
First-in-Class Isonipecotamide-Based Thrombin and Cholinesterase Dual Inhibitors with Potential for Alzheimer Disease

 ,
,  ,
,  ,
,  ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Results
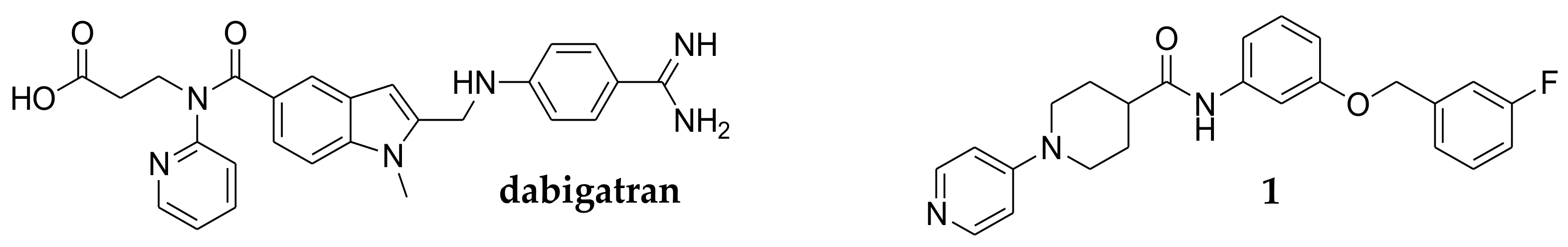
2.1. Target Protein Prediction by Similarity Search
2.2. Chemistry
2.3. Dual Cholinesterase/Coagulation Factor Inhibition by Isonipecotamide Derivatives
3. Materials and Methods
3.1. Chemistry
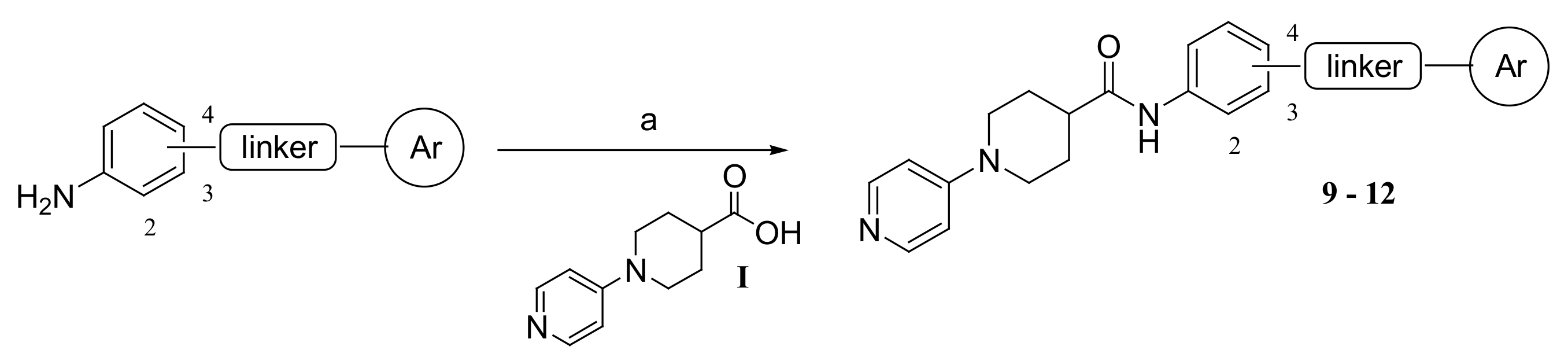
3.1.1. Synthesis of Compounds 9–12
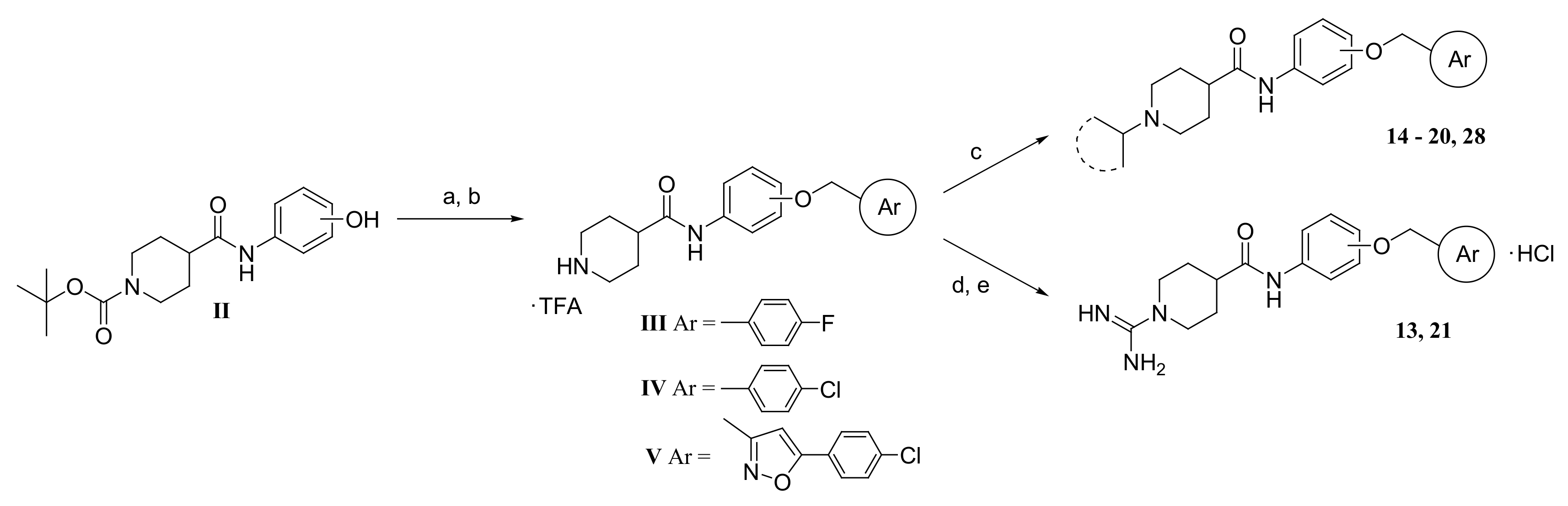
3.1.2. Synthesis of Compounds 14–20, and 28
3.1.3. Synthesis of N-(2-(4-Chlorobenzyloxy)phenyl)-1-amidinopiperidine-4-carboxamide hydrochloride (21)
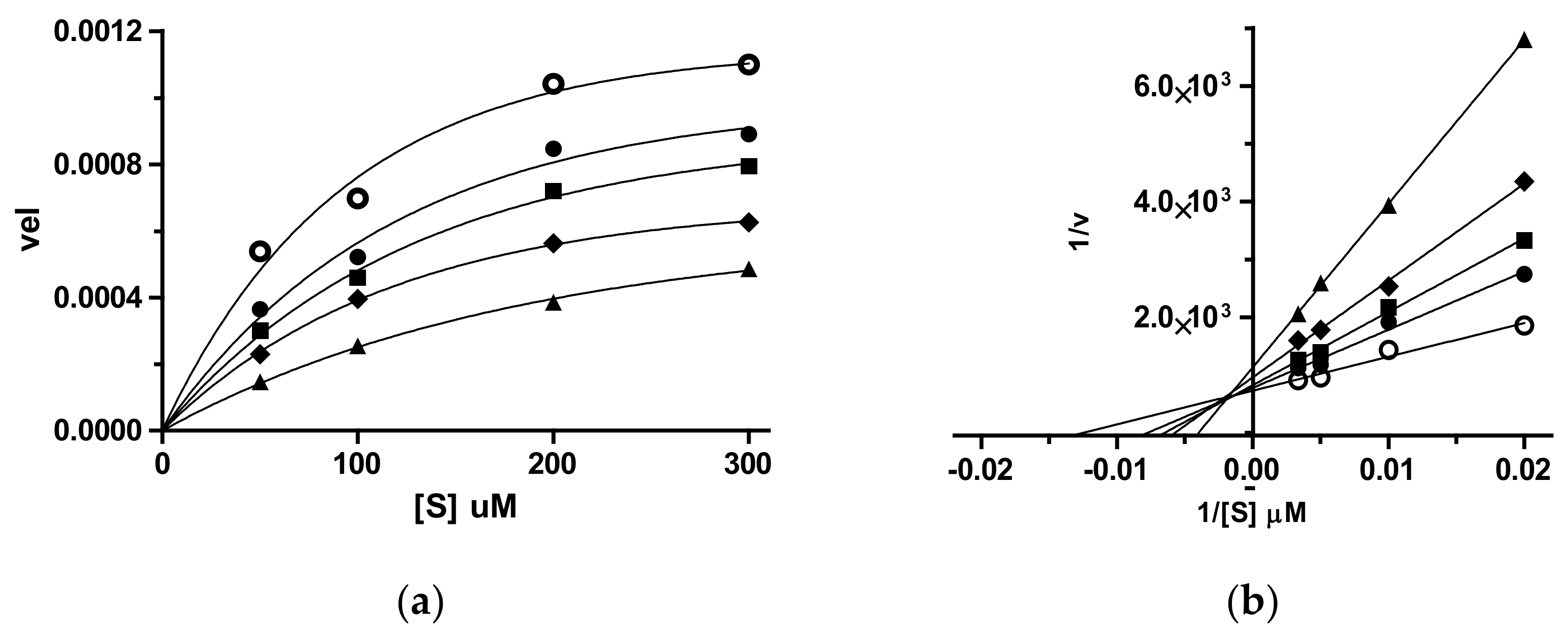
3.2. Enzymatic Assays
3.2.1. Cholinesterases Inhibition Assay
3.2.2. Coagulative Serine Proteases Thrombin and fXa Inhibition Assay
3.3. Chemoinformatics and Computational Chemistry
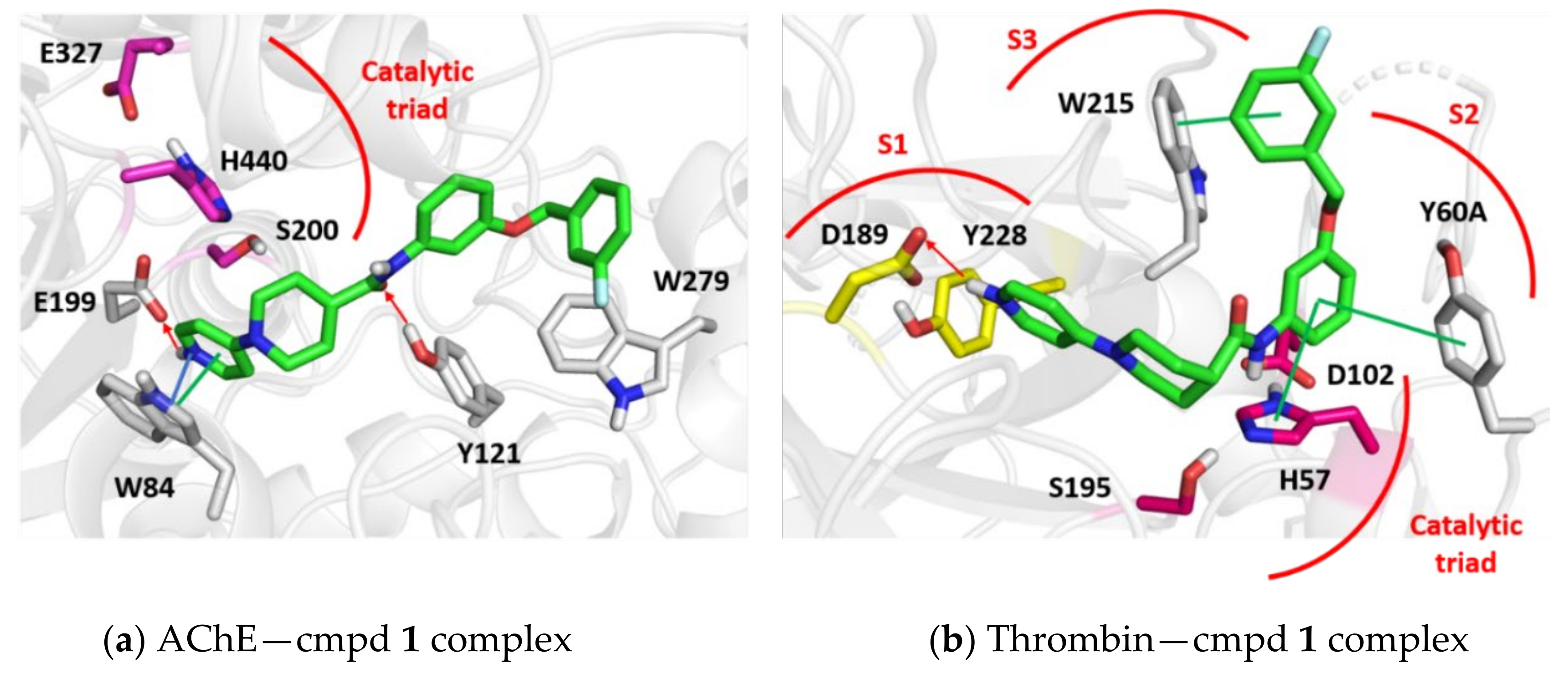
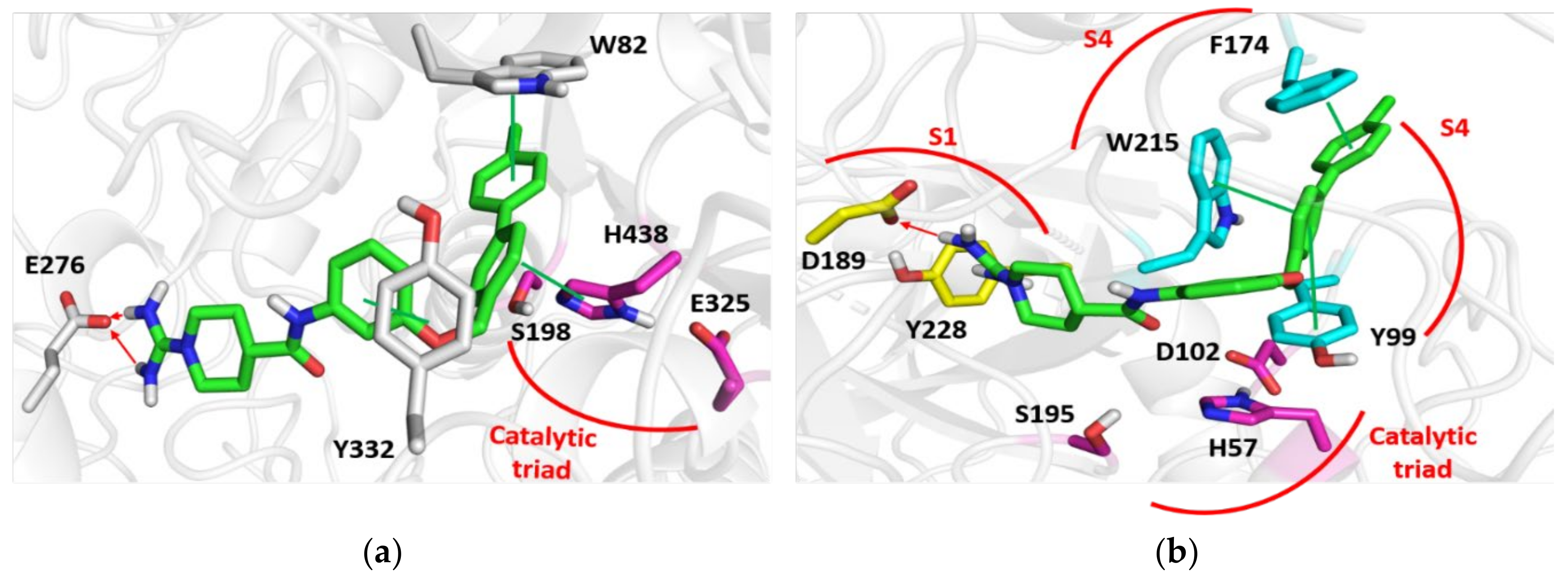
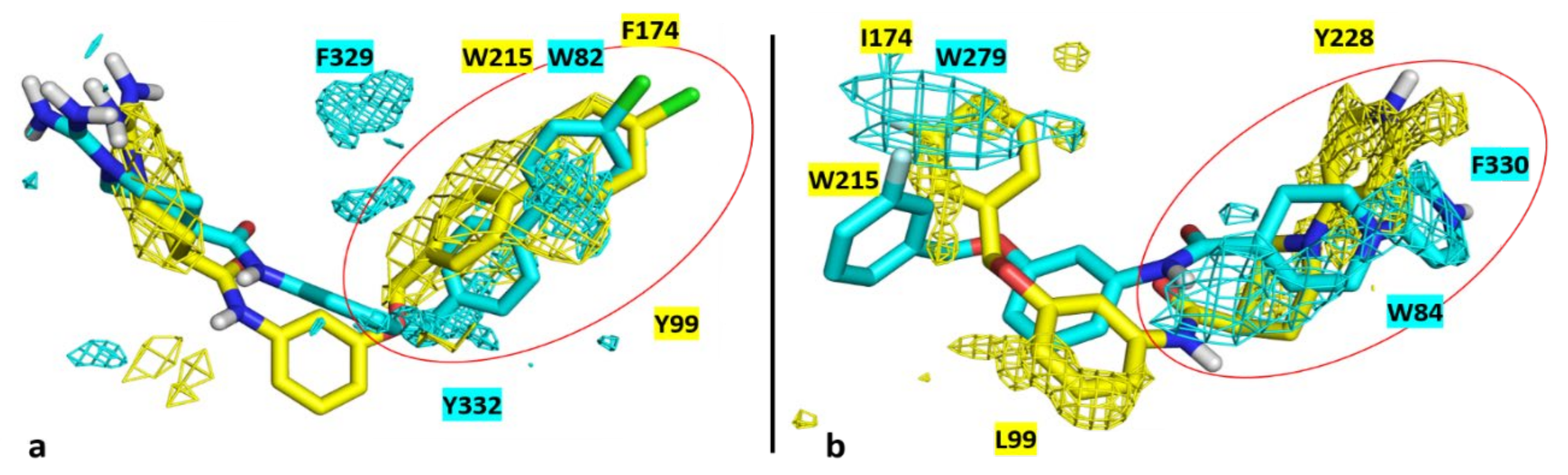
3.4. Molecular Modelling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Patterson, C. World Alzheimer Report 2018: The State of the Art of Dementia Research: New Frontiers. 2018. Available online: https://www.alz.co.uk/research/ (accessed on 21 September 2018).
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chételat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A.; Jones, D.T. Alzheimer disease. Nat. Rev. Dis. Primers 2021, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, A.; Ekavali, A. A review on Alzheimer’s disease pathophysiology and its management: An update. Pharmacol. Rep. 2015, 67, 195–203. [Google Scholar] [CrossRef]
- Geldenhuys, W.J.; Darvesh, A.S. Pharmacotherapy of Alzheimer’s disease: Current and future trends. Expert Rev. Neurother. 2015, 15, 3–5. [Google Scholar] [CrossRef]
- Zupanica, E.; Krambergera, M.G.; von Eulerd, M.; Norrvingf, B.; Winblada, B.; Secniki, J.; Fastbomh, J.; Eriksdotterg, M.; Garcia-Ptaceki, S. Secondary stroke prevention after ischemic stroke in patients with Alzheimer’s disease and other dementia disorders. J. Alzheimer’s Dis. 2020, 73, 1013–1021. [Google Scholar] [CrossRef] [Green Version]
- Scheffer, S.; Hermkens, D.M.A.; van der Weerd, L.; de Vries, H.E.; Daemen, M.J.A.P. Vascular hypothesis of Alzheimer disease. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1265–1283. [Google Scholar] [CrossRef]
- Grossmann, K. Alzheimer’s disease—Rationales for potential treatment withthe thrombin inhibitor dabigatran. Int. J. Mol. Sci. 2021, 22, 4805. [Google Scholar] [CrossRef] [PubMed]
- Marangoni, M.N.; Braun, D.; Situ, A.; Moyano, A.L.; Kalinin, S.; Polak, P.; Givogri, M.I.; Feinstein, D.L. Differential effects on glial activation by a direct versus an indirect thrombin inhibitor. J. Neuroimmunol. 2016, 297, 159–168. [Google Scholar] [CrossRef]
- Iannucci, J.; Renehan, W.; Grammas, P. Thrombin, a mediator of coagulation, inflammation, and neurotoxicity at the neurovascular interface: Implications for Alzheimer’s disease. Front. Neurosci. 2020, 14, 762. [Google Scholar] [CrossRef]
- Cortes-Canteli, M.; Kruyer, A.; Fernandez-Nueda, I.; Marcos-Diaz, A.; Ceron, C.; Richards, A.T.; Jno-Charles, O.C.; Rodriguez, I.; Callejas, S.; Norris, E.; et al. Long-term dabigatran treatment delays Alzheimer’s disease pathogenesis in the TgCRND8 mouse model. J. Am. Coll. Cardiol. 2019, 74, 1910–1923. [Google Scholar] [CrossRef] [PubMed]
- Ihara, M.; Saito, S. Drug repositioning for alzheimer’sdisease: Finding hidden clues in old drugs. J. Alzheimer’s Dis. 2020, 74, 1013–1028. [Google Scholar] [CrossRef]
- de Candia, M.; Liantonio, F.; Carotti, A.; De Cristofaro, R.; Altomare, C. Fluorinated benzyloxyphenyl piperidine-4-carboxamides with dual function against thrombosis: Inhibitors of factor Xa and platelet aggregation. J. Med. Chem. 2009, 52, 1018–1028. [Google Scholar] [CrossRef] [PubMed]
- Lopopolo, G.; Fiorella, F.; de Candia, M.; Nicolotti, O.; Martel, S.; Carrupt, P.A.; Altomare, C. Biarylmethoxy isonipecotanilides as potent and selective inhibitors of blood coagulation factor Xa. Eur. J. Pharm. Sci. 2011, 42, 180–191. [Google Scholar] [CrossRef]
- Lopopolo, G.; de Candia, M.; Panza, L.; Romano, M.R.; Lograno, M.D.; Campagna, F.; Altomare, C. β-D-glucosyl conjugates of highly potent inhibitors of blood coagulation factor Xa bearing 2-chorothiophene as a P1 motif. ChemMedChem 2012, 7, 1669–1677. [Google Scholar] [CrossRef]
- de Candia, M.; Fiorella, F.; Lopopolo, G.; Carotti, A.; Romano, M.R.; Lograno, M.D.; Martel, S.; Carrupt, P.A.; Belviso, B.D.; Caliandro, R.; et al. Synthesis and biological evaluation of direct thrombin inhibitors bearing 4-(piperidin-1-yl)pyridine at the P1 position with potent anticoagulant activity. J. Med. Chem. 2013, 56, 8696–8711. [Google Scholar] [CrossRef]
- Belviso, B.D.; Caliandro, R.; de Candia, M.; Zaetta, G.; Lopopolo, G.; Incampo, F.; Colucci, M.; Altomare, C. How a β-D-glucoside side chain enhances binding affinity to thrombin of inhibitors bearing 2-chlorothiophene as P1 moiety: Crystallography, fragment deconstruction study, and evaluation of antithrombotic properties. J. Med. Chem. 2014, 57, 8563–8575. [Google Scholar] [CrossRef] [PubMed]
- Alberga, D.; Trisciuzzi, D.; Montaruli, M.; Leonetti, F.; Mangiatordi, G.F.; Nicolotti, O. A new approach for drug target and bioactivity prediction: The Multi-fingerprint Similarity Search aLgorithm (MuSSeL). J. Chem. Inf. Model. 2019, 59, 586–596. [Google Scholar] [CrossRef]
- Montaruli, M.; Alberga, D.; Ciriaco, F.; Trisciuzzi, D.; Tondo, A.R.; Mangiatordi, G.F.; Nicolotti, O. Accelerating drug discovery by early protein drug target prediction based on multi-fingerprint similarity search. Molecules 2019, 24, 2233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavalli, A.; Bolognesi, M.L.; Minarini, A.; Rosini, M.; Tumiatti, V.; Recanatini, M.; Melchiorre, C. Multi-target-directed ligands to combat neurodegenerative diseases. J. Med. Chem. 2008, 51, 347–372. [Google Scholar] [CrossRef]
- Maramai, S.; Benchekroun, M.; Gabr, M.T.; Yahiaoui, S. Multitarget therapeutic strategies for Alzheimer’s disease: Review on emerging target combinations. BioMed Res. Int. 2020, 2020, 5120230. [Google Scholar] [CrossRef]
- De Freitas Silva, M.; Dias, K.S.T.; Gontijo, V.S.; Ortiz, C.J.C.; Viegas, C., Jr. Multi-target directed drugs as a modern approach for drug design towards Alzheimer’s disease: An update. Curr. Med. Chem. 2018, 25, 3491–3525. [Google Scholar] [CrossRef]
- Mezeiova, E.; Chalupova, K.; Nepovimova, E.; Gorecki, L.; Prchal, L.; Malinak, D.; Kuca, K.; Soukup, O.; Korabecny, J. Donepezil derivatives targeting amyloid-β cascade in Alzheimer’s disease. Curr. Alzheimer Res. 2019, 16, 772–800. [Google Scholar] [CrossRef] [PubMed]
- Ha, Z.Y.; Mathew, S.; Yeong, K.Y. Butyrylcholinesterase: A multifaced pharmacological target and too. Curr. Prot. Pept. Sci. 2020, 21, 99–109. [Google Scholar] [CrossRef] [PubMed]
- de Candia, M.; Zaetta, G.; Denora, N.; Tricarico, D.; Majellaro, M.; Cellamare, S.; Altomare, C.D. New azepino[4,3-b]indole derivatives as nanomolar selective inhibitors of human butyrylcholinesterase showing protective effects against NMDA-induced neurotoxicity. Eur. J. Med. Chem. 2017, 125, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Nicolotti, O.; Giangreco, I.; Miscioscia, T.F.; Carotti, A. Improving quantitative structure-activity relationships through multiobjective optimization. J. Chem. Inf. Model. 2009, 49, 2290–2302. [Google Scholar] [CrossRef] [PubMed]
- Nicolotti, O.; Miscioscia, T.F.; Carotti, A.; Leonetti, F.; Carotti, A. An integrated approach to ligand- and structure-based drug design: Development and application to a series of serine protease inhibitors. J. Chem. Inf. Model. 2008, 48, 1211–1226. [Google Scholar] [CrossRef] [PubMed]
- Purgatorio, R.; de Candia, M.; Catto, M.; Carrieri, A.; Pisani, L.; De Palma, A.; Toma, M.; Ivanova, O.A.; Voskressensky, L.G.; Altomare, C.D. Investigating 1,2,3,4,5,6-hexahydroazepino[4,3-b]indole as scaffold of butyrylcholinesterase-selective inhibitors with additional neuroprotective activities for Alzheimer’s disease. Eur. J. Med. Chem. 2019, 177, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Prusoff, W.H. Relation between the inhibition constant (Ki) and the concentration of inhibitor which causes fifty per cent inhibition (IC50) of an enzymic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar]
- Gao, X.H.; Liu, L.B.; Liu, H.R.; Tang, J.J.; Kang, L.; Wu, H.; Cui, P.; Yan, J. Structure–activity relationship investigation of benzamide and picolinamide derivatives containing dimethylamine side chain as acetylcholinesterase inhibitors. J. Enzyme Inhib. Med. Chem. 2017, 33, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.E.; Pickett, S.D. Computational methods for the prediction of “drug-likeness”. Drug Discov. Today 2000, 5, 49–58. [Google Scholar] [CrossRef]
- Geldenhuys, W.J.; Mohammad, A.S.; Adkins, C.E.; Lockman, P.R. Molecular determinants of blood-barin-barrier permeation. Ther. Deliv. 2015, 6, 961–971. [Google Scholar] [CrossRef]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y.J. admetSAR: A comprehensive source and free tool for assessment of chemical ADMET properties. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar] [CrossRef] [PubMed]
- Nazaré, M.; Will, D.W.; Matter, H.; Schreuder, H.; Ritter, K.; Urmann, M.; Essrich, M.; Bauer, A.; Wagner, M.; Czech, J.; et al. Probing the subpockets of factor Xa reveals two binding modes for inhibitors based on a 2-carboxyindole scaffold: A study combining structure–activity relationship and X-ray crystallography. J. Med. Chem. 2005, 48, 4511–4525. [Google Scholar] [CrossRef] [PubMed]
- Kryger, G.; Silman, I.; Sussman, J.L. Structure of acetylcholinesterase complexed with E2020 (Aricept®): Implications for the design of new anti-Alzheimer drugs. Structure 1999, 7, 297–307. [Google Scholar] [CrossRef] [Green Version]
- Košak, U.; Strašek, N.; Knez, D.; Jukič, M.; Žakelj, S.; Zahirović, A.; Pišlar, A.; Brazzolotto, X.; Nachon, F.; Kos, J.; et al. N-Alkylpiperidine carbamates as potential anti-Alzheimer’s agents. Eur. J. Med. Chem. 2020, 197, 112282. [Google Scholar] [CrossRef]
- Shi, Y.; Li, C.; O’Connor, S.P.; Zhang, J.; Shi, M.; Bisaha, S.N.; Wang, Y.; Sitkoff, D.; Pudzianowski, A.T.; Huang, C.; et al. Aroylguanidine-based factor Xa inhibitors: The discovery of BMS-344577. Bioorg. Med. Chem. Lett. 2009, 19, 6882–6889. [Google Scholar] [CrossRef]
- Engh, R.A.; Brandstetter, H.; Sucher, G.; Eichinger, A.; Baumann, U.; Bode, W.; Huber, R.; Poll, T.; Rudolph, R.; von der Saal, W. Enzyme flexibility, solvent and ‘weak’ interactions characterize thrombin–ligand interactions: Implications for drug design. Structure 1996, 4, 1353–1362. [Google Scholar] [CrossRef] [Green Version]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Schrödinger Release. Protein Preparation Wizard; Schrödinger LLC: New York, NY, USA, 2016. [Google Scholar]
- Schrödinger Release. LigPrep; Schrödinger LLC: New York, NY, USA, 2020. [Google Scholar]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release. Prime; Schrödinger LLC: New York, NY, USA, 2020. [Google Scholar]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins Struct. Funct. Bioinform. 2004, 55, 351–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 model: A next generation energy model for high resolution protein structure modeling: The VSGB 2.0 energy model. Proteins 2011, 79, 2794–2812. [Google Scholar] [CrossRef] [Green Version]
- Harder, E.; Damm, W.; Maple, J.; Wu, C.; Reboul, M.; Xiang, J.Y.; Wang, L.; Lupyan, D.; Dahlgren, M.K.; Knight, J.L.; et al. OPLS3: A force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput. 2016, 12, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Siragusa, L.; Cross, S.; Baroni, M.; Goracci, L.; Cruciani, G. BioGPS: Navigating biological space to predict polypharmacology, off-targeting, and selectivity. Proteins Struct. Funct. Bioinform. 2015, 83, 517–532. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, K. Anticoagulants for treatment of Alzheimer’s disease. J. Alzheimer’s Dis. 2020, 77, 1373–1382. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Ki (μM) 1 | |||||||
|---|---|---|---|---|---|---|---|
| Cmpd | Linker pos. | Linker | F-pos. | thr 2 | fXa 3 | AChE 4 | BChE 5 |
| 1 6 | 3 | OCH2 | 3′ | 0.006 | 5.60 | 0.058 | 6.95 |
| 2 6 | 3 | OCH2 | 4′ | 0.210 | 1.10 | 0.240 | 1.21 |
| 3 6 | 3 | CH2O | 4′ | 26.8 | 0.774 | 0.165 | 2.60 |
| 4 6 | 4 | OCH2 | 3′ | 6.70 | 25.1 | 3.88 | 0.425 |
| 5 6 | 2 | OCH2 | 3′ | (10 ± 5%) | (2 ± 2%) | (12 ± 8%) | 0.655 |
| 6 6 | 3 | OCH2CH2 | 3′ | 2.14 | 0.488 | 0.369 | 1.75 |
| 7 6 | 3 | OCH2CH2 | 4′ | 4.46 | 0.133 | 0.650 | 5.63 |
| 8 6 | 3 | CH2CH2 | 4′ | 3.72 | 37.7 | 0.495 | 8.20 |
| 9 | 3 | CONH | 4′ | 4.47 | 4.04 | 0.270 | 8.25 |
| 10 | 3 | CONHCH2 | 4′ | 10.6 | 2.40 | 0.255 | 0.370 |
| 11 | 3 | SO2NHCH2 | 4′ | 11.2 | 3.09 | 0.155 | 4.10 |
| 12 | 3 | NHCONH | 4′ | 26.7 | 16.5 | 0.735 | (15 ± 7%) |
| dabigatran | 0.0042 | 5.10 | (36 ± 7%) | ||||
| apixaban | 0.00012 | ||||||
| donepezil | 0.021 | 2.25 | |||||

| Ki (μM) 1 | |||||||
|---|---|---|---|---|---|---|---|
| Compd | OCH2 pos. | R | X | thr 2 | fXa 3 | AChE 4 | BChE 5 |
| 13 | 3 | C(=NH)NH2 | F | 20.0 | 33.0 | 10.0 | 0.260 |
| 14 | 3 | i-Pr | F | (5 ± 5%) | (28 ± 8%) | 15.5 | 0.130 |
| 15 | 3 | c-Pent | F | 30.0 | (40 ± 6%) | (27 ± 9%) | 0.115 |
| 16 | 4 | i-Pr | F | (11 ± 6%) | (5 ± 5%) | 6.25 | 2.36 |
| 17 | 4 | c-Pent | F | (25 ± 6%) | (28 ± 9%) | 3.52 | 0.255 |
| 18 | 2 | i-Pr | F | 5.35 | 10.5 | (15 ± 5%) | 0.378 |
| 19 | 2 | c-Pent | F | (12 ± 3%) | (28 ± 3%) | (35 ± 7%) | 0.215 |
| 20 | 2 | i-Pr | Cl | (20 ± 5%) | 0.484 | (11 ± 2%) | 0.105 |
| 21 | 2 | C(=NH)NH2 | Cl | 25.9 | 60.6 | (38 ± 3%) | 8.20 |

| OCH2 | Ki (μM) 1 | ||||||
|---|---|---|---|---|---|---|---|
| Compd | Pos | R | Ar–Ar | thr 2 | fXa 3 | AChE 4 | BChE 5 |
| 22 | 3 | i-Pr |  | (5 ± 2%) | 0.165 | 3.33 | (35 ± 10%) |
| 23 | 3 | C(=NH)NH2 | 2.04 | 0.031 | 5.80 | 0.090 | |
| 24 | 3 | i-Pr |  | (15 ± 5%) | 0.218 | 6.30 | 3.06 |
| 25 | 3 | C(=NH)NH2 | 0.630 | 0.026 | 11.0 | 7.67 | |
| 26 | 2 | i-Pr | 13.7 | 0.007 | 11.5 | 1.11 | |
| 27 | 2 | C(=NH)NH2 | 21.2 | 0.060 | (16 ± 3%) | 10.0 | |
| 28 | 3 | i-Pr |  | 23.0 | 0.867 | 7.00 | 4.10 |
| 29 | 3 | C(=NH)NH2 | 15.3 | 0.388 | 10.0 | 1.12 | |
| 30 | 3 | i-Pr |  | 16.0 | 0.090 | 5.10 | 2.13 |
| 31 | 3 | C(=NH)NH2 | 15.0 | 0.015 | (31 ± 6%) | 1.62 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Purgatorio, R.; Gambacorta, N.; de Candia, M.; Catto, M.; Rullo, M.; Pisani, L.; Nicolotti, O.; Altomare, C.D. First-in-Class Isonipecotamide-Based Thrombin and Cholinesterase Dual Inhibitors with Potential for Alzheimer Disease. Molecules 2021, 26, 5208. https://doi.org/10.3390/molecules26175208
Purgatorio R, Gambacorta N, de Candia M, Catto M, Rullo M, Pisani L, Nicolotti O, Altomare CD. First-in-Class Isonipecotamide-Based Thrombin and Cholinesterase Dual Inhibitors with Potential for Alzheimer Disease. Molecules. 2021; 26(17):5208. https://doi.org/10.3390/molecules26175208
Chicago/Turabian StylePurgatorio, Rosa, Nicola Gambacorta, Modesto de Candia, Marco Catto, Mariagrazia Rullo, Leonardo Pisani, Orazio Nicolotti, and Cosimo D. Altomare. 2021. "First-in-Class Isonipecotamide-Based Thrombin and Cholinesterase Dual Inhibitors with Potential for Alzheimer Disease" Molecules 26, no. 17: 5208. https://doi.org/10.3390/molecules26175208
APA StylePurgatorio, R., Gambacorta, N., de Candia, M., Catto, M., Rullo, M., Pisani, L., Nicolotti, O., & Altomare, C. D. (2021). First-in-Class Isonipecotamide-Based Thrombin and Cholinesterase Dual Inhibitors with Potential for Alzheimer Disease. Molecules, 26(17), 5208. https://doi.org/10.3390/molecules26175208