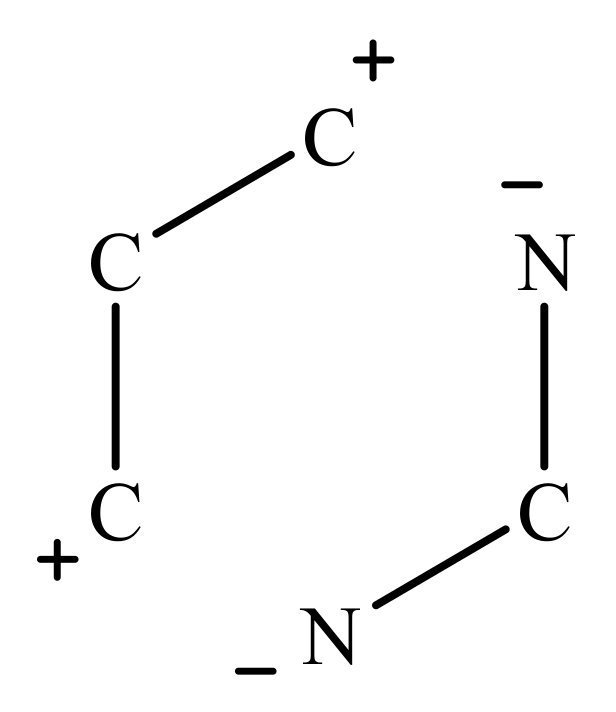
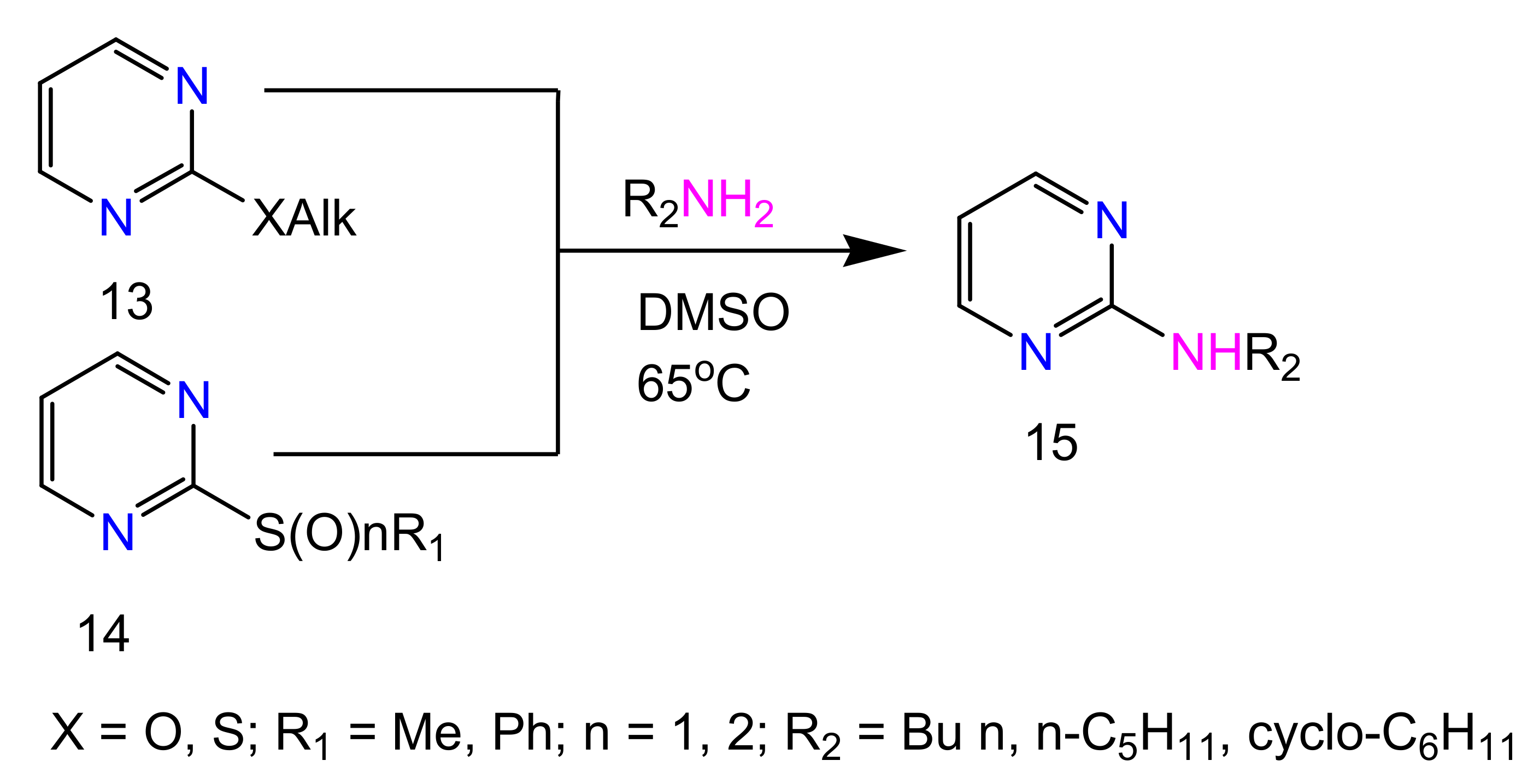
2.1. 2-Aminopyrimidines
2.2. Condensation Reactions
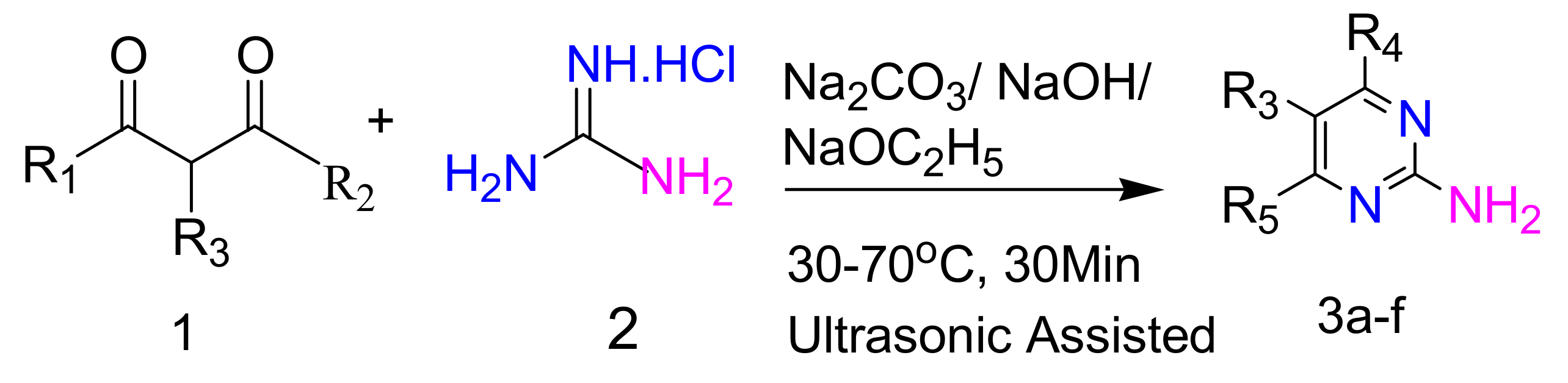
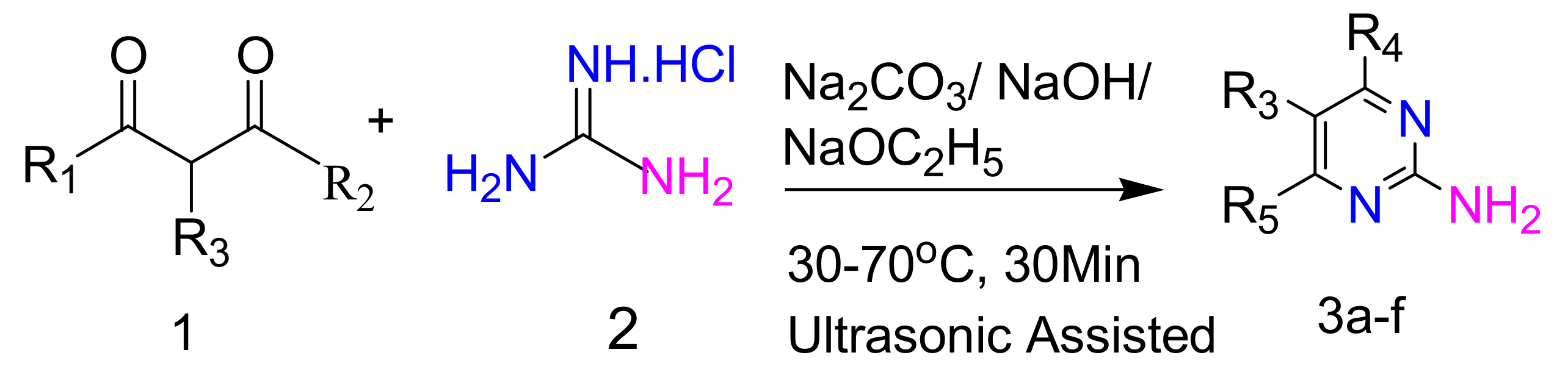
2.2.1. Synthesis of 2-Aminopyrimidines from Guanidine and β-Dicarbonyl Compounds

| Sr. No. | R1 | R2 | R3 | R4 | R5 |
|---|---|---|---|---|---|
| 3a | CH3 | CH3 | H | CH3 | CH3 |
| 3b | CH3 | OC2H5 | H | CH3 | OH |
| 3c | OC2H5 | OC2H5 | H | OH | OH |
| 3d | OC2H5 | OC2H5 | C2H5 | OH | OH |
| 3e | OC2H5 | OC2H5 | C4H9 | OH | OH |
| 3f | OC2H5 | OC2H5 |  | OH | OH |
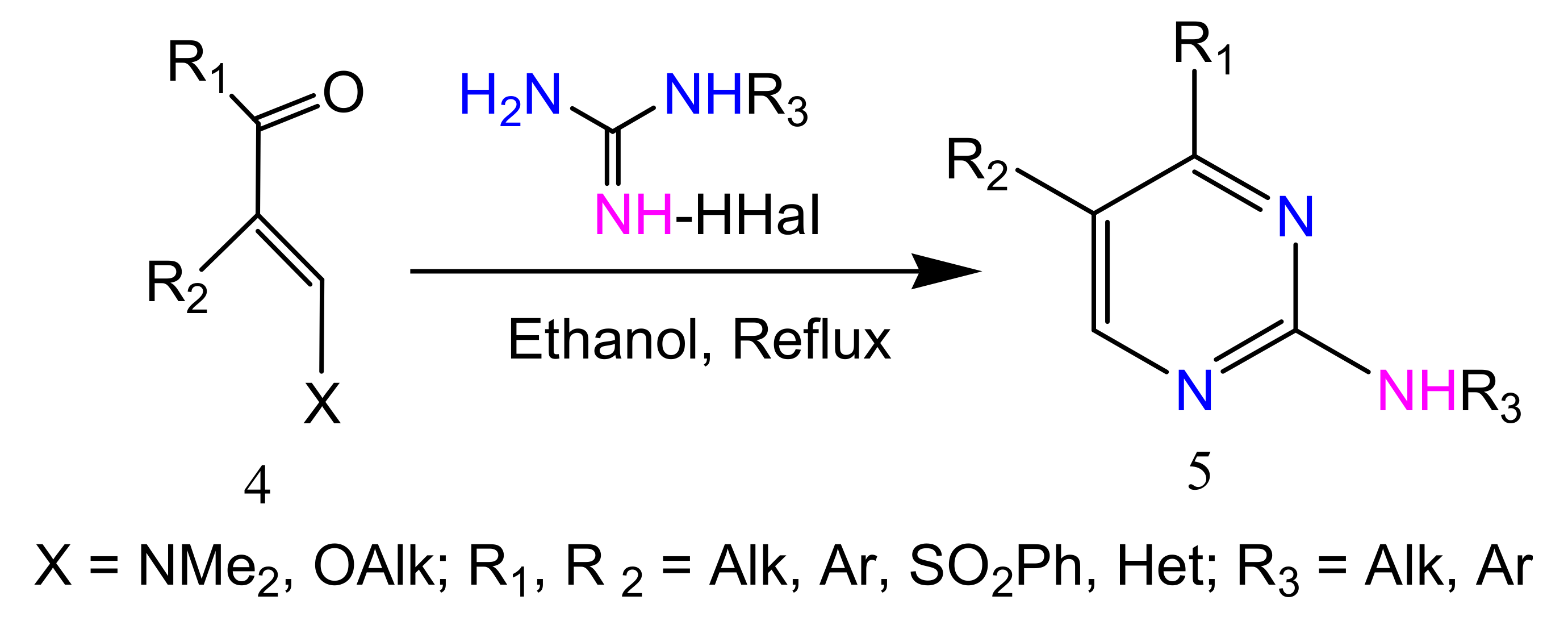
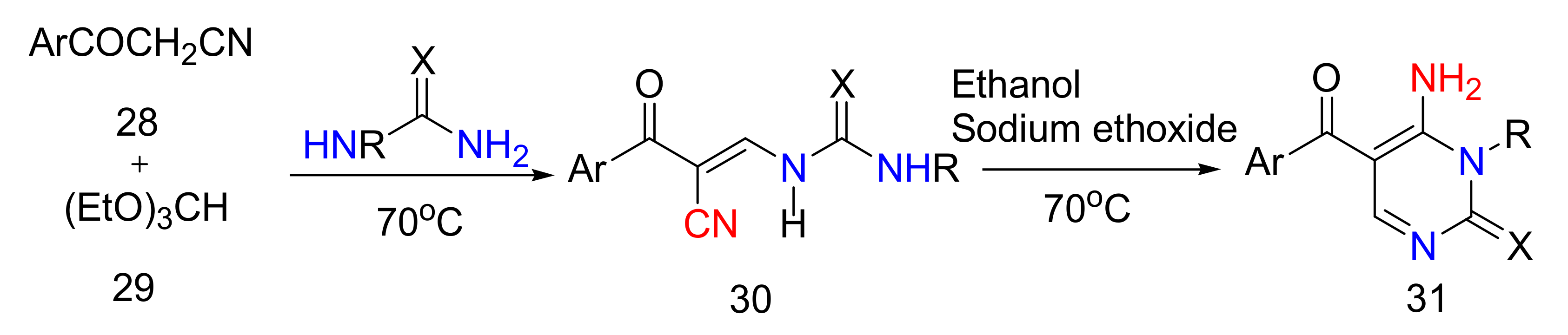
2.2.2. Synthesis of 2-Aminopyrimidines from α, β-Unsaturated Ketones
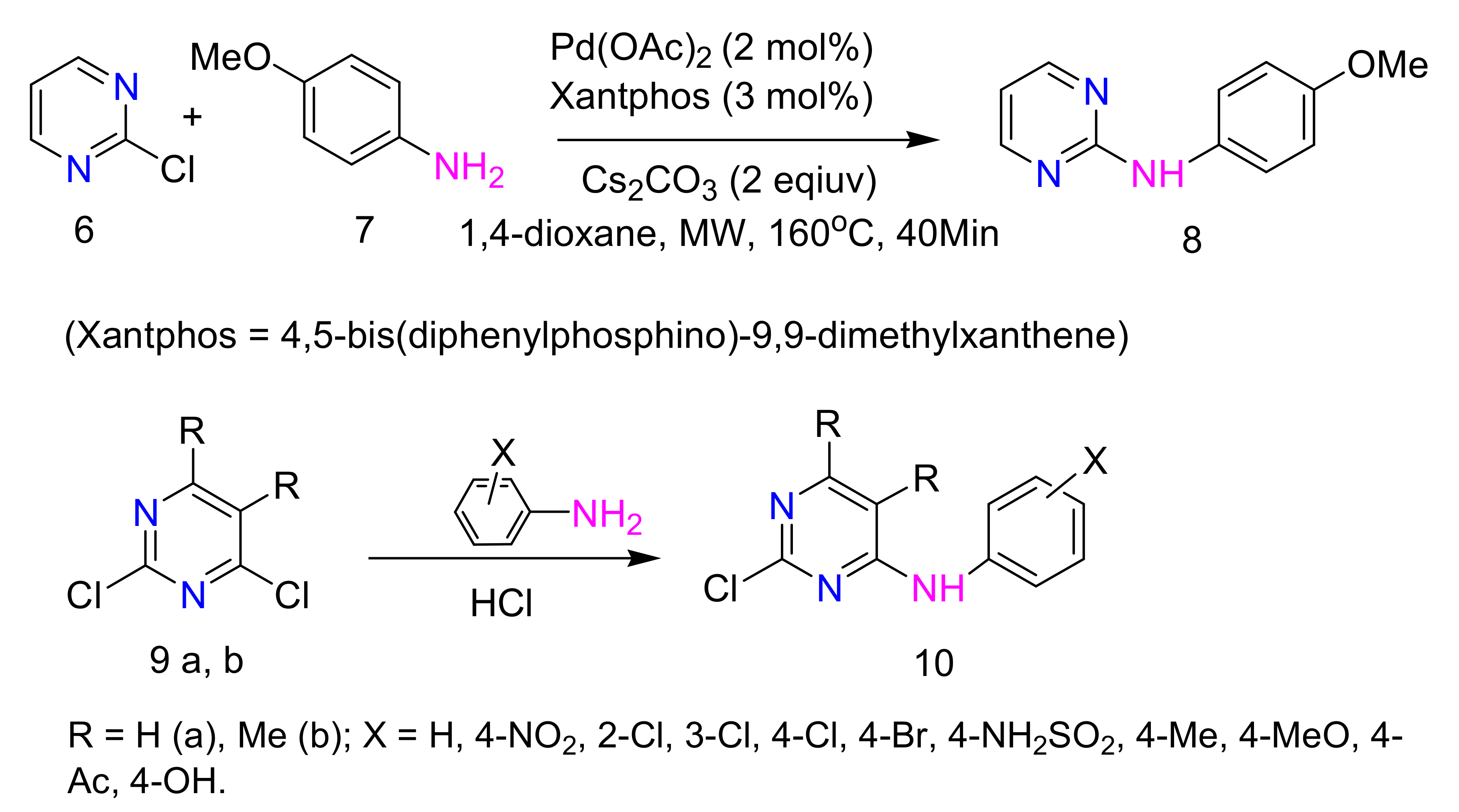
2.3. Substitution Reaction
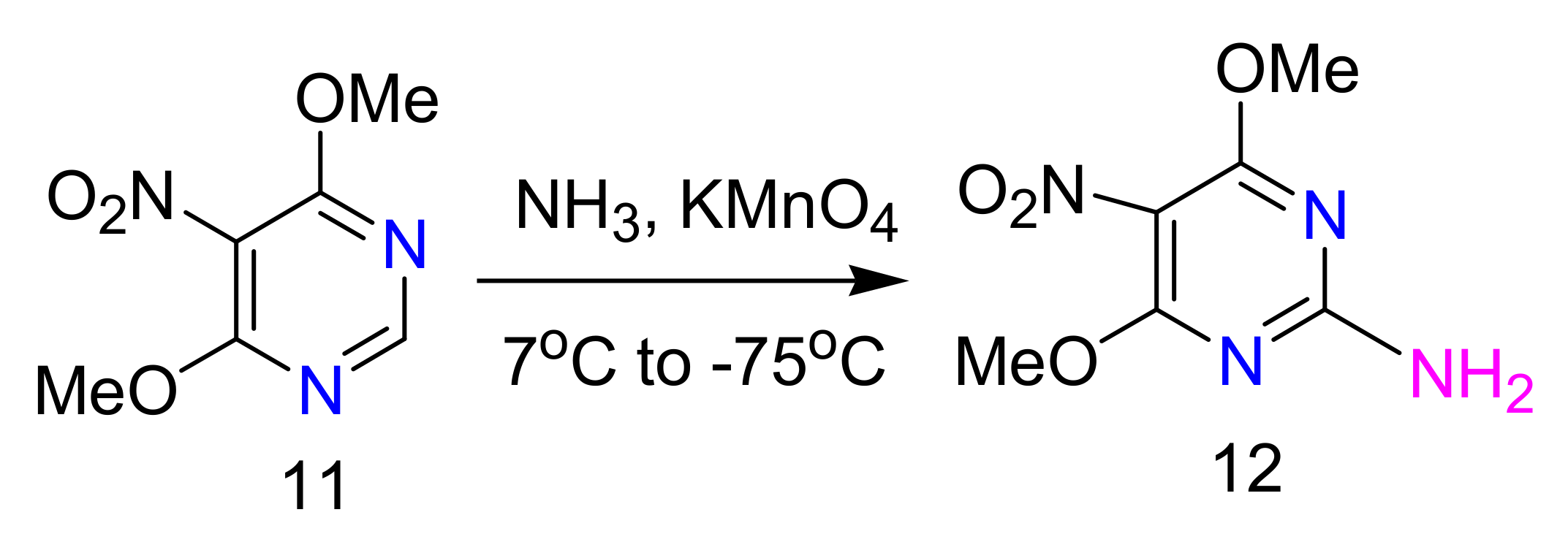
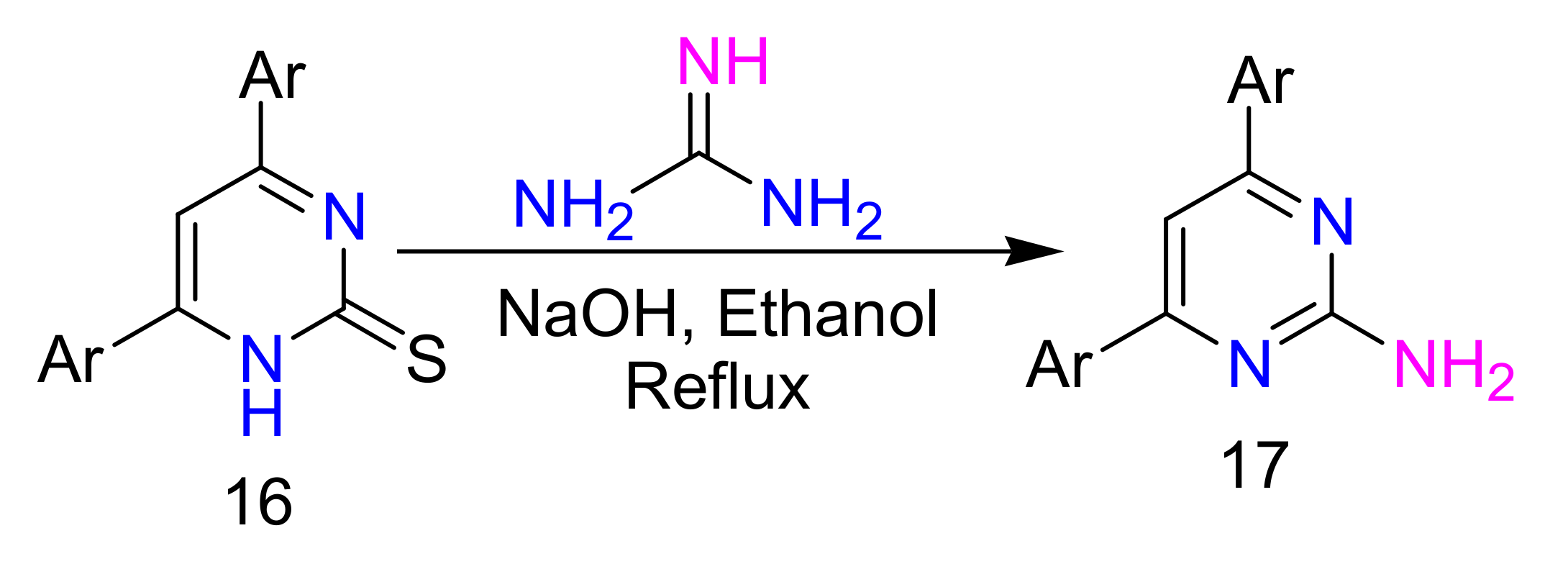
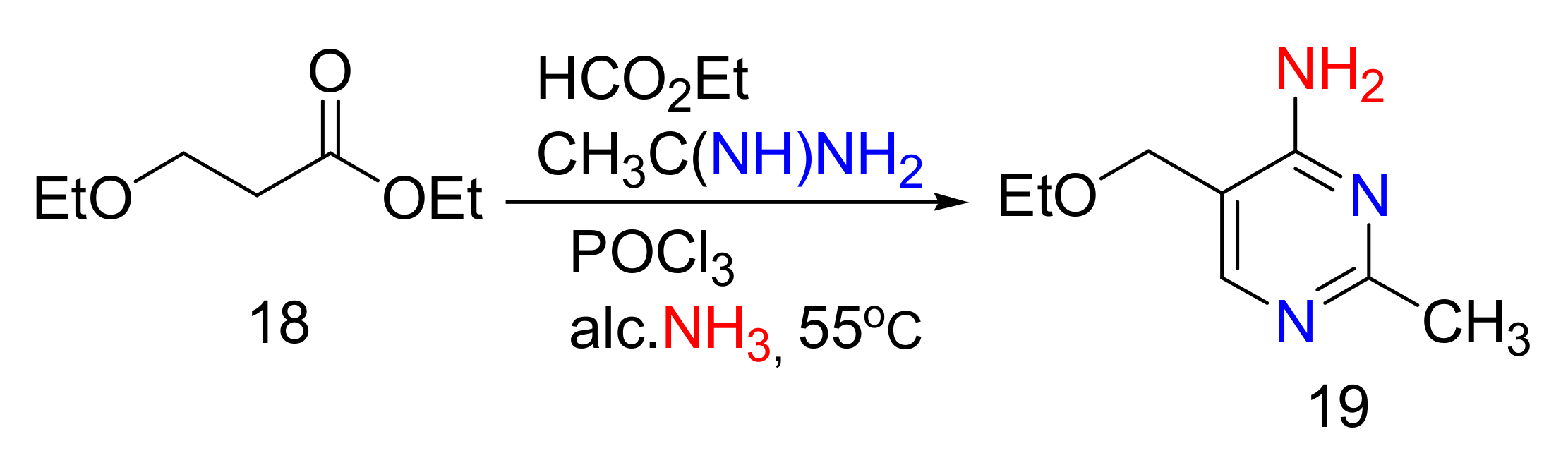
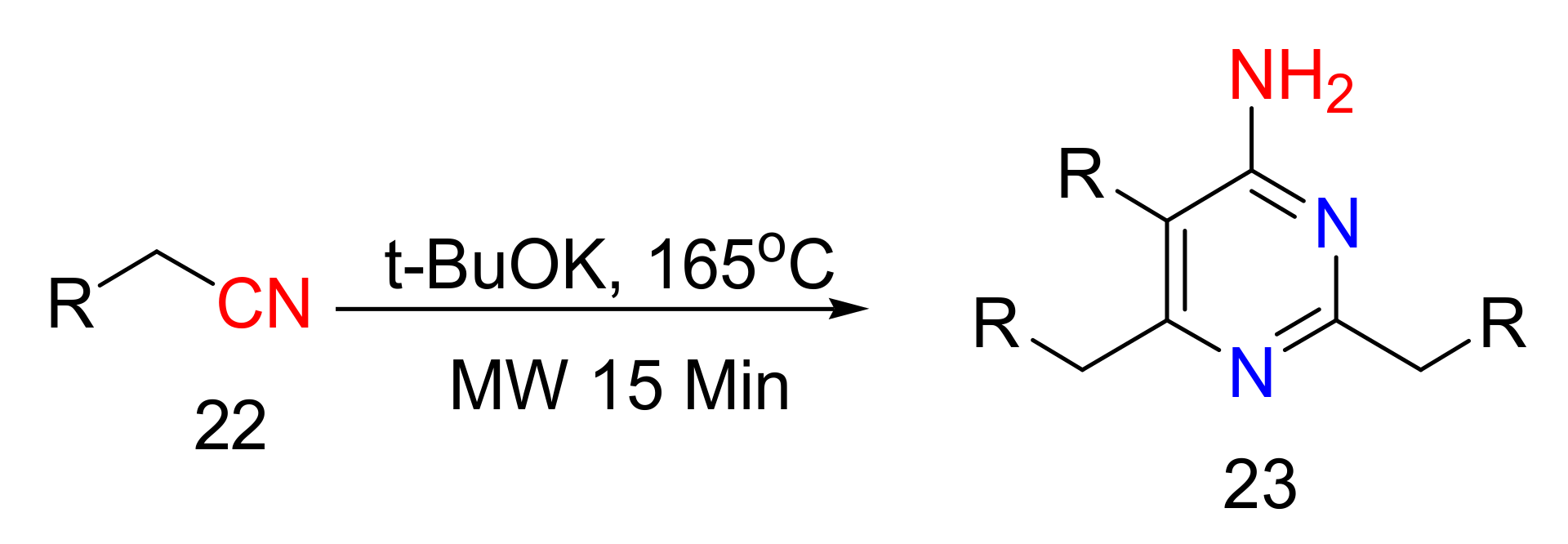
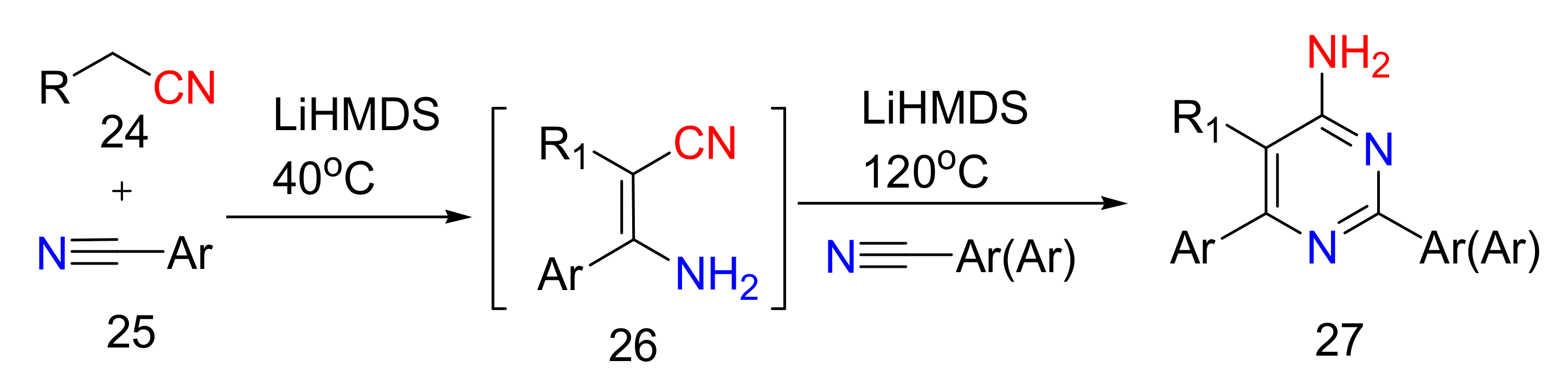
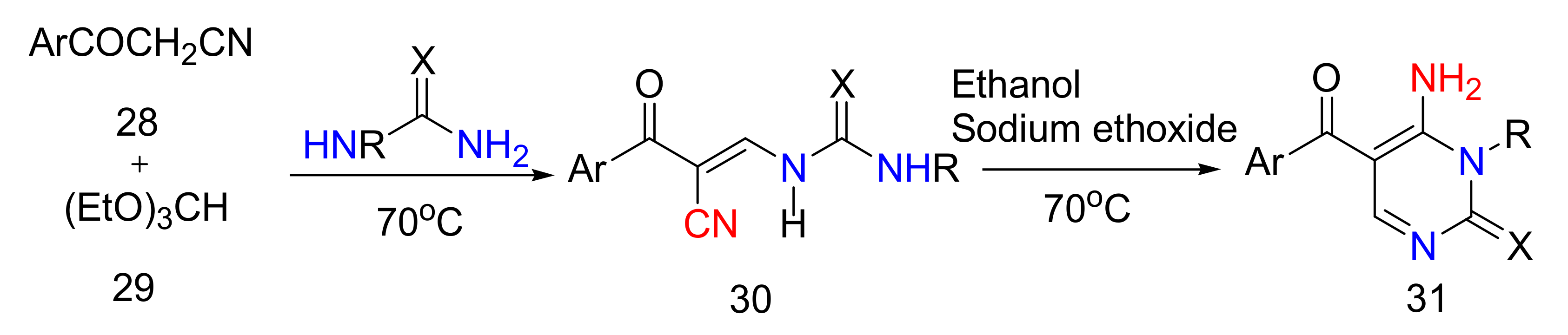
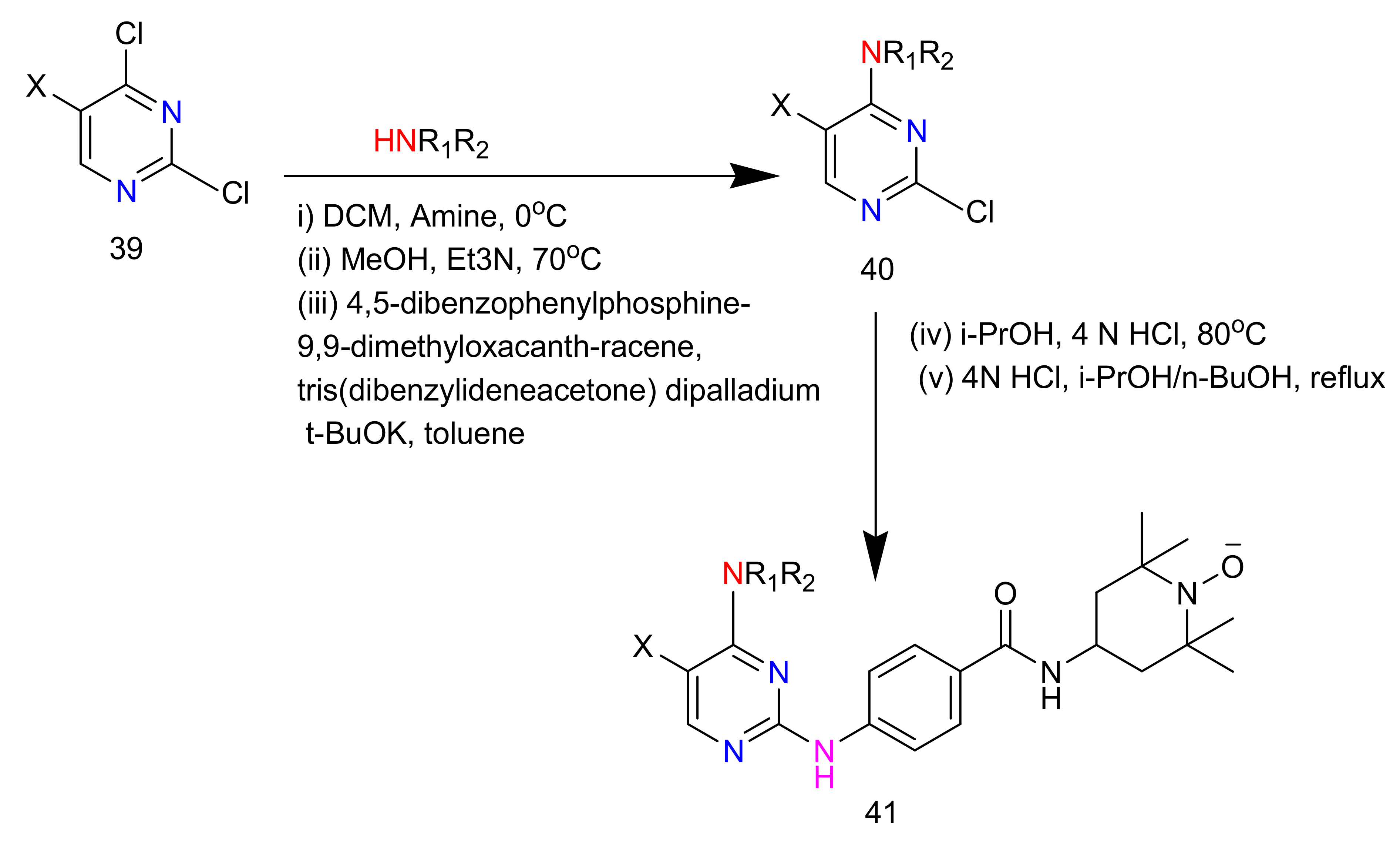
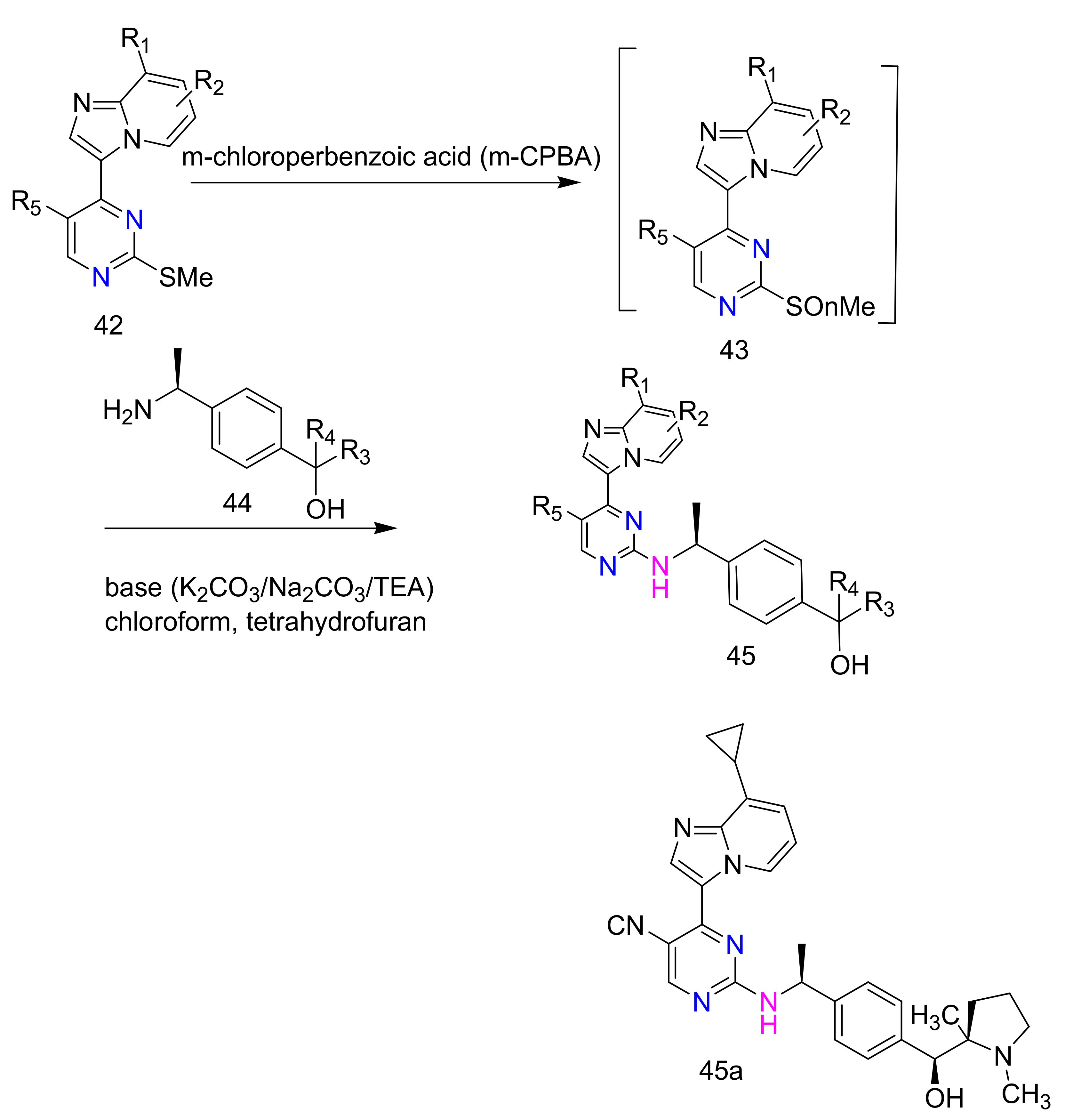
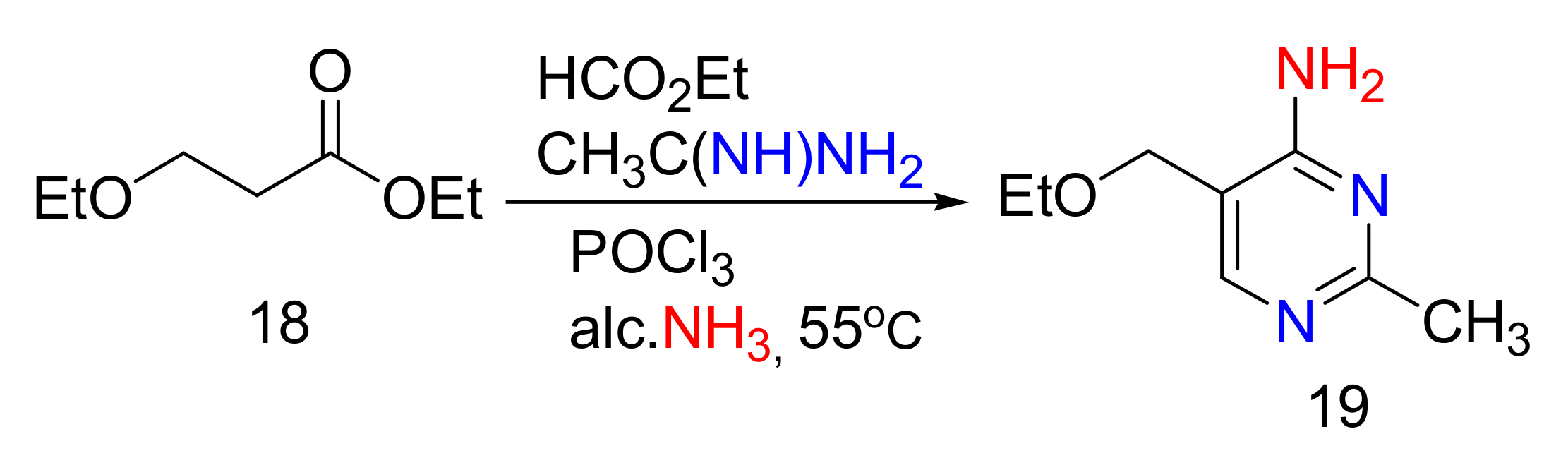
2.4. 4-Aminopyrimidines
2.5. Synthesis of 4-Aminopyrimidines
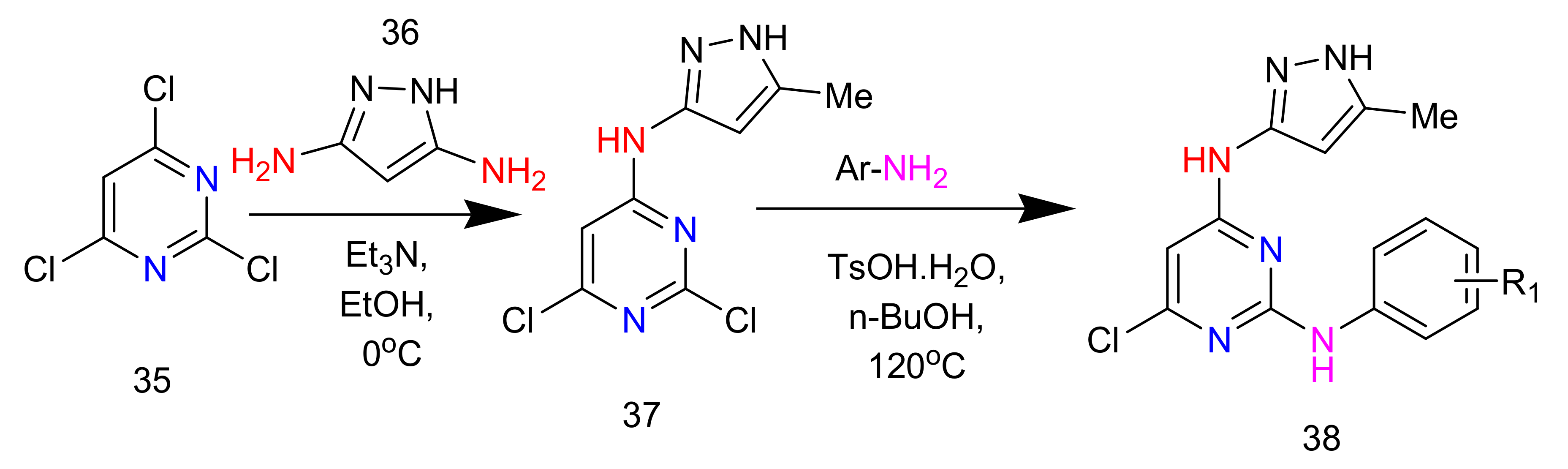
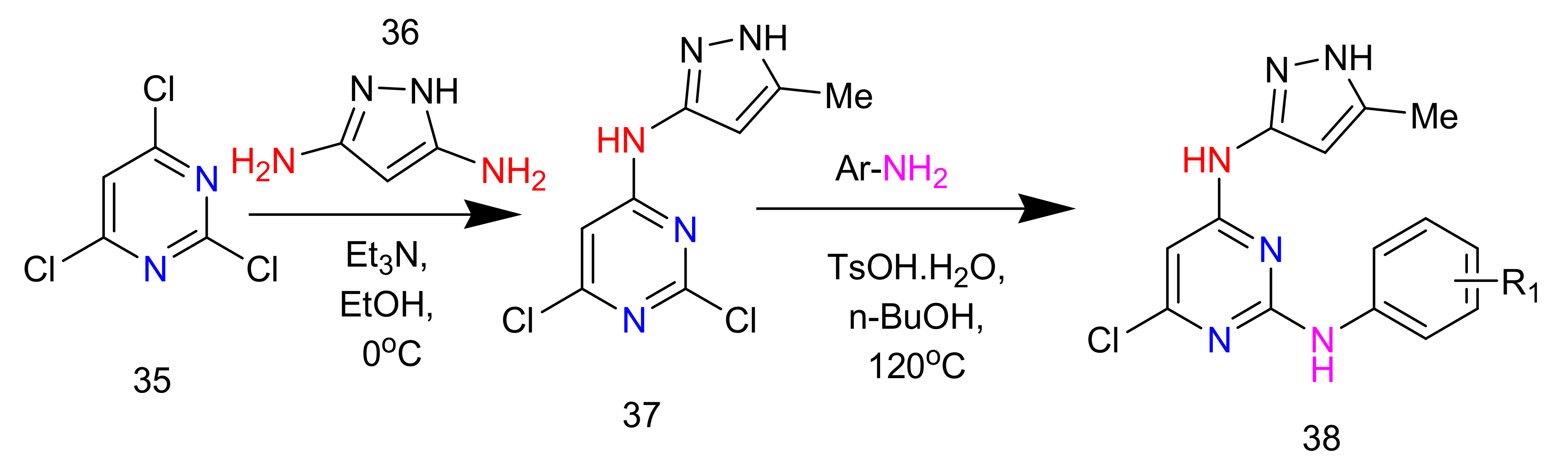
2.6. Synthesis of 2,4-Diaminopyrimidines
2.7. AURK and PLK Inhibition Studies of Aminopyrimidines
3. Conclusions and Future Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nigg, E.A. Mitotic kinases as regulators of cell division and its checkpoints. Nat. Rev. Mol. Cell Biol. 2001, 2, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Bai, L.; He, J.; Zhong, L.; Duan, X.; Ouyang, L.; Zhu, Y.; Wang, T.; Zhang, Y.; Shi, J. Recent advances in discovery and development of natural products as source for anti-Parkinson’s disease lead compounds. Eur. J. Med. Chem. 2017, 141, 257–272. [Google Scholar] [CrossRef]
- Li, J.; Wang, P.; Zhou, B.; Shi, J.; Liu, J.; Li, X.; Fan, L.; Zheng, Y.; Ouyang, L. Development of 4,5-dihydro-benzodiazepinone derivatives as a new chemical series of BRD4 inhibitors. Eur. J. Med. Chem. 2016, 121, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Glover, D.; Leibowitz, M.H.; McLean, A.D.; Parry, H. Mutations in aurora prevent centrosome separation leading to the formation of monopolar spindles. Cell 1995, 81, 95–105. [Google Scholar] [CrossRef] [Green Version]
- Llamazares, S.; Moreira, A.; Tavares, A.; Girdham, C.; Spruce, A.B.; Gonzalez, C.; Karess, E.R.; Glover, D.; Sunkel, C. polo encodes a protein kinase homolog required for mitosis in Drosophila. Genes Dev. 1991, 5, 2153–2165. [Google Scholar] [CrossRef]
- Borisa, A.C.; Bhatt, H.G. A comprehensive review on Aurora kinase: Small molecule inhibitors and clinical trial studies. Eur. J. Med. Chem. 2017, 140, 1–19. [Google Scholar] [CrossRef]
- Carvajal, R.D.; Tse, A.; Schwartz, G.K. Aurora Kinases: New Targets for Cancer Therapy. Clin. Cancer Res. 2006, 12, 6869–6875. [Google Scholar] [CrossRef] [Green Version]
- Bavetsias, V.; Faisal, A.; Crumpler, S.; Brown, N.; Kosmopoulou, M.; Joshi, A.; Atrash, B.; Pérez-Fuertes, Y.; Schmitt, J.A.; Boxall, K.J.; et al. Aurora Isoform Selectivity: Design and Synthesis of Imidazo[4,5-b]pyridine Derivatives as Highly Selective Inhibitors of Aurora-A Kinase in Cells. J. Med. Chem. 2013, 56, 9122–9135. [Google Scholar] [CrossRef]
- Elia, A.E.H.; Cantley, L.C.; Yaffe, M.B. Proteomic screen finds pSer/pThr-binding domain localizing Plk1 to mitotic substrates. Science 2003, 299, 1228–1231. [Google Scholar] [CrossRef]
- Zhan, M.-M.; Yang, Y.; Luo, J.; Zhang, X.-X.; Xiao, X.; Li, S.; Cheng, K.; Xie, Z.; Tu, Z.; Liao, C. Design, synthesis, and biological evaluation of novel highly selective polo-like kinase 2 inhibitors based on the tetrahydropteridin chemical scaffold. Eur. J. Med. Chem. 2018, 143, 724–731. [Google Scholar] [CrossRef]
- Barr, F.; Silljé, H.H.W.; Nigg, E. Polo-like kinases and the orchestration of cell division. Nat. Rev. Mol. Cell Biol. 2004, 5, 429–441. [Google Scholar] [CrossRef] [PubMed]
- Carry, J.-C.; Clerc, F.; Minoux, H.; Schio, L.; Mauger, J.; Nair, A.; Parmantier, E.; Le Moigne, R.; Delorme, C.; Nicolas, J.-P.; et al. SAR156497, an Exquisitely Selective Inhibitor of Aurora Kinases. J. Med. Chem. 2014, 58, 362–375. [Google Scholar] [CrossRef]
- Lens, S.M.A.; Voest, E.E.; Medema, R. Shared and separate functions of polo-like kinases and aurora kinases in cancer. Nat. Rev. Cancer 2010, 10, 825–841. [Google Scholar] [CrossRef] [PubMed]
- Toyoshima-Morimoto, F.; Taniguchi, E.; Shinya, N.; Iwamatsu, A.; Nishida, E. Polo-like kinase 1 phosphorylates cyclin B1 and targets it to the nucleus during prophase. Nat. Cell Biol. 2001, 410, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Degenhardt, Y.; Lampkin, T. Targeting Polo-like Kinase in Cancer Therapy: Fig. 1. Clin. Cancer Res. 2010, 16, 384–389. [Google Scholar] [CrossRef] [Green Version]
- Murga-Zamalloa, C.; Inamdar, K.V.; Wilcox, R.A. The role of aurora A and polo-like kinases in high-risk lymphomas. Blood Adv. 2019, 3, 1778–1787. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Erikson, R.L. Polo-like kinase (Plk)1 depletion induces apoptosis in cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 5789–5794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, B.; Zhong, L.; He, J.; Zhang, H.; Li, F.; Wang, T.; Zou, J.; Lin, Y.-X.; Zhang, C.; Guo, X.; et al. Discovery of Inhibitors of Aurora/PLK Targets as Anticancer Agents. J. Med. Chem. 2019, 62, 7697–7707. [Google Scholar] [CrossRef]
- Macůrek, L.; Lindqvist, A.; Lim, D.; Lampson, M.A.; Klompmaker, R.; Freire, R.; Clouin, C.; Taylor, S.; Yaffe, M.B.; Medema, R.H. Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nat. Cell Biol. 2008, 455, 119–123. [Google Scholar] [CrossRef]
- Asteriti, I.A.; De Mattia, F.; Guarguaglini, G. Cross-Talk between AURKA and Plk1 in Mitotic Entry and Spindle Assembly. Front. Oncol. 2015, 5, 283. [Google Scholar] [CrossRef] [Green Version]
- Joukov, V.; De Nicolo, A. Aurora-PLK1 cascades as key signaling modules in the regulation of mitosis. Sci. Signal. 2018, 11, eaar4195. [Google Scholar] [CrossRef] [Green Version]
- Sankhe, K.; Prabhu, A.; Khan, T. Design strategies, SAR, and mechanistic insight of Aurora kinase inhibitors in cancer. Chem. Biol. Drug Des. 2021, 98, 73–93. [Google Scholar] [CrossRef] [PubMed]
- Suri, A.; Bailey, A.W.; Tavares, M.T.; Gunosewoyo, H.; Dyer, C.P.; Grupenmacher, A.T.; Piper, D.R.; Horton, R.A.; Tomita, T.; Kozikowski, A.P.; et al. Evaluation of Protein Kinase Inhibitors with PLK4 Cross-Over Potential in a Pre-Clinical Model of Cancer. Int. J. Mol. Sci. 2019, 20, 2112. [Google Scholar] [CrossRef] [Green Version]
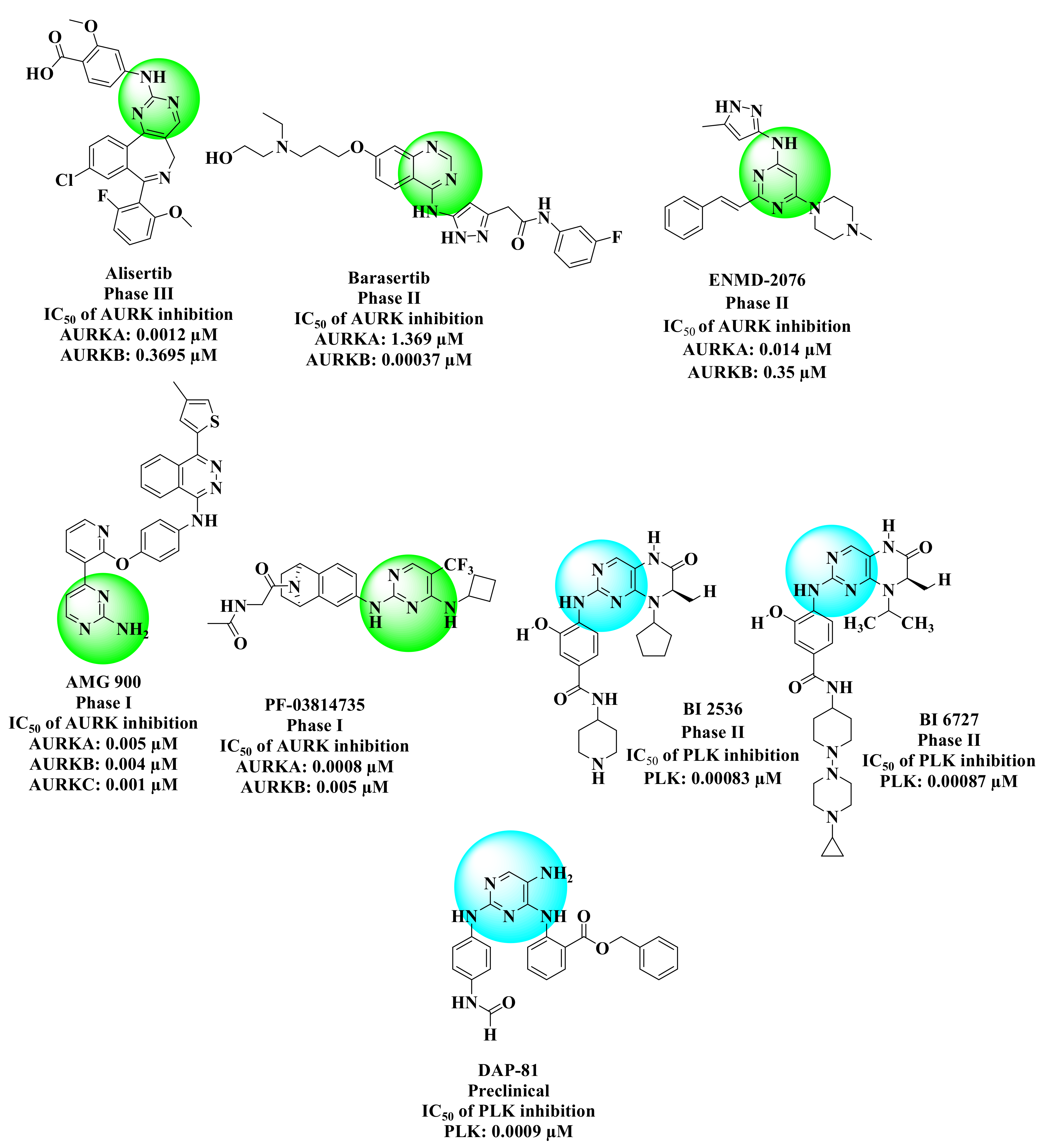
- Gjertsen, B.T.; Schöffski, P. Discovery and development of the Polo-like kinase inhibitor volasertib in cancer therapy. Leukemia 2015, 29, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Alferez, D.G.; Goodlad, R.A.; Odedra, R.; Sini, P.; Crafter, C.; Ryan, A.J.; Wedge, S.R.; Wright, N.A.; Anderson, E.; Wilkinson, R.W. Inhibition of Aurora-B kinase activity confers antitumor efficacy in preclinical mouse models of early and advanced gastrointestinal neoplasia. Int. J. Oncol. 2012, 41, 1475–1485. [Google Scholar] [CrossRef]
- Manfredi, M.G.; Ecsedy, J.A.; Chakravarty, A.; Silverman, L.; Zhang, M.; Hoar, K.M.; Stroud, S.G.; Chen, W.; Shinde, V.; Huck, J.J.; et al. Characterization of Alisertib (MLN8237), an Investigational Small-Molecule Inhibitor of Aurora A Kinase Using Novel In Vivo Pharmacodynamic Assays. Clin. Cancer Res. 2011, 17, 7614–7624. [Google Scholar] [CrossRef] [Green Version]
- Helfrich, B.A.; Kim, J.; Gao, D.; Chan, D.C.; Zhang, Z.; Tan, A.C.; Bunn, P.A. Barasertib (AZD1152), a Small Molecule Aurora B Inhibitor, Inhibits the Growth of SCLC Cell Lines In Vitro and In Vivo. Mol. Cancer Ther. 2016, 15, 2314–2322. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, G.C.; Brokx, R.D.; Denny, T.A.; Hembrough, T.A.; Plum, S.M.; Fogler, W.E.; Sidor, C.F.; Bray, M. ENMD-2076 Is an Orally Active Kinase Inhibitor with Antiangiogenic and Antiproliferative Mechanisms of Action. Mol. Cancer Ther. 2011, 10, 126–137. [Google Scholar] [CrossRef] [Green Version]
- Payton, M.; Bush, T.L.; Chung, G.; Ziegler, B.; Eden, P.; McElroy, P.; Ross, S.; Cee, V.J.; Deak, H.L.; Hodous, B.L.; et al. Preclinical Evaluation of AMG 900, a Novel Potent and Highly Selective Pan-Aurora Kinase Inhibitor with Activity in Taxane-Resistant Tumor Cell Lines. Cancer Res. 2010, 70, 9846–9854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jani, J.P.; Arcari, J.; Bernardo, V.; Bhattacharya, S.K.; Briere, D.; Cohen, B.D.; Coleman, K.; Christensen, J.G.; Emerson, E.O.; Jakowski, A.; et al. PF-03814735, an Orally Bioavailable Small Molecule Aurora Kinase Inhibitor for Cancer Therapy. Mol. Cancer Ther. 2010, 9, 883–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutteridge, R.E.A.; Ndiaye, M.A.; Liu, X.; Ahmad, N. Plk1 Inhibitors in Cancer Therapy: From Laboratory to Clinics. Mol. Cancer Ther. 2016, 15, 1427–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, A.; Wang, L.; Xu, S.; Xu, J. Aurora-A kinase inhibitor scaffolds and binding modes. Drug Discov. Today 2011, 16, 260–269. [Google Scholar] [CrossRef]
- Koroleva, E.V.; Gusak, K.N.; Ignatovich, Z.V. Synthesis and applications of 2-aminopyrimidine derivatives as key intermediates in chemical synthesis of biomolecules. Russ. Chem. Rev. 2010, 79, 655–681. [Google Scholar] [CrossRef]
- Price, R.W.; Moos, A. A New Synthesis of 2-Aminopyrimidine. J. Am. Chem. Soc. 1945, 67, 207–208. [Google Scholar] [CrossRef]
- Fellows, E.J. Substituted Sulfanilamidopyrimidines. Exp. Biol. Med. 1941, 48, 680–684. [Google Scholar] [CrossRef]
- Brown, D.J.; England, B.T. The Dimroth rearrangement. Part IX. The formation and isomerisations of propynyl (and related)-iminopyrimidines. J. Chem. Soc. C 1967, 1922–1927. [Google Scholar] [CrossRef]
- Jansa, P.; Holý, A.; Dračínský, M.; Kolman, V.; Janeba, Z.; Kostecká, P.; Kmoníčková, E.; Zídek, Z. 5-Substituted 2-amino-4,6-dihydroxypyrimidines and 2-amino-4,6-dichloropyrimidines: Synthesis and inhibitory effects on immune-activated nitric oxide production. Med. Chem. Res. 2014, 23, 4482–4490. [Google Scholar] [CrossRef]
- Bayramoğlu, D.; Kurtay, G.; Güllü, M. Ultrasound-assisted rapid synthesis of 2-aminopyrimidine and barbituric acid derivatives. Synth. Commun. 2020, 50, 649–658. [Google Scholar] [CrossRef]
- Bennett, G.B.; Mason, R.B.; Alden, L.J.; Roach, J.B. Synthesis and antiinflammatory activity of trisubstituted pyrimidines and triazines. J. Med. Chem. 1978, 21, 623–628. [Google Scholar] [CrossRef]
- Zhang, H.Q.; Xia, Z.; Vasudevan, A.; Djuric, S.W. Efficient Pd-catalyzed synthesis of 2-arylaminopyrimidines via microwave irradiation. Tetrahedron Lett. 2006, 47, 4881–4884. [Google Scholar] [CrossRef]
- Deng, X.; Mani, N.S. An Efficient Route to 4-Aryl-5-pyrimidinylimidazoles via Sequential Functionalization of 2,4-Dichloropyrimidine. Org. Lett. 2006, 8, 269–272. [Google Scholar] [CrossRef]
- Yoshida, K.; Taguchi, M. Reaction of N-substituted cyclic amines with 2,4-dichloroquinazoline, 2,4-dichloropyrimidine, and its 5-methyl derivative. J. Chem. Soc. Perkin Trans. 1992, 919–922. [Google Scholar] [CrossRef]
- Gao, X.; Fu, H.; Qiao, R.; Jiang, Y.; Zhao, Y. Copper-Catalyzed Synthesis of Primary Arylamines via Cascade Reactions of Aryl Halides with Amidine Hydrochlorides. J. Org. Chem. 2008, 73, 6864–6866. [Google Scholar] [CrossRef]
- Narayan, S.; Seelhammer, T.; Gawley, R.E. Microwave assisted solvent free amination of halo-(pyridine or pyrimidine) without transition metal catalyst. Tetrahedron Lett. 2004, 45, 757–759. [Google Scholar] [CrossRef]
- Collins, I. Rapid analogue syntheses of heteroaromatic compounds. J. Chem. Soc. Perkin Trans. 2002, 34, 1921–1940. [Google Scholar] [CrossRef]
- Soldatenkov, A.T.; Temesgen, A.V.; Kolyadina, N.M. Oxidation of Heterocyclic Compounds by Permanganate Anion. (Review). Chem. Heterocycl. Compd. 2004, 40, 537–560. [Google Scholar] [CrossRef]
- Breuker, K.; Van der Plas, H.C. Occurrence of an SN(ANRORC) mechanism in the Chichibabin amination of 4-phenylpyrimidine. J. Org. Chem. 1979, 44, 4677–4680. [Google Scholar] [CrossRef]
- Brown, D.J.; Ford, P.W. Simple pyrimidines. Part X. The formation and reactivity of 2-, 4-, and 5-pyrimidinyl sulphones and sulphoxides. J. Chem. Soc. C 1967, 568–572. [Google Scholar] [CrossRef]
- Williams, R.R.; Cline, J.K. Synthesis of vitamin B1. J. Am. Chem. Soc. 1936, 58, 1504–1505. [Google Scholar] [CrossRef]
- Karad, S.N.; Liu, R. Regiocontrolled Gold-Catalyzed [2 + 2 + 2] Cycloadditions of Ynamides with Two Discrete Nitriles to Construct 4-Aminopyrimidine Cores. Angew. Chem. Int. Ed. 2014, 53, 9072–9076. [Google Scholar] [CrossRef]
- Baxendale, I.R.; Ley, S.V. Formation of 4-Aminopyrimidines via the Trimerization of Nitriles Using Focused Microwave Heating. J. Comb. Chem. 2005, 7, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Li, Y.; Xiang, S.; Fan, W.; Jin, J.; Huang, D. Utilization of nitriles as the nitrogen source: Practical and economical construction of 4-aminopyrimidine and β-enaminonitrile skeletons. Org. Chem. Front. 2019, 6, 3071–3077. [Google Scholar] [CrossRef]
- Jachak, M.N.; Tantak, C.D.; Toche, R.B.; Badgujar, N.S. A Convenient Route for the Synthesis of 4-Aryl- and 4-Aminopyrimidines. Mon. Chem. 2004, 135, 1529–1538. [Google Scholar] [CrossRef]
- Létinois, U.; Schütz, J.; Härter, R.; Stoll, R.; Huffschmidt, F.; Bonrath, W.; Karge, R. Lewis Acid-Catalyzed Synthesis of 4-Aminopyrimidines: A Scalable Industrial Process. Org. Process. Res. Dev. 2013, 17, 427–431. [Google Scholar] [CrossRef]
- Long, L.; Luo, Y.; Hou, Z.-J.; Ma, H.-J.; Long, Z.-J.; Tu, Z.-C.; Huang, L.-J.; Liu, Q.; Lu, G. Synthesis and biological evaluation of aurora kinases inhibitors based on N -trisubstituted pyrimidine scaffold. Eur. J. Med. Chem. 2018, 145, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.-Z.; Tang, Z.-B.; Sang, C.-Y.; Qi, Z.-Y.; Hui, L.; Chen, S.-W. Synthesis and biological evaluation of nitroxide labeled pyrimidines as Aurora kinase inhibitors. Bioorg. Med. Chem. Lett. 2019, 29, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Hashihayata, T.; Kawamura, M.; Mitsuya, M.; Satoh, Y. Novel Aminopyrimidine Derivatives as plk1. Inhibitors. Patent No. WO2008081910A1, 10 July 2008. [Google Scholar]
- Chi, Y.-H.; Yeh, T.-K.; Ke, Y.-Y.; Lin, W.-H.; Tsai, C.-H.; Wang, W.-P.; Chen, Y.-T.; Su, Y.-C.; Wang, P.-C.; Chen, Y.-F.; et al. Discovery and synthesis of a Pyrimidine-based Aurora kinase inhibitor to reduce levels of MYC Oncoproteins. J. Med. Chem. 2021, 64, 7312–7330. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
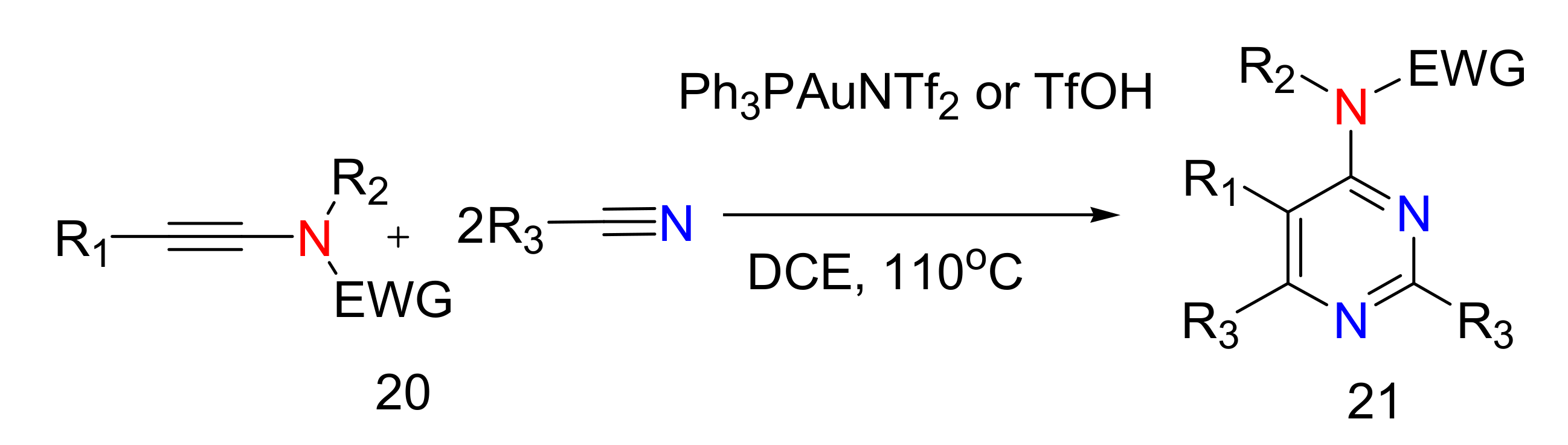{kind=link}
{kind=link}
{kind=link}
{kind=link}
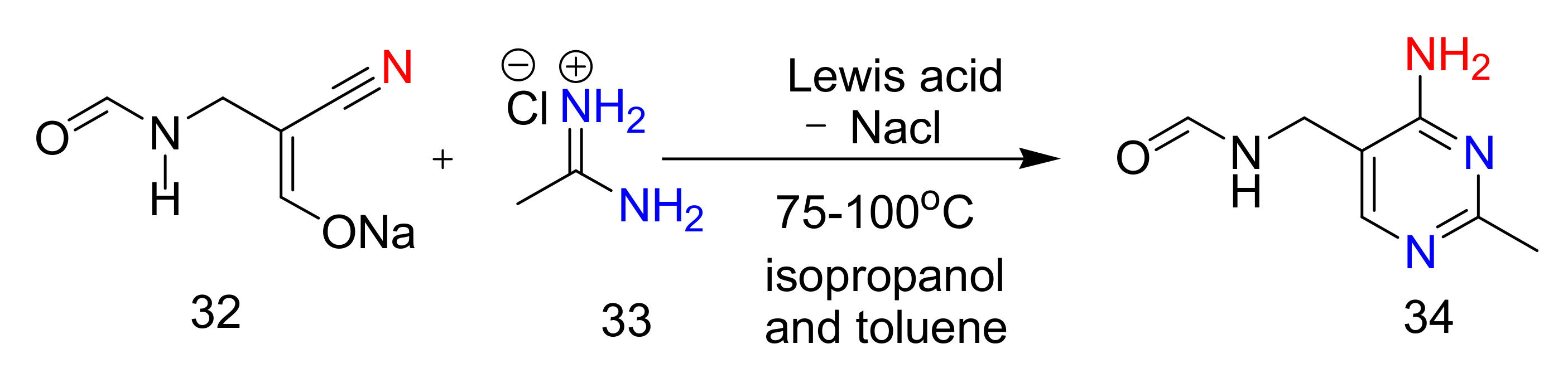{kind=link}
{kind=link}
{kind=link}
{kind=link}



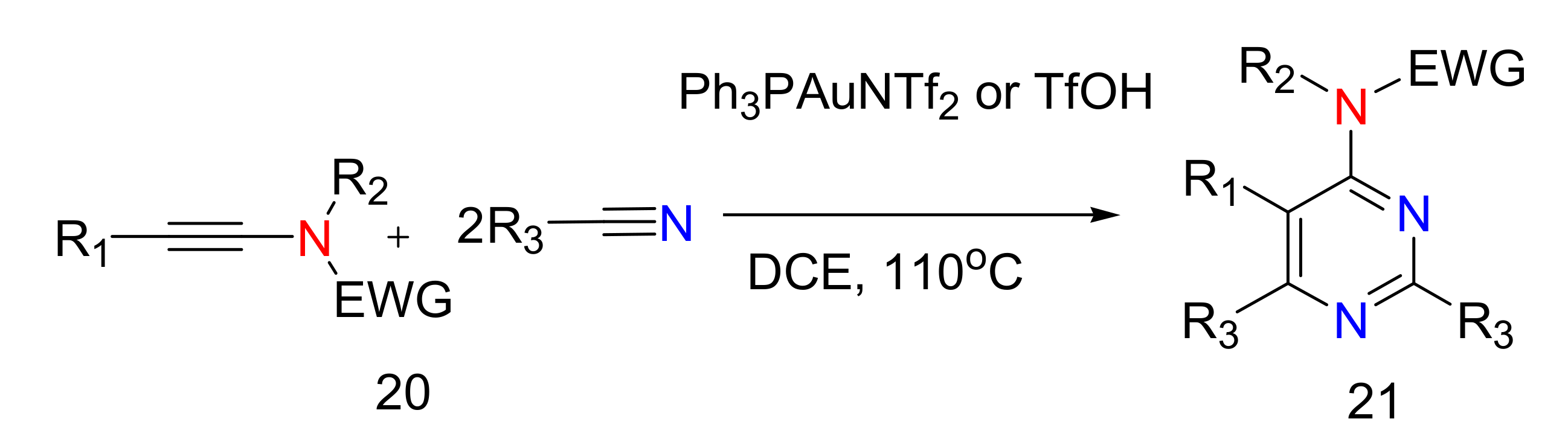


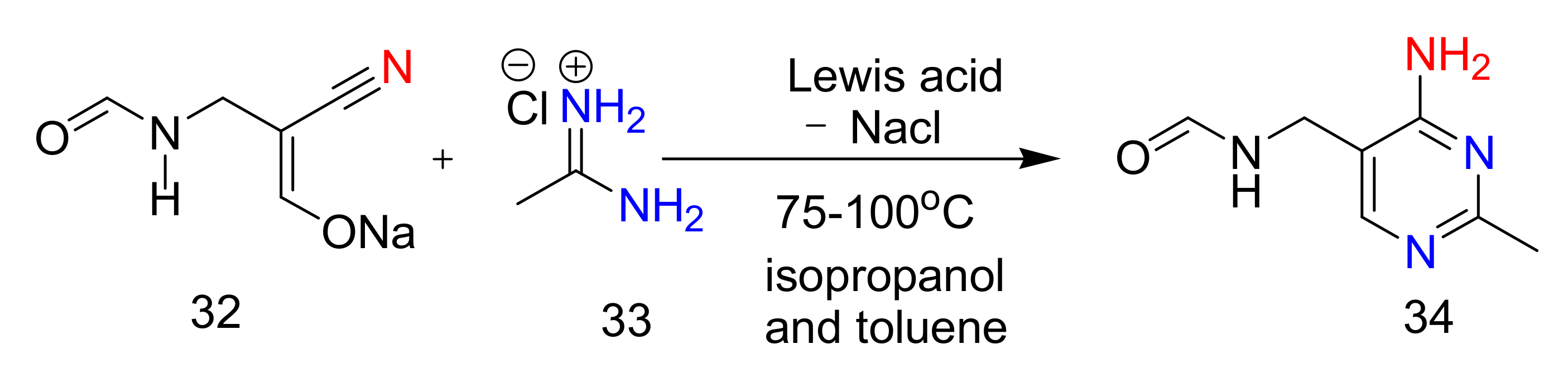


| Compounds | R1 | IC50 (µM) | |
|---|---|---|---|
| AURKA | AURKB | ||
| 38a | 3,4-diOMe | 0.038 | 0.452 |
| 38b | 3,4-methylenedioxy | 0.020 | 0.091 |
| 38c | 3-OMe and 4-COOMe | 0.067 | 0.442 |
| 38d | 2,3,4-triOMe | 0.094 | 0.188 |
| 38e | 3,4-diCl | 0.033 | 0.050 |
| 38f | 3-F | 0.025 | 0.102 |
| 38g | 4-F | 0.023 | 0.0751 |
| 38h | 3,4-diF | 0.017 | 0.0892 |
| 38i | 3-F and 4-Cl | 0.031 | 0.101 |
| 38j | 3-Cl and 4-F | 0.0071 | 0.0257 |
| 38k | 3-F and 4- COOMe | 0.035 | 0.145 |
| Compounds | X | NR1R2 | IC50 (µM) | |||
|---|---|---|---|---|---|---|
| HeLa | A-549 | Hep-G2 | LoVo | |||
| 41a | NO2 | 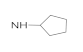 | 55.56 ± 4.01 | 33.16 ± 3.08 | 28.73 ± 1.95 | 41.24 ± 3.52 |
| 41b | NO2 |  | 17.49 ± 0.82 | 1.96 ± 0.12 | 56.83 ± 4.39 | 39.73 ± 2.95 |
| 41c | NO2 | 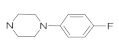 | 25.33 ± 2.33 | 10.25 ± 1.08 | 13.08 ± 1.21 | 27.82 ± 2.47 |
| 41d | NO2 | 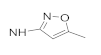 | 7.36 ± 0.82 | 1.14 ± 0.09 | 7.36 ± 0.92 | 26.81 ± 2.54 |
| 41e | NO2 | 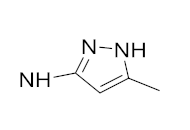 | 29.13 ± 1.21 | 15.32 ± 1.21 | 37.51 ± 1.21 | >100 |
| 41f | NO2 | 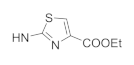 | 3.79 ± 1.21 | 0.74 ± 0.08 | 6.87 ± 0.58 | 12.53 ± 1.02 |
| 41g | NO2 | 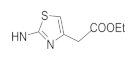 | 11.95 ± 1.05 | 1.04 ± 0.13 | 26.44 ± 2.3 | 27.01 ± 2.57 |
| 41h | F | 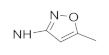 | 5.68 ± 0.62 | 0.88 ± 0.07 | 10.82 ± 0.89 | 11.97 ± 1.33 |
| 41i | F |  | 11.20 ± 1.07 | 2.12 ± 0.18 | 10.80 ± 1.28 | 42.18 ± 3.91 |
| 41j | F | 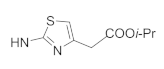 | 11.20 ± 1.07 | 2.12 ± 0.18 | 10.80 ± 1.28 | 42.18 ± 3.91 |
| 41k | F |  | 5.23 ± 0.47 | 2.31 ± 0.31 | 11.53 ± 1.25 | 21.91 ± 2.03 |
| 41l | F |  | 2.72 ± 0.25 | 0.89 ± 0.05 | 5.73 ± 0.39 | 11.41 ± 1.08 |
| 41m | F | 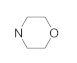 | 14.35 ± 1.09 | 13.17 ± 1.13 | 22.61 ± 2.05 | 35.52 ± 2.71 |
| 41n | F | 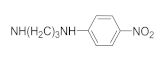 | 31.73 ± 2.86 | 12.37 ± 1.19 | 19.82 ± 1.56 | 53.62 ± 4.96 |
| VX-680 | 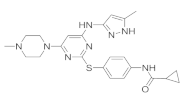 | 46.20 ± 4.08 | 35.80 ± 3.33 | 53.30 ± 5.82 | 45.30 ± 4.97 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jadhav, M.; Sankhe, K.; Bhandare, R.R.; Edis, Z.; Bloukh, S.H.; Khan, T.A. Synthetic Strategies of Pyrimidine-Based Scaffolds as Aurora Kinase and Polo-like Kinase Inhibitors. Molecules 2021, 26, 5170. https://doi.org/10.3390/molecules26175170
Jadhav M, Sankhe K, Bhandare RR, Edis Z, Bloukh SH, Khan TA. Synthetic Strategies of Pyrimidine-Based Scaffolds as Aurora Kinase and Polo-like Kinase Inhibitors. Molecules. 2021; 26(17):5170. https://doi.org/10.3390/molecules26175170
Chicago/Turabian StyleJadhav, Mrunal, Kaksha Sankhe, Richie R. Bhandare, Zehra Edis, Samir Haj Bloukh, and Tabassum Asif Khan. 2021. "Synthetic Strategies of Pyrimidine-Based Scaffolds as Aurora Kinase and Polo-like Kinase Inhibitors" Molecules 26, no. 17: 5170. https://doi.org/10.3390/molecules26175170
APA StyleJadhav, M., Sankhe, K., Bhandare, R. R., Edis, Z., Bloukh, S. H., & Khan, T. A. (2021). Synthetic Strategies of Pyrimidine-Based Scaffolds as Aurora Kinase and Polo-like Kinase Inhibitors. Molecules, 26(17), 5170. https://doi.org/10.3390/molecules26175170