
Sulphur-Bridged BAl5S5+ with 17 Counting Electrons: A Regular Planar Pentacoordinate Boron System


Abstract
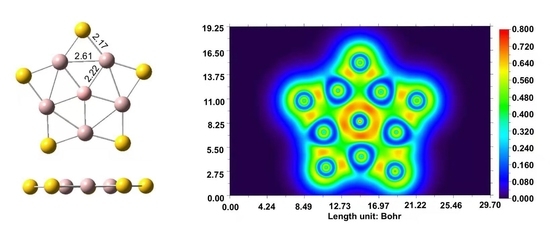
:
1. Introduction
2. Results and Discussion
2.1. Structure and Stability of XAl5S5n (X = C, B; n = 0, +1, +2) Clusters
2.2. Electronic Properties and NBO Analysis on CAl5S5+, CAl5S52+, BAl5S5, BAl5S5+
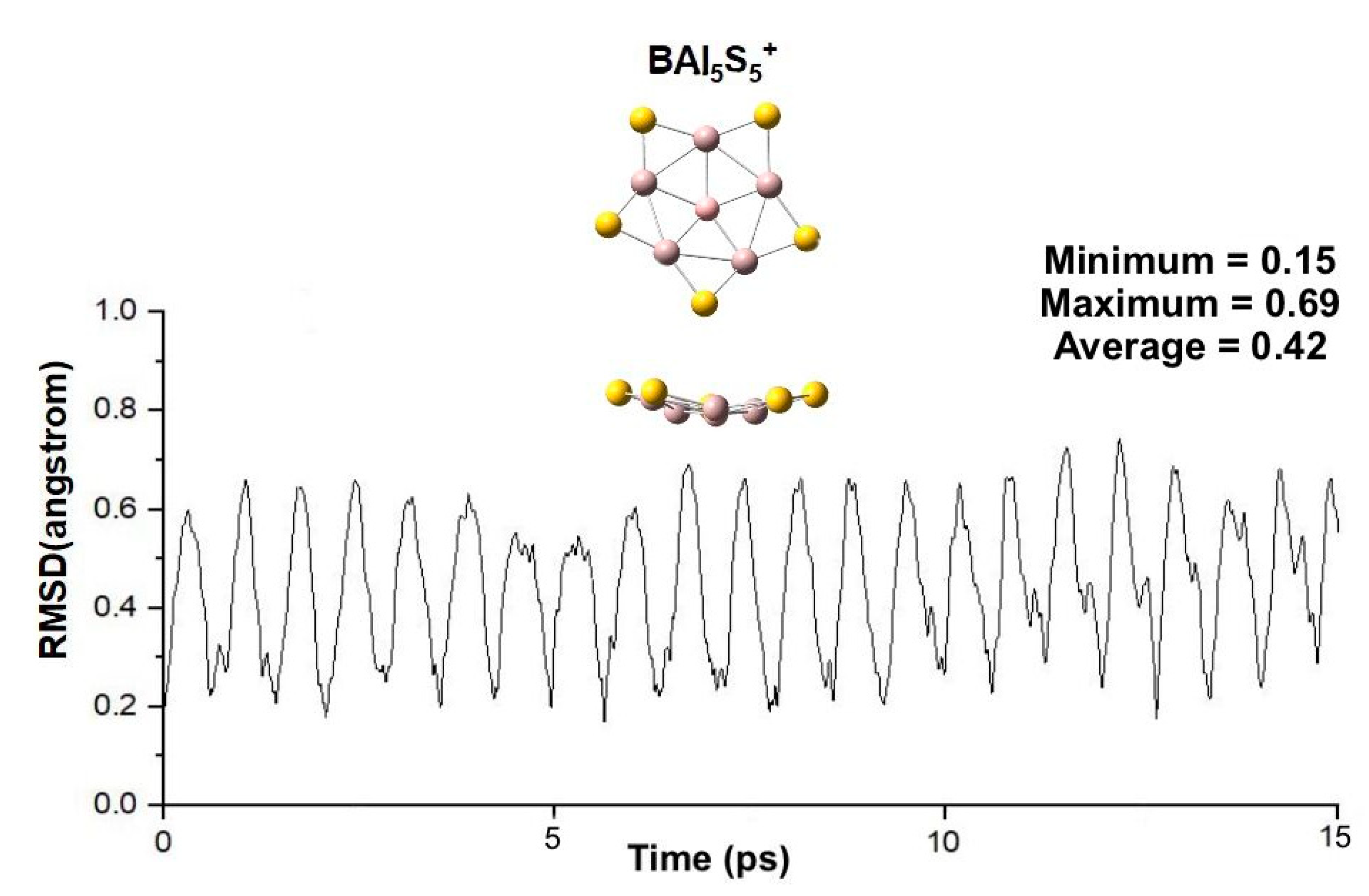
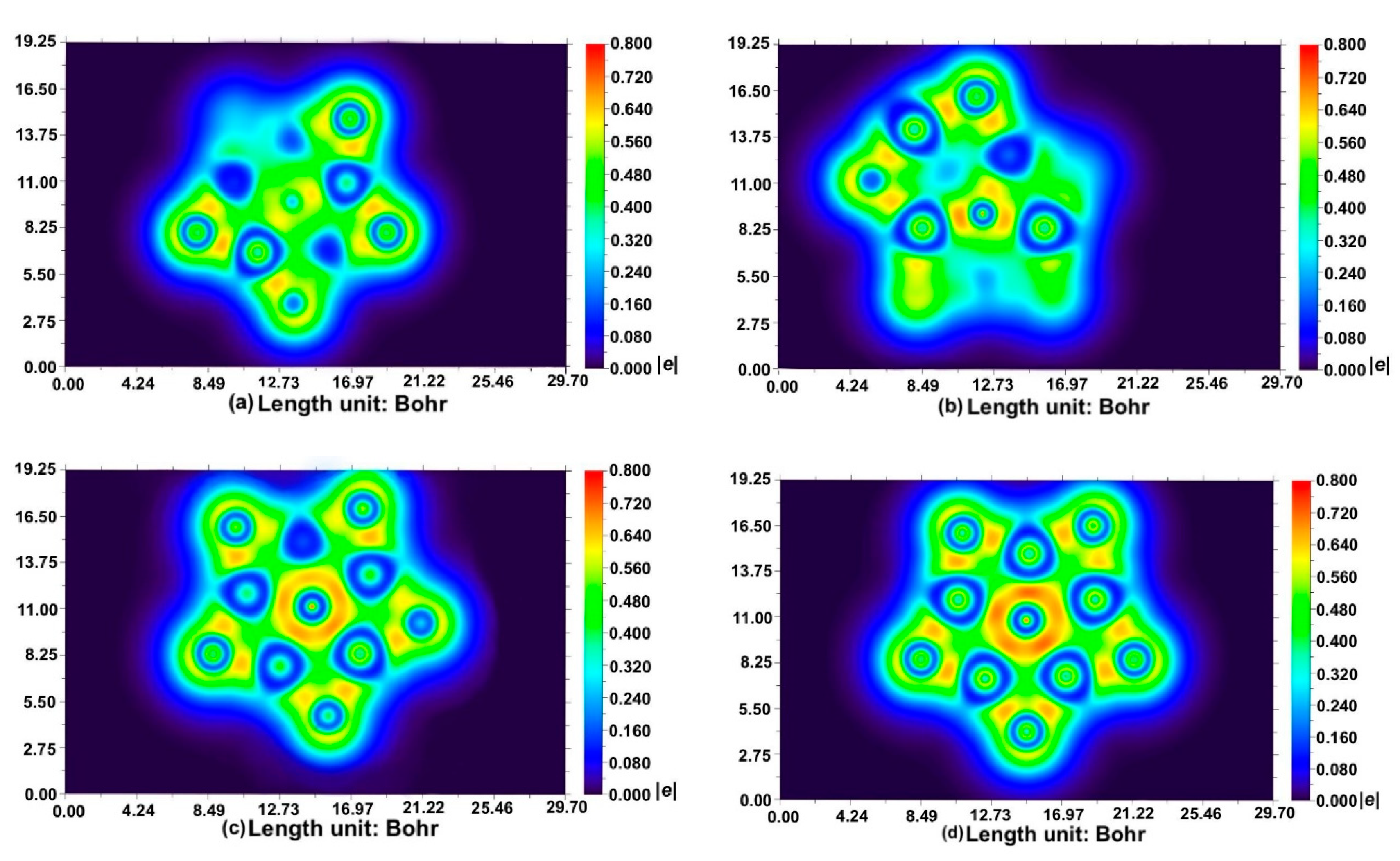
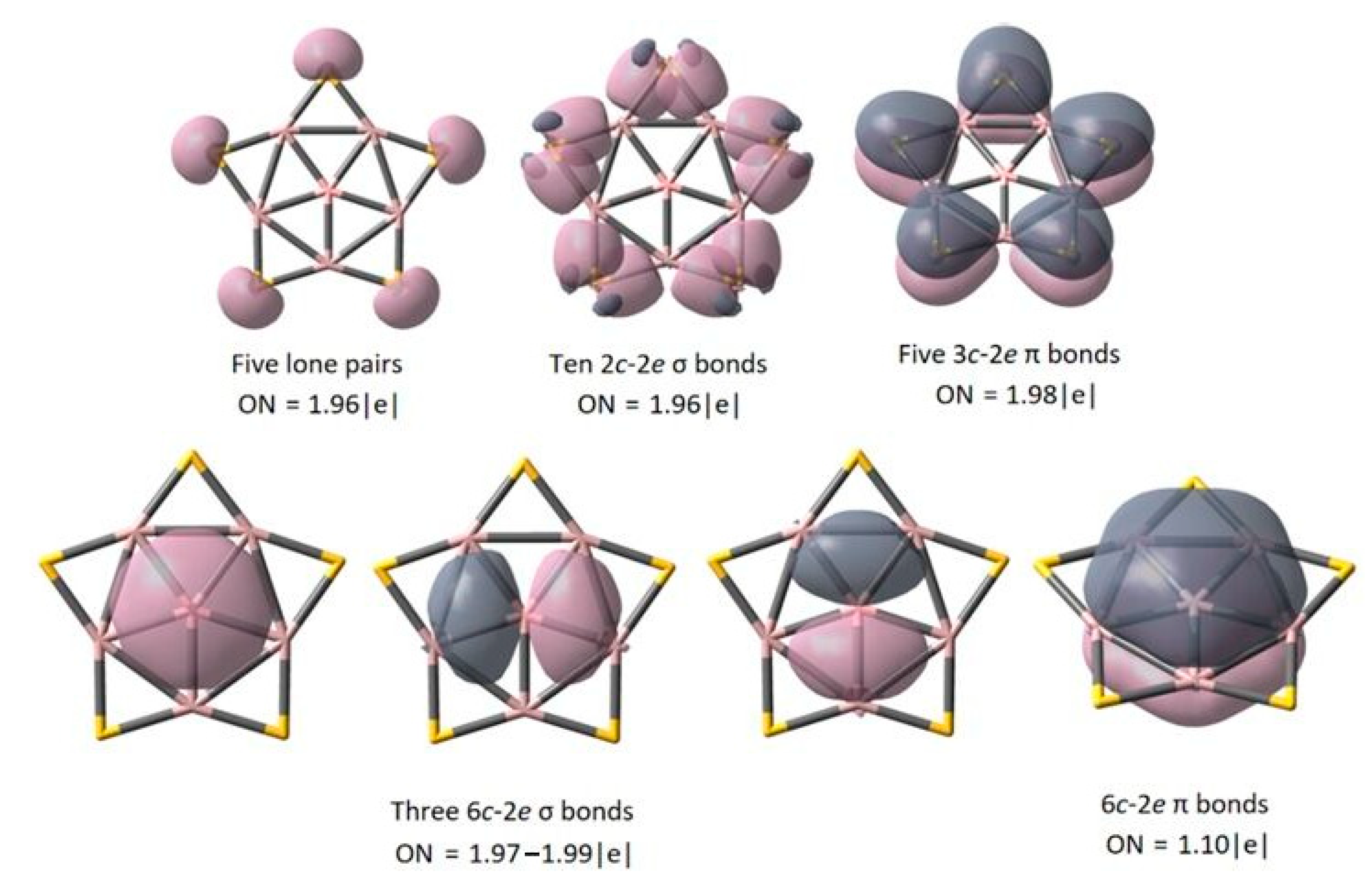
2.3. Chemical Bonding on the PPB BAl5S5+ Cluster
3. Computational Methodology
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Wang, Y.; Li, Y.F.; Chen, Z.F. Planar Hypercoordinate Motifs in Two-Diemensional Materials. Acc. Chem. Res. 2020, 1, 887–895. [Google Scholar] [CrossRef]
- Vassilev-Galindo, V.; Pan, S.J.; Donald, K.; Merino, G. Planar pentacoordinate carbons. Nat. Rev. Chem. 2018, 2, 0114. [Google Scholar] [CrossRef]
- Zhu, C.Y.; Lv, H.F.; Qu, X.; Zhang, M.; Wang, J.Y.; Wen, S.Z.; Li, Q.; Geng, Y.; Su, Z.M.; Wu, X.J.; et al. TMC (TM = Co, Ni, and Cu) monolayers with planar pentacoordinate carbon and their potential applications. J. Mater. Chem. C 2019, 7, 6406–6413. [Google Scholar] [CrossRef]
- Hoffmann, R.; Alder, R.W.; Wilcox, C.F. Planar tetracoordinate carbon. J. Am. Chem. Soc. 1970, 92, 4992–4993. [Google Scholar] [CrossRef]
- Collins, J.B.; Dill, J.D.; Jemmis, E.D.; Apeloig, Y.; von Rague Schleyer, P.; Seeger, R.; Pople, J.A. Stabilization of planar tetracoordinate carbon. J. Am. Chem. Soc. 1976, 98, 5419–5427. [Google Scholar] [CrossRef]
- Cui, Z.-H.; Shao, C.-B.; Gap, S.-M.; Ding, Y.-H. Pentaatomic planar tetracoordinate carbon molecules [XCAl3]q [(X,q) = (B,−2), (C,−1), (N,0)] with C–X multiple bonding. Phys. Chem. Chem. Phys. 2010, 12, 13637–13645. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, K.; Ogoshi, S.; Nishiguchi, S.; Kurosawa, H. Synthesis, Structure, and Reactivity of Neutral η3-Propargylpalladium Complexes. J. Am. Chem. Soc. 1998, 120, 1938–1939. [Google Scholar] [CrossRef]
- Wang, Z.X.; Schleyer, P.R. Construction principles of „hyparenes”: Families of molecules with planar pentacoordinate carbons. Science 2001, 292, 2465–2469. [Google Scholar] [CrossRef]
- Wang, L.-S.; Boldyrev, A.I.; Li, X.; Simon, J. Experimental Observation of Pentaatomic Tetracoordinate Planar Carbon-Containing Molecules. J. Am. Chem. Soc. 2000, 122, 7681–7687. [Google Scholar] [CrossRef]
- Wang, L.-M.; Huang, W.; Averkiev, B.B.; Boldyrev, A.I.; Wang, L.-S. CB7−: Experimental and Theoretical Evidence against Hypercoordinate Planar Carbon. Angew. Chem. Int. Ed. 2007, 46, 4550–4553. [Google Scholar] [CrossRef] [PubMed]
- Averkiev, B.B.; Dmitry, Y.Z.; Wang, L.-M.; Huang, W.; Wang, L.-S.; Boldyrev, A.I. Carbon Avoids Hypercoordination in CB6−, CB62−, and C2B5− Planar Carbon−Boron Clusters. J. Am. Chem. Soc. 2008, 130, 9248–9250. [Google Scholar] [CrossRef]
- Averkiev, B.B.; Wang, L.-M.; Huang, W.; Wang, L.-S.; Boldyrev, A.I. Experimental and theoretical investigations of CB8-: Towards rational design of hypercoordinated planar chemical species. Phys. Chem. Chem. Phys. 2009, 11, 9840–9849. [Google Scholar] [CrossRef]
- Wu, Y.B.; Jiang, J.L.; Lu, H.G.; Wang, Z.X.; Perez-Peralta, N.; Islas, R.; Contreras, M.; Merino, G.; Wu, I.C.; von Rague Schleyer, P. Starlike Aluminum–Carbon Aromatic Species. Chem. Eur. J. 2011, 17, 714–719. [Google Scholar] [CrossRef]
- Wu, Y.B.; Yuan, C.-X.; Yang, P. The C6B122− complex: A beautiful molecular wheel containing a ring of six planar tetracoordinate carbon atoms. J. Mol. Struc. Theochem 2006, 765, 35–38. [Google Scholar] [CrossRef]
- Aldridge, S.; Downs, A.J. Hydrides of the Main-Group Metals: New Variations on an Old Theme. Chem. Rev. 2001, 101, 3305–3366. [Google Scholar] [CrossRef] [PubMed]
- Minkin, V.I.; Minyaev, R.M.; Hoffmann, R. Non-classical structures of organic compounds: Unusual stereochemistry and hypercoordination. Russ. Chem. Rev. 2002, 71, 869–892. [Google Scholar] [CrossRef] [Green Version]
- Erhardt, S.; Frenking, G.; Chen, Z. Aromatic Boron Wheels with More than One Carbon Atom in the Center: C2B8, C3B93+, and C5B11+. Angew. Chem. 2005, 117, 1102–1106. [Google Scholar] [CrossRef]
- Keese, R. Carbon Flatland: Planar Tetracoordinate Carbon and Fenestranes. Chem. Rev. 2006, 106, 4787–4808. [Google Scholar] [CrossRef] [PubMed]
- Islas, R.; Heine, T.; Ito, K.; Schleyer, P.R.; Merino, G. Boron Rings Enclosing Planar Hypercoordinate Group 14 Elements. J. Am. Chem. Soc. 2007, 129, 14767–14774. [Google Scholar] [CrossRef]
- Liao, Y.; Cruz, C.L.; Schleyer, P.R.; Chen, Z. Many M©Bn boron wheels are local, but not global minima. Phys. Chem. Chem. Phys 2012, 14, 14898–14904. [Google Scholar] [CrossRef] [PubMed]
- Sergeeva, A.P.; Popov, I.A.; Piazza, Z.A.; Li, W.-L.; Romanescu, C.; Wang, L.-S.; Boldyrev, A.I. Understanding Boron through Size-Selected Clusters: Structure, Chemical Bonding, and Fluxionality. Acc. Chem. Res. 2014, 47, 1349–1358. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.-M.; Ganz, E.; Chen, Z.; Wang, Z.-X.; von Rague Schleyer, P. Four Decades of the Chemistry of Planar Hypercoordinate Compounds. Angew. Chem. Int. Ed. 2015, 54, 9468–9501. [Google Scholar] [CrossRef]
- Wang, L.-S. Photoelectron spectroscopy of size-selected boron clusters: From planar structures to borophenes and borospherenes. Int. Rev. Phys. Chem. 2016, 35, 69–142. [Google Scholar] [CrossRef]
- Guo, J.-C.; Feng, L.-Y.; Dong, C.; Zhai, H.-J. Planar Pentacoordinate versus Tetracoordinate Carbons in Ternary CBe4Li4 and CBe4Li42– Clusters. J. Phys. Chem. A 2018, 122, 8370–8376. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.-C.; Feng, L.-Y.; Zhai, H.-J. Ternary CBe4Au4 cluster: A 16-electron system with quasi-planar tetracoordinate carbon. Phys. Chem. Chem. Phys. 2018, 20, 6299–6306. [Google Scholar] [CrossRef]
- Miao, C.Q.; Guo, J.C.; Li, S.D. M@B9 and M@B10 molecular wheels containing planar nona-and deca-coordinate heavy group 11, 12, and 13 metals (M=Ag, Au, Cd, Hg, In, Tl). Sci. China Chem. 2009, 52, 900–904. [Google Scholar] [CrossRef]
- Li, S.-D.; Miao, C.-Q.; Ren, G.-M. D5h Cu5H5X: Pentagonal Hydrocopper Cu5H5 Containing Pentacoordinate Planar Nonmetal Centers (X = B, C, N, O). Eur. J. Inorg. Chem. 2004, 2004, 2232–2234. [Google Scholar] [CrossRef]
- Aihara, J.-I.; Kanno, A.H.; Ishida, T. Aromaticity of Planar Boron Clusters Confirmed. J. Am. Chem. Soc. 2005, 127, 13324–13330. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Zeng, X.C. Probing the Planar Tetra-, Penta-, and Hexacoordinate Carbon in Carbon−Boron Mixed Clusters. J. Am. Chem. Soc. 2008, 130, 2580–2592. [Google Scholar] [CrossRef]
- Yu, H.-L.; Sang, R.-L.; Wu, Y.-Y. Structure and Aromaticity of B6H5+ Cation: A Novel Borhydride System Containing Planar Pentacoordinated Boron. J. Phys. Chem. A 2009, 113, 3382–3386. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Halla, J.O.C.; Wu, Y.-B.; Wang, Z.-X.; Islas, R.; Heine, T.; Merino, G. CAl4Be and CAl3Be2−: Global minima with a planar pentacoordinate carbon atom. Chem. Commun. 2009, 46, 8776–8778. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-B.; Duan, Y.; Lu, H.-G.; Li, S.-D. CAl2Be32– and Its Salt Complex LiCAl2Be3–: Anionic Global Minima with Planar Pentacoordinate Carbon. J. Phys. Chem. A 2012, 116, 3290–3294. [Google Scholar] [CrossRef]
- Li, S.-D.; Guo, Q.-L.; Miao, C.-Q.; Ren, G.-M. Investigation on Transition-Metal Hydrometal Complexes MnHnC with Planar Coordinate Carbon Centers by Density Functional Theory. Wuli Huaxue Xuebao 2007, 23, 743–745. [Google Scholar]
- Cui, Z.-H.; Sui, J.-J.; Ding, Y.-H. How can carbon favor planar multi-coordination in boron-based clusters? Global structures of CBxEy2−(E = Al, Ga, x + y = 4). Phys. Chem. Chem. Phys. 2015, 17, 32016–32022. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-Y.; Ding, Y.-H. Computational prediction of a global planar penta-coordinate carbon structure CAl4Ga+. Comput. Theor. Chem. 2014, 1048, 18–24. [Google Scholar] [CrossRef]
- Merino, G.; Mendez-Rojas, M.; Vela, A.; Heine, T. Recent advances in planar tetracoordinate carbon chemistry. J. Comput. Chem. 2006, 28, 362–372. [Google Scholar] [CrossRef]
- Wang, Y.Q.; Wang, C.; Liu, X.M.; Zhang, M.; Geng, Y.; Zhao, L. Planar Tetracoordinate Boron and Pentacoordinate Boron in B6S5 Clusters. Chem. J. Chin. Univ. 2020, 41, 1625–1630. [Google Scholar]
- Zhang, X.; Wang, Y.; Geng, Y.; Zhang, M.; Su, Z. Theoretical study on the stable plane CAl4 structure of sulfur—aluminum bridge bond. Chem. J. Chin. Univ. 2018, 39, 2485–2491. [Google Scholar]
- Wang, Y.; Lv, J.; Zhu, L.; Ma, Y. CALYPSO: A method for crystal structure prediction. Comput. Phys. Commun. 2012, 183, 2063–2070. [Google Scholar] [CrossRef] [Green Version]
- VandeVondele, J.; Krack, M.; Mohamed, F.; Parrinello, M.; Chassaing, T.; Hutter, J. Quickstep: Fast and accurate density functional calculations using a mixed Gaussian and plane waves approach. Comput. Phys. Commun. 2005, 167, 103–128. [Google Scholar] [CrossRef] [Green Version]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lv, J.; Zhu, L.; Ma, Y. Crystal structure prediction via particle-swarm optimization. Phys. Rev. B 2010, 82, 094116. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A.; Vreven, J.; Kudin, T.K.N.; Burant, J.C.; et al. Gaussian 09, Revision A.02; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CAl5S5+ (18e) | CAl5S52+ (17e) | BAl5S5 (18e) | BAl5S5+ (17e) | ||
|---|---|---|---|---|---|
| QC | −2.50 | −1.87 | QB | −2.56 | −0.63 |
| QAl | 1.42/1.43/1.44 | 1.40 | QAl | 1.31 | 0.66 |
| QS | −0.72/−0.73/−0.74 | −0.63 | QS | −0.80 | −0.34 |
| WBIC | 2.43 | 2.06 | WBIB | 3.36 | 2.80 |
| WBIAl | 2.47–2.51 | 2.51 | WBIAl | 2.66 | 2.61 |
| WBIS | 1.99–2.02 | 2.10 | WBIS | 1.92 | 1.99 |
| WBIC-Al | 0.40–0.45 | 0.36 | WBIB-Al | 0.60 | 0.50 |
| WBIAl-Al | 0.10 | 0.10 | WBIAl-Al | 0.14 | 0.12 |
| WBIAl-S | 0.89–0.93 | 0.96 | WBIAl-S | 0.85 | 0.91 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, Y.; Wang, Y.; Zhang, M.; Geng, Y.; Su, Z. Sulphur-Bridged BAl5S5+ with 17 Counting Electrons: A Regular Planar Pentacoordinate Boron System. Molecules 2021, 26, 5205. https://doi.org/10.3390/molecules26175205
Ye Y, Wang Y, Zhang M, Geng Y, Su Z. Sulphur-Bridged BAl5S5+ with 17 Counting Electrons: A Regular Planar Pentacoordinate Boron System. Molecules. 2021; 26(17):5205. https://doi.org/10.3390/molecules26175205
Chicago/Turabian StyleYe, Yuhan, Yiqiao Wang, Min Zhang, Yun Geng, and Zhongmin Su. 2021. "Sulphur-Bridged BAl5S5+ with 17 Counting Electrons: A Regular Planar Pentacoordinate Boron System" Molecules 26, no. 17: 5205. https://doi.org/10.3390/molecules26175205