
Anharmonic DFT Study of Near-Infrared Spectra of Caffeine: Vibrational Analysis of the Second Overtones and Ternary Combinations





Abstract
:1. Introduction
2. Results and Discussion
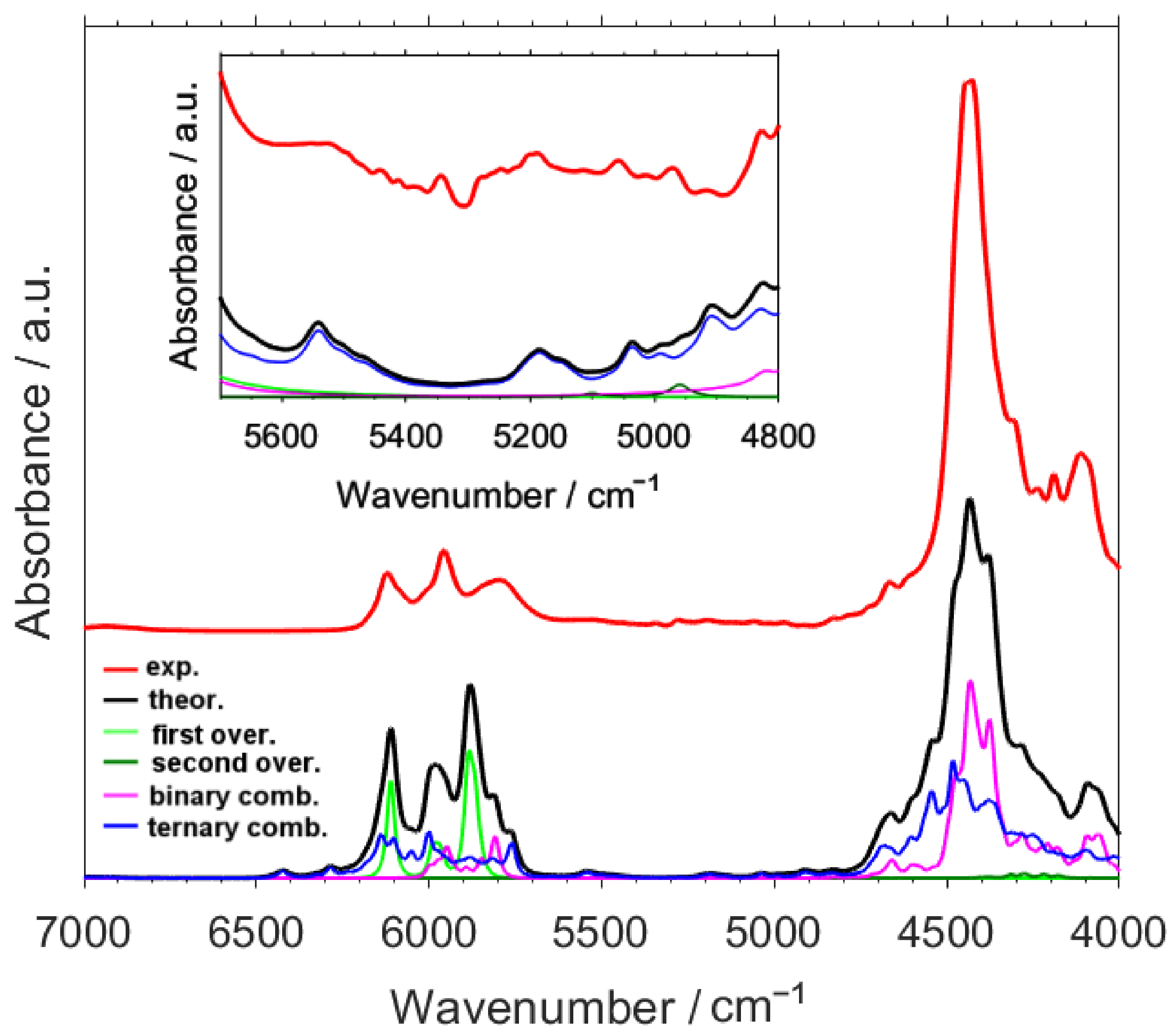
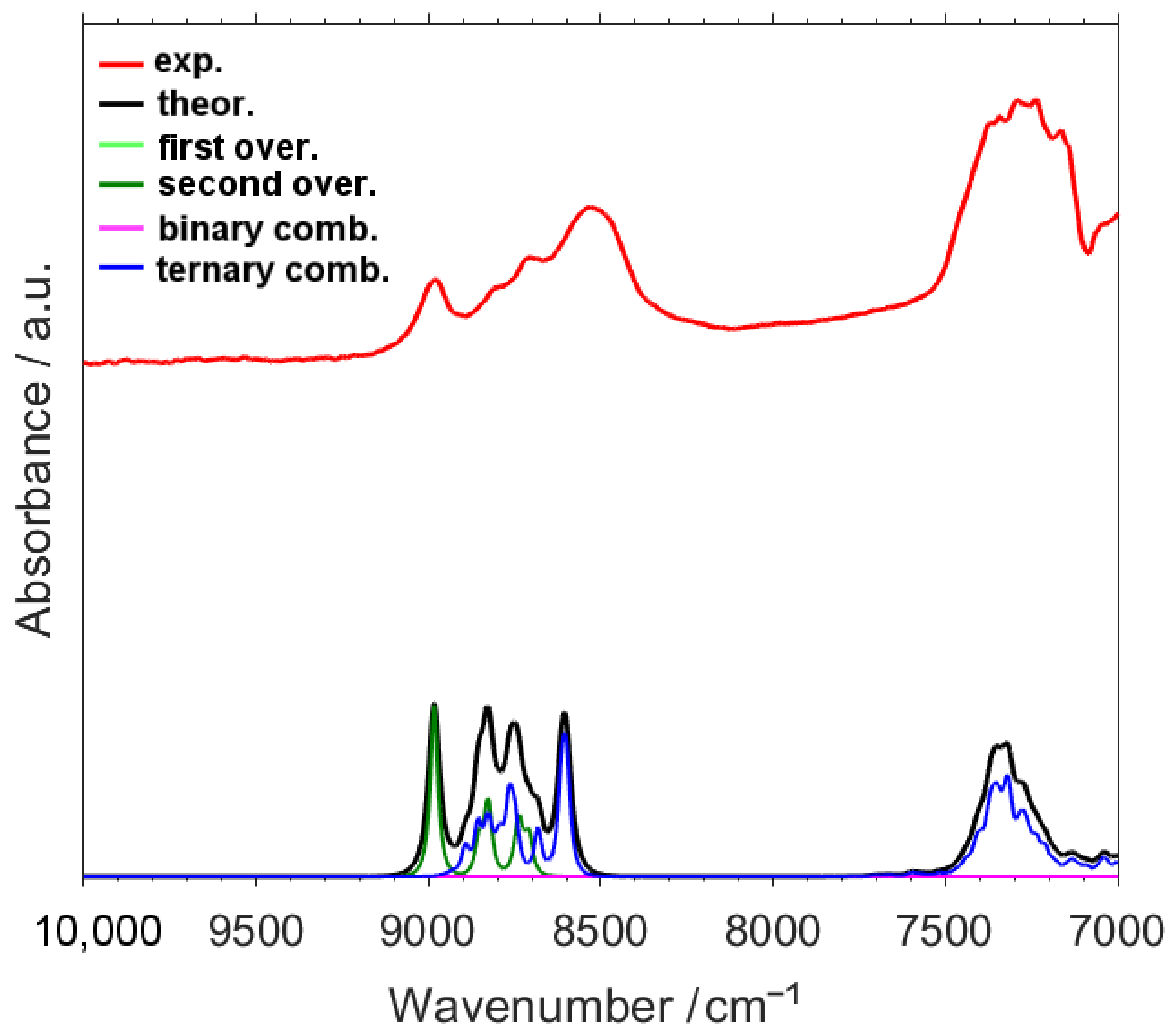
2.1. Analysis of a NIR Spectrum of Caffeine
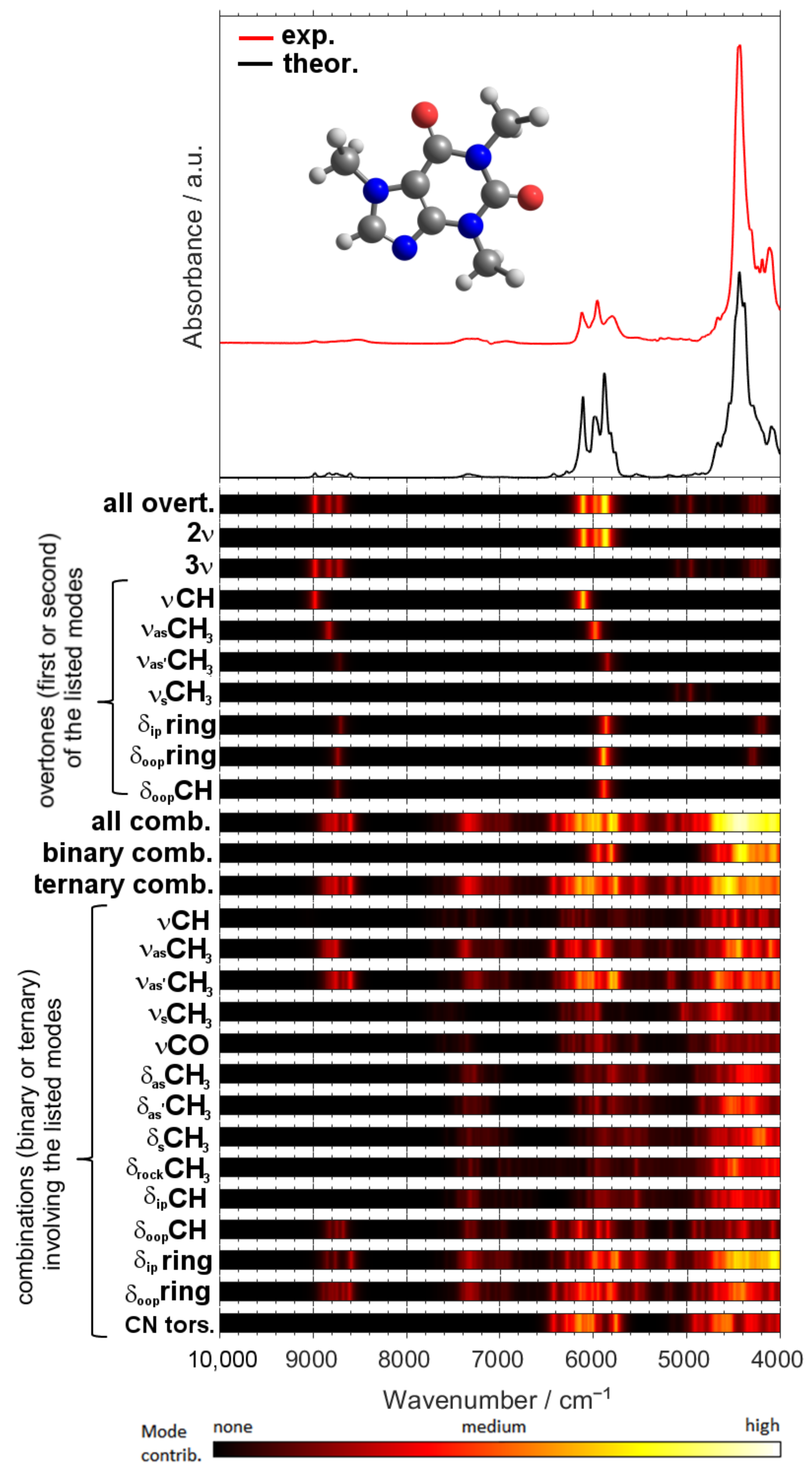
2.2. Detailed Interpretation of NIR Bands of Caffeine
2.3. Linking the Interpreted NIR Absorption Regions of Caffeine with Wavenumber Ranges Found to Be Meaningful in Quantitative Analytical Applications
3. Materials and Methods
3.1. Experimental
3.2. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Ozaki, Y.; Morisawa, Y. Principles and characteristics of NIR spectroscopy. In Near-Infrared Spectroscopy; Ozaki, Y., Huck, C.W., Tsuchikawa, S., Engelsen, S.B., Eds.; Springer: Singapore, 2021; pp. 37–60. [Google Scholar] [CrossRef]
- Ozaki, Y.; Huck, C.W.; Beć, K.B. Near infrared spectroscopy and its applications. In Molecular and Laser Spectroscopy. Advances and Applications; Gupta, V.P., Ed.; Elsevier: San Diego, CA, USA, 2018; pp. 11–38. [Google Scholar] [CrossRef]
- Cozzolino, D. Infrared Spectroscopy: Theory, Developments and Applications; Nova Science Publishers: New York, NY, USA, 2014. [Google Scholar]
- Dorrepaal, R.; Gowen, A. Identification of magnesium oxychloride cement biomaterial heterogeneity using Raman chemical mapping and NIR hyperspectral chemical imaging. Sci. Rep. 2018, 8, 13034. [Google Scholar] [CrossRef] [PubMed]
- Baeten, V.; Dardenne, P. Application of NIR in agriculture. In Near-Infrared Spectroscopy; Ozaki, Y., Huck, C.W., Tsuchikawa, S., Engelsen, S.B., Eds.; Springer: Singapore, 2021; pp. 331–345. [Google Scholar] [CrossRef]
- Baeten, V.; Rogez, H.; Fernández Pierna, J.A.; Vermeulen, P.; Dardenne, P. Vibrational spectroscopy methods for the rapid control of agro-food products. In Handbook of Food Analysis, 3rd ed.; Toldra, F., Nollet, L.M.L., Eds.; CRC Press: Boca Raton, FL, USA, 2015; Volume 2, pp. 591–614. [Google Scholar]
- Manley, M.; Williams, P.J. Applications: Food Science. In Near-Infrared Spectroscopy; Ozaki, Y., Huck, C.W., Tsuchikawa, S., Engelsen, S.B., Eds.; Springer: Singapore, 2021; pp. 347–359. [Google Scholar] [CrossRef]
- Cozzolino, D. From consumers’ science to food functionality—Challenges and opportunities for vibrational spectroscopy. Adv. Food Nutr. Res. 2021, 97, 119–146. [Google Scholar] [CrossRef]
- Kessler, R.W.; Kessler, W. Inline and online process analytical technology with an outlook for the petrochemical industry. In Near-Infrared Spectroscopy; Ozaki, Y., Huck, C.W., Tsuchikawa, S., Engelsen, S.B., Eds.; Springer: Singapore, 2021; pp. 553–593. [Google Scholar] [CrossRef]
- Scherzer, T. Applications of NIR techniques in polymer coatings and synthetic textiles. In Near-Infrared Spectroscopy; Ozaki, Y., Huck, C.W., Tsuchikawa, S., Engelsen, S.B., Eds.; Springer: Singapore, 2021; pp. 475–516. [Google Scholar] [CrossRef]
- Igne, B.; Ciurczak, E.W. Near-infrared spectroscopy in the pharmaceutical industry. In Near-Infrared Spectroscopy; Ozaki, Y., Huck, C.W., Tsuchikawa, S., Engelsen, S.B., Eds.; Springer: Singapore, 2021; pp. 391–412. [Google Scholar] [CrossRef]
- Huck, C.W. Infrared spectroscopy in near-infrared/ infrared bioanalysis including imaging. In Encyclopedia of Analytical Chemistry: Applications, Theory and Instrumentation; Meyers, R.A., Ed.; John Wiley & Sons, Ltd.: New York, NY, USA, 2016. [Google Scholar] [CrossRef]
- Ishigaki, M.; Ozaki, Y. Near-infrared spectroscopy and imaging in protein research. In Vibrational Spectroscopy in Protein Research; Ozaki, Y., Baranska, M., Lednev, I.K., Wood, B.R., Eds.; Academic Press: London, UK, 2020; pp. 143–176. [Google Scholar]
- Ozaki, Y.; Huck, C.W.; Ishigaki, M.; Ishikawa, D.; Ikehata, A.; Shinzawa, H. Near-infrared spectroscopy in biological molecules and tissues. In Encyclopedia of Biophysics; Roberts, G., Watts, A., European Biophysical Societies, Eds.; Springer: Berlin/Heidelberg, Germany, 2018; pp. 1681–1780. [Google Scholar] [CrossRef]
- Heise, H.M. Medical applications of NIR spectroscopy. In Near-Infrared Spectroscopy; Ozaki, Y., Huck, C.W., Tsuchikawa, S., Engelsen, S.B., Eds.; Springer: Singapore, 2021; pp. 437–473. [Google Scholar] [CrossRef]
- Türker-Kaya, S.; Huck, C.W. A review of mid-infrared and near-infrared imaging: Principles, concepts and applications in plant tissue analysis. Molecules 2017, 22, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czarnecki, M.A.; Morisawa, Y.; Futami, Y.; Ozaki, Y. Advances in molecular structure and interaction studies using near-infrared spectroscopy. Chem. Rev. 2015, 115, 9707–9744. [Google Scholar] [CrossRef]
- Czarnecki, M.A. Frequency shift or intensity shift? The origin of spectral changes in vibrational spectra. Vib. Spectrosc. 2012, 58, 193–198. [Google Scholar] [CrossRef]
- Czarnecki, M.A.; Beć, K.B.; Grabska, J.; Hofer, T.S.; Ozaki, Y. Overview of application of NIR spectroscopy to physical chemistry. In Near-Infrared Spectroscopy; Ozaki, Y., Huck, C.W., Tsuchikawa, S., Engelsen, S.B., Eds.; Springer: Singapore, 2021; pp. 297–330. [Google Scholar] [CrossRef]
- Beć, K.B.; Grabska, J.; Huck, C.W. Near-infrared spectroscopy in bio-applications. Molecules 2020, 25, 2948. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, A.S. IR spectral group frequencies of organic compounds. In Encyclopedia of Spectroscopy and Spectrometry, 2nd ed.; Lindon, J.C., Ed.; Academic Press: London, UK, 2010; pp. 1187–1198. [Google Scholar] [CrossRef]
- Larkin, P. IR and Raman spectra-structure correlations: Characteristic group frequencies. In Infrared and Raman Spectroscopy. Principles and Spectral Interpretation; Larkin, P., Ed.; Elsevier: San Diego, CA, USA, 2011; pp. 73–115. [Google Scholar] [CrossRef]
- Weyer, L.G.; Lo, S.C. Spectra-structure correlations in the near-infrared. In Handbook of Vibrational Spectroscopy; Chalmers, J.M., Griffiths, P.R., Eds.; John Wiley & Sons, Ltd.: Chichester, UK, 2006; Volume 3, pp. 1817–1837. [Google Scholar] [CrossRef]
- Barone, V.; Alessandrini, S.; Biczysko, M.; Cheeseman, J.R.; Clary, D.C.; McCoy, A.B.; DiRisio, R.J.; Neese, F.; Melosso, M.; Puzzarini, C. Computational molecular spectroscopy. Nat. Rev. Methods Primers 2021, 1, 38. [Google Scholar] [CrossRef]
- Bloino, J.; Baiardi, A.; Biczysko, M. Aiming at an accurate prediction of vibrational and electronic spectra for medium-to-large molecules: An overview. Int. J. Quantum Chem. 2016, 116, 1543–1574. [Google Scholar] [CrossRef] [Green Version]
- Beć, K.B.; Huck, C.W. Breakthrough potential in near-infrared spectroscopy: Spectra simulation. A review of recent developments. Front. Chem. 2019, 7, 48. [Google Scholar] [CrossRef] [Green Version]
- Beć, K.B.; Grabska, J.; Huck, C.W. Current and future research directions in computer-aided near-infrared spectroscopy: A perspective. Spectrochim. Acta A 2021, 254, 119625. [Google Scholar] [CrossRef]
- Paoloni, L.; Mazzeo, G.; Longhi, G.; Abbate, S.; Fusè, M.; Bloino, J.; Barone, V. Toward fully unsupervised anharmonic computations complementing experiment for robust and reliable assignment and interpretation of IR and VCD spectra from Mid-IR to NIR: The case of 2,3-butanediol and trans-1,2-cyclohexanediol. J. Phys. Chem. A 2020, 124, 1011–1024. [Google Scholar] [CrossRef]
- Beć, K.B.; Grabska, J.; Ozaki, Y. Advances in anharmonic methods and their applications to vibrational spectroscopies. In Frontiers of Quantum Chemistry; Wójcik, M.J., Nakatsuji, H., Kirtman, B., Ozaki, Y., Eds.; Springer: Singapore, 2017; pp. 438–512. [Google Scholar] [CrossRef]
- Beć, K.B.; Grabska, J.; Huck, C.W.; Ozaki, Y. Quantum mechanical simulation of near-infrared spectra. Applications in physical and analytical chemistry. In Molecular Spectroscopy: A Quantum Chemistry Approach; Ozaki, Y., Wójcik, M.J., Popp, J., Eds.; Wiley-VCH: Weinheim, Germany, 2019; Volume 2, pp. 353–388. [Google Scholar] [CrossRef]
- Beć, K.B.; Futami, Y.; Wójcik, M.J.; Ozaki, Y. A spectroscopic and theoretical study in the near-infrared region of low concentration aliphatic alcohols. Phys. Chem. Chem. Phys. 2016, 18, 13666–13682. [Google Scholar] [CrossRef]
- Grabska, J.; Beć, K.B.; Ozaki, Y.; Huck, C.W. Temperature drift of conformational equilibria of butyl alcohols studied by near-infrared spectroscopy and fully anharmonic DFT. J. Phys. Chem. A 2017, 121, 1950–1961. [Google Scholar] [CrossRef] [PubMed]
- Beć, K.B.; Grabska, J.; Huck, C.W.; Czarnecki, M.A. Effect of conformational isomerism on NIR spectra of ethanol isotopologues. Spectroscopic and anharmonic DFT study. J. Mol. Liq. 2020, 310, 113271. [Google Scholar] [CrossRef]
- Barone, V.; Biczysko, M.; Bloino, J.; Cimino, P.; Penocchio, E.; Puzzarini, C. CC/DFT route toward accurate structures and spectroscopic features for observed and elusive conformers of flexible molecules: Pyruvic acid as a case study. J. Chem. Theory Comput. 2015, 11, 4342–4363. [Google Scholar] [CrossRef] [Green Version]
- Grabska, J.; Ishigaki, M.; Beć, K.B.; Wójcik, M.J.; Ozaki, Y. Correlations between Structure and Near-Infrared Spectra of Saturated and Unsaturated Carboxylic Acids. Insight from Anharmonic Density Functional Theory Calculations. J. Phys. Chem. A 2017, 121, 3437–3451. [Google Scholar] [CrossRef]
- Grabska, J.; Beć, K.B.; Ishigaki, M.; Wójcik, M.J.; Ozaki, Y. Spectra-structure correlations of saturated and unsaturated medium-chain fatty acids. Near-infrared and anharmonic DFT study of hexanoic acid and sorbic acid. Spectrochim. Acta A 2017, 185, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Grabska, J.; Beć, K.B.; Ishigaki, M.; Huck, C.W.; Ozaki, Y. NIR spectra simulations by anharmonic DFT-saturated and unsaturated long-chain fatty acids. J. Phys. Chem. B 2018, 122, 6931–6944. [Google Scholar] [CrossRef]
- Barone, V.; Biczyszko, M.; Bloino, J.; Puzzarini, C. Glycine conformers: A never-ending story? Phys. Chem. Chem. Phys. 2013, 15, 1358–1363. [Google Scholar] [CrossRef]
- Shu, C.; Jiang, Z.; Biczysko, M. Toward accurate prediction of amino acid derivatives structure and energetics from DFT: Glycine conformers and their interconversions. J. Mol. Model. 2020, 26, 129. [Google Scholar] [CrossRef]
- Beć, K.B.; Grabska, J.; Ozaki, Y.; Czarnecki, M.A.; Huck, C.W. Simulated NIR spectra as sensitive markers of the structure and interactions in nucleobases. Sci. Rep. 2019, 9, 17398. [Google Scholar] [CrossRef] [PubMed]
- Kirchler, C.G.; Pezzei, C.K.; Beć, K.B.; Mayr, S.; Ishigaki, M.; Ozaki, Y.; Huck, C.W. Critical evaluation of spectral information of benchtop vs. portable near-infrared spectrometers: Quantum chemistry and two-dimensional correlation spectroscopy for a better understanding of PLS regression models of the rosmarinic acid content in Rosmarini folium. Analyst 2017, 142, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Beć, K.B.; Grabska, J.; Kirchler, C.G.; Huck, C.W. NIR spectra simulation of thymol for better understanding of the spectra forming factors, phase and concentration effects and PLS regression features. J. Mol. Liq. 2018, 268, 895–902. [Google Scholar] [CrossRef]
- Mayr, S.; Schmelzer, J.; Kirchler, C.G.; Pezzei, C.K.; Beć, K.B.; Grabska, J.; Huck, C.W. Theae nigrae folium: Comparing the analytical performance of benchtop and handheld near-infrared spectrometers. Talanta 2020, 221, 121165. [Google Scholar] [CrossRef]
- Mayr, S.; Beć, K.B.; Grabska, J.; Schneckenreiter, E.; Huck, C.W. Near-infrared spectroscopy in quality control of Piper nigrum: A Comparison of performance of benchtop and handheld spectrometers. Talanta 2020, 223, 121809. [Google Scholar] [CrossRef]
- Grabska, J.; Beć, K.B.; Mayr, S.; Huck, C.W. Theoretical simulation of near-infrared spectrum of piperine. Insight into band origins and the features of regression models. App. Spectrosc. 2021, 75, 1022–1032. [Google Scholar] [CrossRef]
- Singh, S.; Czarnecki, M.A. How much anharmonicity is in vibrational spectra of CH3I and CD3I? Spectrochim. Acta A 2021, 248, 119176. [Google Scholar] [CrossRef]
- Singh, S.; Szostak, R.; Czarnecki, M.A. Vibrational intensities and anharmonicity in MIR, NIR and Raman spectra of liquid CHCl3, CDCl3, CHBr3 and CDBr3: Spectroscopic and theoretical study. J. Mol. Liq. 2021, 336, 116277. [Google Scholar] [CrossRef]
- Grabska, J.; Beć, K.B.; Kirchler, C.G.; Ozaki, Y.; Huck, C.W. Distinct diference in sensitivity of NIR vs. IR bands of melamine to inter-molecular interactions with impact on analytical spectroscopy explained by anharmonic quantum mechanical study. Molecules 2019, 24, 1402. [Google Scholar] [CrossRef] [Green Version]
- Crocombe, R.A. Portable spectroscopy. Appl. Spectr. 2018, 72, 1701–1751. [Google Scholar] [CrossRef] [PubMed]
- Huck, C.W.; Guggenbichler, W.; Bonn, G.K. Analysis of caffeine, theobromine and theophylline in coffee by nearinfrared spectroscopy (NIRS) compared to high-performance liquidchromatography (HPLC) coupled to mass spectrometry. Anal. Chim. Acta 2005, 538, 195–203. [Google Scholar] [CrossRef]
- Barbin, D.F.; de Souza Madureira Felicio, A.L.; Sun, D.-W.; Nixdorf, S.L.; Hirooka, E.Y. Application of infrared spectral techniques on quality and compositional attributes of coffee: An overview. Food Res. Int. 2014, 61, 23–32. [Google Scholar] [CrossRef] [Green Version]
- Paradkar, M.M.; Irudayaraj, J. A rapid FTIR spectroscopic method for estimation of caffeine in soft drinks and total methylxanthines in tea and coffee. J. Food Sci. 2002, 67, 2507–2511. [Google Scholar] [CrossRef]
- Weldegebreal, B.; Redi-Abshiro, M.; Chandravanshi, B.S. Development of new analytical methods for the determination of caffeine content in aqueous solution of green coffee beans. Chem. Cent. J. 2017, 11, 126. [Google Scholar] [CrossRef] [Green Version]
- Sinija, V.R.; Mishra, H.N. FT-NIR spectroscopy for caffeine estimation in instant green tea powder and granules. LWT Food Sci. Technol. 2009, 42, 998–1002. [Google Scholar] [CrossRef]
- Wanyika, H.N.; Gatebe, E.G.; Gitu, L.M.; Ngumba, E.K.; Maritim, C.W. Determination of caffeine content of tea and instant coffee brands found in the Kenyan market. Afr. J. Food Sci. 2010, 4, 353–358. [Google Scholar]
- De Lima, G.G.; Zavattieri Ruiz, H.; Matos, M.; Helm, C.V.; de Liz, M.V.; Magalhães, W.L.E. Prediction of yerba mate caffeine content using near infrared spectroscopy. Spectrosc. Lett. 2019, 52, 282–287. [Google Scholar] [CrossRef]
- Oellig, C.; Schunck, J.; Schwack, W. Determination of caffeine, theobromine and theophylline in Mate beer and Mate soft drinks by high-performance thin-layer chromatography. J. Chromatogr. A 2018, 1533, 208–212. [Google Scholar] [CrossRef]
- Russo, M.; Dugo, P.; Fanali, C.; Dugo, L.; Zoccali, M.; Mondello, L.; De Gara, L. Use of an online extraction technique coupled to liquid chromatography for determination of caffeine in coffee, tea, and xocoa. Food Anal. Methods 2018, 11, 2637. [Google Scholar] [CrossRef]
- QiuChi, C.; Yan, C.; XiaoYun, S.; Chang, L.; Ke, W. Simultaneous determination of benzoic acid, sodium saccharin, acesulfame potassium and caffeine in cola soft drink by high performance liquid chromatography. J. Food Saf. Qual. 2016, 7, 617–622. [Google Scholar]
- Tavares, C.; Sakata, R.K. Caffeine in the treatment of pain. Rev. Bras. Anestesiol. 2012, 62, 387–401. [Google Scholar] [CrossRef] [Green Version]
- Dobson, N.R.; Hunt, C.E. Pharmacology review: Caffeine use in neonates: Indications, pharmacokinetics, clinical effects, outcomes. NeoReviews 2013, 14, 540–550. [Google Scholar] [CrossRef] [Green Version]
- Pohanka, M. The perspective of caffeine and caffeine derived compounds in therapy. Bratisl. Med. J. 2015, 116, 520–530. [Google Scholar] [CrossRef] [Green Version]
- Belay, A.; Ture, K.; Redi, M.; Asfaw, A. Measurement of caffeine in coffee beans with UV/vis spectrometer. Food Chem. 2008, 108, 310–315. [Google Scholar] [CrossRef]
- Atomssa, T.; Gholap, A.V. Characterization of caffeine and determination of caffeine in tea leaves using uv-visible spectrometer. Afr. J. Pure Appl. Chem. 2011, 5, 1–8. [Google Scholar]
- Franeta, J.F.; Agbaba, D.; Pavkov, S.E.; Aleksic, M.; Vladimirov, S. HPLC assay of acetylsalicylic acid, paracetamol, caffeine and phenobarbital in tablets. Il Farm. 2002, 57, 709–713. [Google Scholar] [CrossRef]
- Kent, J.E.; Bell, N.G.A. Molecular properties of caffeine explored by NMR: A benchtop NMR experiment for undergraduate physical-chemistry laboratories. J. Chem. Educ. 2019, 96, 786–791. [Google Scholar] [CrossRef]
- Woodcock, T.; Downey, G.; O’Donnell, C.P. Better quality food and beverages: The role of near infrared spectroscopy. J. Near Infrared Spectrosc. 2008, 16, 1–29. [Google Scholar] [CrossRef]
- Hédoux, A.; Decroix, A.A.; Guinet, Y.; Paccou, L.; Derollez, P.; Descamps, M. Low- and high-frequency Raman investigations on caffeine: Polymorphism, disorder and phase transformation. J. Phys. Chem. B. 2011, 115, 5746–5753. [Google Scholar] [CrossRef]
- Edwards, H.G.; Munshi, T.; Anstis, M. Raman spectroscopic characterisations and analytical discrimination between caffeine and demethylated analogues of pharmaceutical relevance. Spectrochim. Acta A 2005, 61, 1453–1459. [Google Scholar] [CrossRef] [PubMed]
- Downey, G.; Boussion, J. Authentication of coffee bean variety by near-infrared reflectance spectroscopy of dried extract. J. Sci. Food Agric. 1996, 71, 41–49. [Google Scholar] [CrossRef]
- Budiastra, I.W.; Sutrisno; Widyotomo, S.; Ayu, P.C. Prediction of caffeine content in java preanger coffee beans by NIR spectroscopy using PLS and MLR method. IOP Conf. Ser. Earth Environ. Sci. 2018, 147, 012004. [Google Scholar] [CrossRef] [Green Version]
- Ito, M.; Suzuki, T.; Yada, S.; Nakagami, H.; Teramoto, H.; Yonemochi, E.; Terada, K. Development of a method for nondestructive NIR transmittance spectroscopic analysis of acetaminophen and caffeine anhydrate in intact bilayer tablets. J. Pharm. Biomed. Anal. 2010, 53, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Tugnolo, A.; Beghi, R.; Giovenzana, V.; Guidetti, R. Characterization of green, roasted beans, and ground coffee using near infrared spectroscopy: A comparison of two devices. J. Near Infrared Spectrosc. 2019, 27, 93–104. [Google Scholar] [CrossRef]
- Ribeiro, J.S.; Ferreira, M.M.C.; Salva, T.J.G. Chemometric models for the quantitative descriptive sensory analysis of Arabica coffee beverages using near infrared spectroscopy. Talanta 2011, 83, 1352–1358. [Google Scholar] [CrossRef] [Green Version]
- Osborne, B.G.; Fearn, T.; Hindle, P.H. Practical NIR Spectroscopy with Applications in Food and Beverages Analysis, 2nd ed.; Longman Scientific and Technical: Harlow, UK, 1993. [Google Scholar]
- Chen, Y.; Morisawa, Y.; Futami, Y.; Czarnecki, M.A.; Wang, H.-S.; Ozaki, Y. Combined IR/NIR and density functional theory calculations analysis of the solvent effects on frequencies and intensities of the fundamental and overtones of the C=O stretching vibrations of acetone and 2-hexanone. J. Phys. Chem. A 2014, 118, 2576–2583. [Google Scholar] [CrossRef] [PubMed]
- Schuler, M.J.; Hofer, T.S.; Morisawa, Y.; Futami, Y.; Huck, C.W.; Ozaki, Y. Solvation effects on wavenumbers and absorption intensities of the OH-stretch vibration in phenolic compounds—Electrical- and mechanical anharmonicity via a combined DFT/Numerov approach. Phys. Chem. Chem. Phys. 2020, 22, 13017–13029. [Google Scholar] [CrossRef]
- Beć, K.B.; Grabska, J.; Ozaki, Y.; Hawranek, J.P.; Huck, C.W. Influence of non-fundamental modes on mid-infrared spectra. Anharmonic DFT study of aliphatic ethers. J. Phys. Chem. A 2017, 121, 1412–1424. [Google Scholar] [CrossRef]
- Grabska, J.; Czarnecki, M.A.; Beć, K.B.; Ozaki, Y. Spectroscopic and quantum mechanical calculation study of the effect of isotopic substitution on NIR spectra of methanol. J. Phys. Chem. A 2017, 121, 7925–7936. [Google Scholar] [CrossRef]
- Beć, K.B.; Karczmit, D.; Kwaśniewicz, M.; Ozaki, Y.; Czarnecki, M.A. Overtones of νCN vibration as a probe of structure of liquid CH3CN, CD3CN, and CCl3CN: Combined infrared, near-infrared, and Raman spectroscopic studies with anharmonic density functional theory calculations. J. Phys. Chem. A 2019, 123, 4431–4442. [Google Scholar] [CrossRef]
- Gonjo, T.; Futami, Y.; Morisawa, Y.; Wojcik, M.J.; Ozaki, Y. Hydrogen bonding effects on the wavenumbers and absorption intensities of the OH fundamental and the first, second, and third overtones of phenol and 2,6-dihalogenated phenols studied by visible/near-infrared/infrared spectroscopy. J. Phys. Chem. A 2011, 115, 9845–9853. [Google Scholar] [CrossRef]
- Beć, K.B.; Futami, Y.; Wójcik, M.J.; Nakajima, T.; Ozaki, Y. Spectroscopic and computational study of acetic acid and its cyclic dimer in the near-infrared region. J. Phys. Chem. A 2016, 120, 6170–6183. [Google Scholar] [CrossRef]
- Beć, K.B.; Grabska, J.; Huck, C.W.; Czarnecki, M.A. Spectra–structure correlations in isotopomers of ethanol (CX3CX2OX.; X = H, D): Combined near-infrared and anharmonic computational study. Molecules 2019, 24, 2189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizarro, C.; Esteban-Díez, I.; González-Sáiz, J.M. Mixture resolution according to the percentage of robusta variety in order to detect adulteration in roasted coffee by near infrared spectroscopy. Anal. Chim. Acta 2007, 585, 266–276. [Google Scholar] [CrossRef]
- Hu, Y.; Zhang, J.; Sato, H.; Futami, Y.; Noda, I.; Ozaki, Y. C-H…O=C Hydrogen bonding and isothermal crystallization kinetics of poly(3-hydroxybutyrate) investigated by near-infrared spectroscopy. Macromolecules 2006, 39, 3841–3847. [Google Scholar] [CrossRef]
- Zhang, X.; Li, W.; Yin, B.; Chen, W.; Kelly, D.P.; Wang, X.; Zheng, K.; Du, Y. Improvement of near infrared spectroscopic (NIRS) analysis of caffeine in roasted Arabica coffee by variable selection method of stability competitive adaptive reweighted sampling (SCARS). Spectrochim. Acta A 2013, 114, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Esteban-Díez, I.; González-Sáiz, J.M.; Pizarro, C. Prediction of sensory properties of espresso from roasted coffee samples by near-infrared spectroscopy. Anal. Chim. Acta 2004, 525, 171–182. [Google Scholar] [CrossRef]
- Santos, J.R.; Sarraguça, M.C.; Rangel, A.O.S.S.; Lopes, J.A. Evaluation of green coffee beans quality using near infrared spectroscopy: A quantitative approach. Food Chem. 2012, 135, 1828–1835. [Google Scholar] [CrossRef]
- Wang, R.; Xue, J.; Meng, L.; Lee, J.-W.; Zhao, Z.; Sun, P.; Cai, L.; Huang, T.; Wang, Z.; Wang, Z.-K.; et al. Caffeine improves the performance and thermal stability of perovskite solar cells. Joule 2019, 3, 1464–1477. [Google Scholar] [CrossRef]
- Barone, V. Anharmonic vibrational properties by a fully automated second-order perturbative approach. J. Chem. Phys. 2005, 122, 014108. [Google Scholar] [CrossRef]
- Piccardo, M.; Bloino, J.; Barone, V. Generalized vibrational perturbation theory for rotovibrational energies of linear, symmetric and asymmetric tops: Theory, approximations, and automated approaches to deal with medium-to-large molecular systems. Int. J. Quantum Chem. 2015, 115, 948–982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parameterization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barone, V.; Biczysko, M.; Bloino, J. Fully anharmonic IR and Raman spectra of medium-size molecular systems: Accuracy and interpretation. Phys. Chem. Chem. Phys. 2014, 16, 1759–1787. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision, B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Bradley, M.S. Lineshapes in IR and Raman spectroscopy: A primer. Spectroscopy 2015, 30, 42–46. [Google Scholar]
- Pulay, P.; Forgasi, G.; Pang, F.; Boggs, J.E. Systematic ab initio gradient calculation of molecular geometries, force constants, and dipole moment derivatives. J. Am. Chem. Soc. 1979, 101, 2550–2560. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Region [cm−1] | First Overt. | Second Overt. | Binary Comb. | Ternary Comb. |
|---|---|---|---|---|
| 10,000–4000 | 11.57 | 1.28 | 36.91 | 50.24 |
| 10,000–7000 | 0.02 | 26.78 | 0.00 | 73.20 |
| 7000–4000 | 11.85 | 0.64 | 37.83 | 49.67 |
| 5000–4000 | 0.01 | 0.88 | 48.10 | 51.01 |
| 6500–5500 | 40.52 | 0.00 | 14.77 | 44.70 |
| 7700–7000 | 0.05 | 0.00 | 0.01 | 99.95 |
| 9200–8400 | 0.00 | 42.27 | 0.00 | 57.73 |
| Position [cm−1] | Assignments | |
|---|---|---|
| Exp. | Calc. | |
| 4092 | 4062 | δrockCH3 + νsCH3; (δipring, δipCH, δrockCH3) + νasCH3 |
| 4112 | 4096 | δrockCH3 + νasCH3; CNtors + νasCH3 |
| 4183 | 4181 | (δipCH, δipring) + νsCH3; CNtors + δipring + νCH |
| 4236 | 4227 | (δipring, δipCH) + νasCH3; δrockCH3 + νCH; δipring + νsCH3 |
| 4308 | 4282 | (δipCH, δipring) + νsCH3; (δipCH, δipring) + νCH |
| 4428 | 4376 | νas’CH3 + δsCH3; δasCH3 + νas’CH3; δsCH3 + νsCH3 |
| 4444 | 4434 | δsCH3 + νasCH3; νas’CH3 + δipring + δrockCH3; (δipring, δipCH) + νCH |
| 5760 | 5761 | CNtors + νas’CH3 + νsCH3 |
| 5800 | 5815 | νas’CH3 + νsCH3 |
| 5840 | 5881 | 2νasCH3 |
| 5956 | 5973 | νas’CH3 + νasCH3; CNtors + νas’CH3 + νsCH3 |
| 6008 | 5991 | 2νas’CH3 |
| 6120 | 6111 | 2νCH |
| 7140 | 7211 | νas’CH3 + δasCH3 + νsCH3 |
| 7236 | 7239 | νas’CH3 + δasCH3 + νsCH3 |
| 7300 | 7282 | δipCH, δipring + 2νCH |
| 7344 | 7321 | 2νas’CH3 + δasCH3 |
| 7372 | 7355 | δasCH3 + 3νasCH3; δsCH3 + 3νasCH3 |
| 8532 | 8610 | 2νas’CH3 + vsCH3; νas’CH3 + 2νsCH3 |
| 8716 | 8752 | νas’CH3 + νsCH3 + νasCH3; νas’CH3 + νsCH3 + νCO; 3νas’CH3; νsCH3 + 2νasCH3 |
| 8812 | 8830 | 3νasCH3 |
| 8980 | 8984 | 3νCH |
| ν1C = O | 2ν1C = O | 3ν1C = O | ||||
|---|---|---|---|---|---|---|
| Position [cm−1] | Intensity [km mol−1] | Position [cm−1] | Intensity [km mol−1] | Position [cm−1] | Intensity [km mol−1] | |
| relative intensity | 1740 | 206.4 | 3409.3 | 0.72 | 5100.9 | 0.064 |
| 1 | 0.0035 | 0.00031 | ||||
| 1 | 0.089 | |||||
| ν2C = O | 2ν2C = O | 3ν2C = O | ||||
| relative intensity | 1694 | 83.4 | 3313.6 | 0.99 | 4959.9 | 0.02127 |
| 1 | 0.011871 | 0.00026 | ||||
| 1 | 0.021 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grabska, J.; Beć, K.B.; Ozaki, Y.; Huck, C.W. Anharmonic DFT Study of Near-Infrared Spectra of Caffeine: Vibrational Analysis of the Second Overtones and Ternary Combinations. Molecules 2021, 26, 5212. https://doi.org/10.3390/molecules26175212
Grabska J, Beć KB, Ozaki Y, Huck CW. Anharmonic DFT Study of Near-Infrared Spectra of Caffeine: Vibrational Analysis of the Second Overtones and Ternary Combinations. Molecules. 2021; 26(17):5212. https://doi.org/10.3390/molecules26175212
Chicago/Turabian StyleGrabska, Justyna, Krzysztof B. Beć, Yukihiro Ozaki, and Christian W. Huck. 2021. "Anharmonic DFT Study of Near-Infrared Spectra of Caffeine: Vibrational Analysis of the Second Overtones and Ternary Combinations" Molecules 26, no. 17: 5212. https://doi.org/10.3390/molecules26175212
APA StyleGrabska, J., Beć, K. B., Ozaki, Y., & Huck, C. W. (2021). Anharmonic DFT Study of Near-Infrared Spectra of Caffeine: Vibrational Analysis of the Second Overtones and Ternary Combinations. Molecules, 26(17), 5212. https://doi.org/10.3390/molecules26175212