
Prediction of Drug–Target Interactions by Combining Dual-Tree Complex Wavelet Transform with Ensemble Learning Method


Abstract
:1. Introduction
2. Results
2.1. Evaluation Metrics
2.2. Parameter Discussion
2.3. Performance Evaluations on Four Golden Standard Datasets
2.4. Comparison Results between LPQ-Based Model and the Proposed Method
2.5. Comparison with SVM and KNN Classifier
2.6. Comparison with Different Methods on the Same Dataset
2.7. Performance on the Independent Dataset
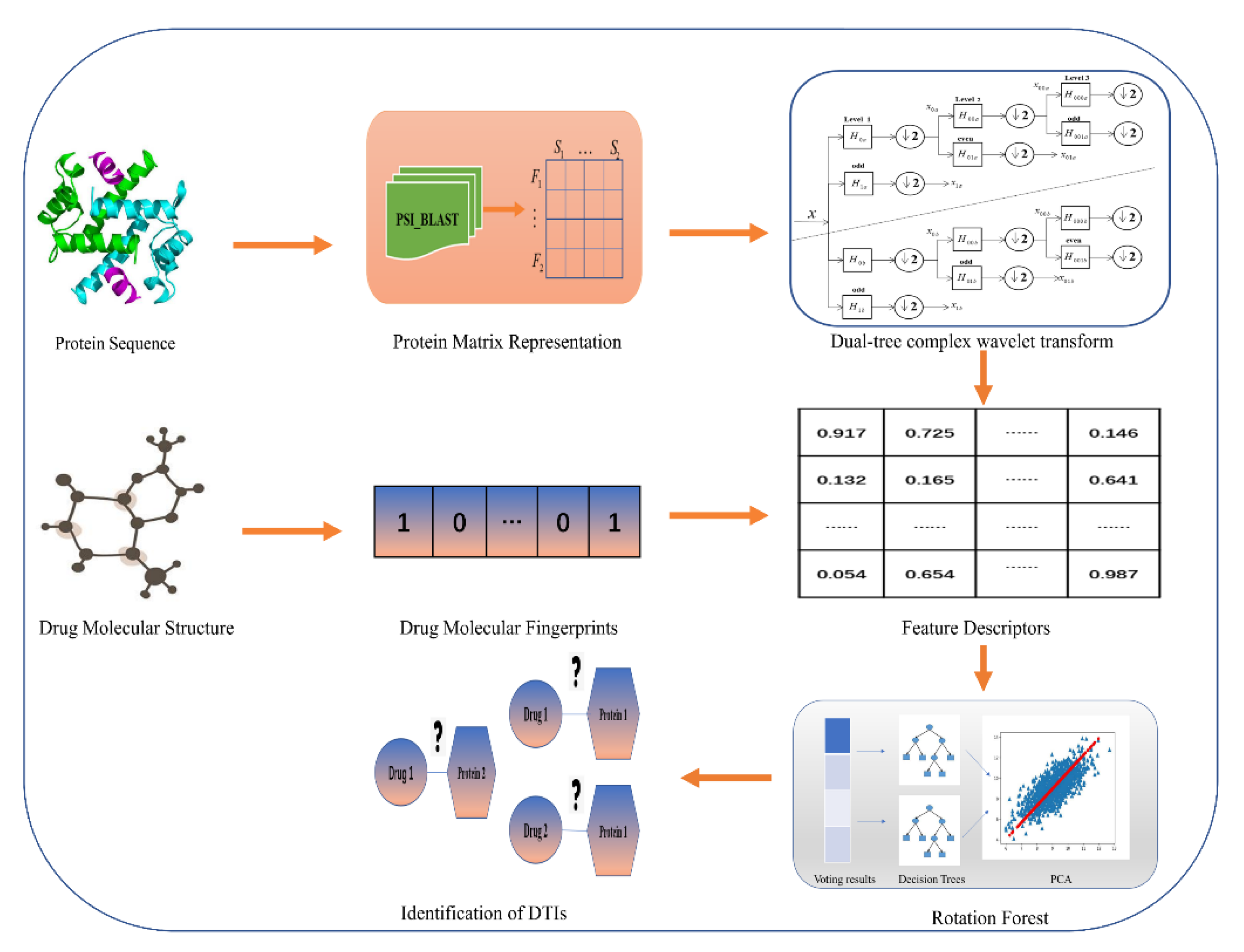
3. Materials and Methods
3.1. Data Collection
3.2. Characterization of Drug Molecules
3.3. Representation of Target Proteins
3.4. Feature Extraction Method
3.5. Rotation Forest Classifier (RoF)
- (1)
- Choose a suitable parameter M for which U can be randomly split into M disjointed subsets, with the number of features contained in the feature subset being equal to L/M.
- (2)
- Let represent the jth feature subset and use it to train the classifier . The sample subset is constructed by a non-empty subset, which is randomly selected from a certain proportion.
- (3)
- Apply PCA [52] on to order the coefficients stored in matrix .
- (4)
- The coefficients obtained from the matrix are used to construct a sparse rotation matrix , which can be defined as follows:
4. Case Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Yuan, Q.; Gao, J.; Wu, D.; Zhang, S.; Mamitsuka, H.; Zhu, S. DrugE-Rank: Improving drug–target interaction prediction of new candidate drugs or targets by ensemble learning to rank. Bioinformatics 2016, 32, i18–i27. [Google Scholar] [CrossRef]
- Kneller, R. The importance of new companies for drug discovery: Origins of a decade of new drugs. Nat. Rev. Drug Discov. 2010, 9, 867–882. [Google Scholar] [CrossRef]
- Mizutani, S.; Pauwels, E.; Stoven, V.; Goto, S.; Yamanishi, Y. Relating drug-protein interaction network with drug side effects. Bioinformatics 2012, 28, i522–i528. [Google Scholar] [CrossRef] [Green Version]
- Brouwers, L.; Iskar, M.; Zeller, G.; Van Noort, V.; Bork, P. Network neighbors of drug targets contribute to drug side-effect similarity. PLoS ONE 2011, 6, e22187. [Google Scholar] [CrossRef]
- De Azevedo, J.; Walter, F.; Dias, R. Experimental approaches to evaluate the thermodynamics of protein-drug interactions. Curr. Drug Targets 2008, 9, 1071–1076. [Google Scholar] [CrossRef]
- Finan, C.; Gaulton, A.; Kruger, F.A.; Lumbers, R.T.; Shah, T.; Engmann, J.; Galver, L.; Kelley, R.; Karlsson, A.; Santos, R. The druggable genome and support for target identification and validation in drug development. Sci. Transl. Med. 2017, 9, 383. [Google Scholar] [CrossRef]
- Šink, R.; Gobec, S.; Pečar, S.; Zega, A. False positives in the early stages of drug discovery. Curr. Med. Chem. 2010, 17, 4231–4255. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Li, Z.; Yang, J.; Tian, G.; Liu, F.; Wen, H.; Peng, L.; Chen, M.; Xiang, J.; Peng, L. Revealing drug-target interactions with computational models and algorithms. Molecules 2019, 24, 1714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2017, 46, D1074–D1082. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, F.; Shi, Z.; Qin, C.; Tao, L.; Liu, X.; Xu, F.; Zhang, L.; Song, Y.; Zhang, J.-X.; Han, B.-C.; et al. Therapeutic target database update 2012: A resource for facilitating target-oriented drug discovery. Nucleic Acids Res. 2011, 40, D1128–D1136. [Google Scholar] [CrossRef]
- Günther, S.; Kuhn, M.; Dunkel, M.; Campillos, M.; Senger, C.; Petsalaki, E.; Ahmed, J.; Urdiales, E.G.; Gewiess, A.; Jensen, L.J.; et al. SuperTarget and Matador: Resources for exploring drug-target relationships. Nucleic Acids Res. 2007, 36 (Suppl. 1), D919–D922. [Google Scholar] [CrossRef]
- Ma, D.-L.; Chan, D.S.-H.; Leung, C.-H. Drug repositioning by structure-based virtual screening. Chem. Soc. Rev. 2013, 42, 2130–2141. [Google Scholar] [CrossRef]
- Waszkowycz, B.; Clark, D.E.; Gancia, E. Outstanding challenges in protein–ligand docking and structure-based virtual screening. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 229–259. [Google Scholar] [CrossRef]
- Wilson, G.L.; Lill, M.A. Integrating structure-based and ligand-based approaches for computational drug design. Future Med. Chem. 2011, 3, 735–750. [Google Scholar] [CrossRef] [PubMed]
- Percha, B.; Garten, Y.; Altman, R.B. Discovery and explanation of drug-drug interactions via text mining. Biocomputing 2012, 2011, 410–421. [Google Scholar] [CrossRef] [Green Version]
- Yamanishi, Y.; Araki, M.; Gutteridge, A.; Honda, W.; Kanehisa, M. Prediction of drug-target interaction networks from the integration of chemical and genomic spaces. Bioinformatics 2008, 24, i232–i240. [Google Scholar] [CrossRef]
- Wang, L.; You, Z.; Chen, X.; Xia, S.-X.; Liu, F.; Yan, X.; Zhou, Y.; Song, K.-J. A computational-based method for predicting drug–target interactions by using stacked autoencoder deep neural network. J. Comput. Biol. 2018, 25, 361–373. [Google Scholar] [CrossRef]
- Hao, M.; Bryant, S.H.; Wang, Y. Predicting drug-target interactions by dual-network integrated logistic matrix factorization. Sci. Rep. 2017, 7, 40376. [Google Scholar] [CrossRef] [Green Version]
- Wen, M.; Zhang, Z.; Niu, S.; Sha, H.; Yang, R.; Yun, Y.; Lu, H. Deep-learning-based drug–target interaction prediction. J. Proteome Res. 2017, 16, 1401–1409. [Google Scholar] [CrossRef]
- Ezzat, A.; Wu, M.; Li, X.-L.; Kwoh, C.-K. Drug-target interaction prediction using ensemble learning and dimensionality reduction. Methods 2017, 129, 81–88. [Google Scholar] [CrossRef]
- Huang, K.; Xiao, C.; Glass, L.M.; Sun, J. MolTrans: Molecular Interaction Transformer for drug–target interaction prediction. Bioinformatics 2020, 37, 830–836. [Google Scholar] [CrossRef]
- Zhang, Y.-F.; Wang, X.; Kaushik, A.C.; Chu, Y.; Shan, X.; Zhao, M.-Z.; Xu, Q.; Wei, D.-Q. SPVec: A Word2vec-inspired feature representation method for drug-target interaction prediction. Front. Chem. 2020, 7, 895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Yang, S.; Li, J. Drug target predictions based on heterogeneous graph inference. Biocomputing 2013 2012, 53–64. [Google Scholar] [CrossRef] [Green Version]
- Redkar, S.; Mondal, S.; Joseph, A.; Hareesha, K.S. A machine learning approach for drug-target interaction prediction using wrapper feature selection and class balancing. Mol. Inform. 2020, 39, 1900062. [Google Scholar] [CrossRef] [PubMed]
- Zweig, M.H.; Campbell, G. Receiver-operating characteristic (ROC) plots: A fundamental evaluation tool in clinical medicine. Clin. Chem. 1993, 39, 561–577. [Google Scholar] [CrossRef]
- Ojansivu, V.; Heikkilä, J. In Blur insensitive texture classification using local phase quantization. In Proceedings of the International Conference on Image and Signal Processing, Cherbourg-Octeville, France, 1–3 July 2008; Springer: Berlin/Heidelberg, Germany, 2008; pp. 236–243. [Google Scholar]
- Björne, J.; Kaewphan, S.; Salakoski, T. UTurku: Drug named entity recognition and drug-drug interaction extraction using SVM classification and domain knowledge. In Proceedings of the Second Joint Conference on Lexical and Computational Semantics (* SEM). Seventh International Workshop on Semantic Evaluation (SemEval 2013), Turku, Finland, Atlanta, GA, USA, 14–15 June 2013; Volume 2, pp. 651–659. [Google Scholar]
- Shi, J.-Y.; Li, J.-X.; Lu, H.-M.; Zhang, Y. Predicting Drug-Target Interactions Between New Drugs and New Targets via Pairwise K-nearest Neighbor and Automatic Similarity Selection; Springer: Cham, Switzerland, 2015; pp. 477–486. [Google Scholar] [CrossRef]
- Chang, C.-C.; Lin, C.-J. LIBSVM: A library for support vector machines. ACM Trans. Intell. Syst. Technol. (TIST) 2011, 2, 1–27. [Google Scholar] [CrossRef]
- Yamanishi, Y.; Kotera, M.; Kanehisa, M.; Goto, S. Drug-target interaction prediction from chemical, genomic and pharmacological data in an integrated framework. Bioinformatics 2010, 26, i246–i254. [Google Scholar] [CrossRef]
- Gönen, M. Predicting drug-target interactions from chemical and genomic kernels using Bayesian matrix factorization. Bioinformatics 2012, 28, 2304–2310. [Google Scholar] [CrossRef]
- Pliakos, K.; Vens, C.; Tsoumakas, G. Predicting drug-target interactions with multi-label classification and label partitioning. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019, 18, 1596–1607. [Google Scholar] [CrossRef] [PubMed]
- Nanni, L.; Lumini, A.; Brahnam, S. A set of descriptors for identifying the protein–drug interaction in cellular networking. J. Theor. Biol. 2014, 359, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Öztürk, H.; Ozkirimli, E.; Özgür, A. A comparative study of SMILES-based compound similarity functions for drug-target interaction prediction. BMC Bioinform. 2016, 17, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, F.; Liu, C.; Jiang, J.; Lu, W.; Li, W.; Liu, G.; Zhou, W.-X.; Huang, J.; Tang, Y. Prediction of drug-target interactions and drug repositioning via network-based inference. PLoS Comput. Biol. 2012, 8, e1002503. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Zhang, Z. A semi-supervised method for drug-target interaction prediction with consistency in networks. PLoS ONE 2013, 8, e62975. [Google Scholar] [CrossRef] [Green Version]
- Schomburg, I.; Chang, A.; Ebeling, C.; Gremse, M.; Heldt, C.; Huhn, G.; Schomburg, D. BRENDA, the enzyme database: Updates and major new developments. Nucleic Acids Res. 2004, 32 (Suppl. 1), D431–D433. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2007, 36 (Suppl. 1), D480–D484. [Google Scholar] [CrossRef]
- Hecker, N.; Ahmed, J.; Von Eichborn, J.; Dunkel, M.; Macha, K.; Eckert, A.; Gilson, M.K.; Bourne, P.E.; Preissner, R. SuperTarget goes quantitative: Update on drug-target interactions. Nucleic Acids Res. 2011, 40, D1113–D1117. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Cheng, D.; Shrivastava, S.; Tzur, D.; Gautam, B.; Hassanali, M. DrugBank: A knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2007, 36 (Suppl. 1), D901–D906. [Google Scholar] [CrossRef]
- Ba-Alawi, W.; Soufan, O.; Essack, M.; Kalnis, P.; Bajic, V.B. DASPfind: New efficient method to predict drug–target interactions. J. Chemin. 2016, 8, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Cheng, F.; Xu, Y.; Li, W.; Tang, Y. Estimation of ADME properties with substructure pattern recognition. J. Chem. Inf. Model. 2010, 50, 1034–1041. [Google Scholar] [CrossRef]
- Gribskov, M.; McLachlan, A.D.; Eisenberg, D. Profile analysis: Detection of distantly related proteins. Proc. Natl. Acad. Sci. USA 1987, 84, 4355–4358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raicar, G.; Saini, H.; Dehzangi, A.; Lal, S.; Sharma, A. Improving protein fold recognition and structural class prediction accuracies using physicochemical properties of amino acids. J. Theor. Biol. 2016, 402, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Koonin, E.V. Iterated profile searches with PSI-BLAST—A tool for discovery in protein databases. Trends Biochem. Sci. 1998, 23, 444–447. [Google Scholar] [CrossRef]
- Wang, S.; Liu, S. Protein sub-nuclear localization based on effective fusion representations and dimension reduction algorithm LDA. Int. J. Mol. Sci. 2015, 16, 30343–30361. [Google Scholar] [CrossRef] [Green Version]
- Selesnick, I.W.; Baraniuk, R.G.; Kingsbury, N.C. The dual-tree complex wavelet transform. IEEE Signal Process. Mag. 2005, 22, 123–151. [Google Scholar] [CrossRef] [Green Version]
- Shensa, M.J. The discrete wavelet transform: Wedding the a trous and Mallat algorithms. IEEE Trans. Signal Process. 1992, 40, 2464–2482. [Google Scholar] [CrossRef] [Green Version]
- Yin, M.; Duan, P.; Liu, W.; Liang, X. A novel infrared and visible image fusion algorithm based on shift-invariant dual-tree complex shearlet transform and sparse representation. Neurocomputing 2017, 226, 182–191. [Google Scholar] [CrossRef]
- Rodríguez, J.J.; Kuncheva, L.; Alonso, C.J. Rotation forest: A new classifier ensemble method. IEEE Trans. Pattern Anal. Mach. Intell. 2006, 28, 1619–1630. [Google Scholar] [CrossRef] [PubMed]
- Abdi, H.; Williams, L.J. Principal component analysis. Wiley Interdiscip. Rev. Comput. Stat. 2010, 2, 433–459. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Test Set | ACC. (%) | PR. (%) | Sen. (%) | MCC (%) | AUC |
|---|---|---|---|---|---|
| 1 | 89.40 | 89.80 | 87.88 | 81.00 | 0.9452 |
| 2 | 88.72 | 89.02 | 88.72 | 79.98 | 0.9516 |
| 3 | 88.80 | 90.84 | 85.96 | 80.07 | 0.9499 |
| 4 | 88.80 | 91.68 | 85.22 | 80.06 | 0.9435 |
| 5 | 90.34 | 92.49 | 88.71 | 82.53 | 0.9585 |
| Average | 89.21 ± 0.69 | 90.77 ± 1.40 | 87.30 ± 1.62 | 80.73 ± 1.09 | 0.9498 ± 0.0059 |
| Test Set | ACC. (%) | PR. (%) | Sen. (%) | MCC (%) | AUC |
|---|---|---|---|---|---|
| 1 | 85.08 | 85.08 | 85.08 | 74.62 | 0.9324 |
| 2 | 84.41 | 86.26 | 80.14 | 73.49 | 0.9154 |
| 3 | 84.92 | 87.06 | 84.59 | 74.27 | 0.9233 |
| 4 | 85.59 | 83.59 | 89.40 | 75.22 | 0.9200 |
| 5 | 87.46 | 85.92 | 87.77 | 78.01 | 0.9440 |
| Average | 85.49 ± 1.18 | 85.58 ± 1.32 | 85.40 ± 3.54 | 75.12 ± 1.73 | 0.9270 ± 0.0113 |
| Test Set | ACC. (%) | PR. (%) | Sen. (%) | MCC (%) | AUC |
|---|---|---|---|---|---|
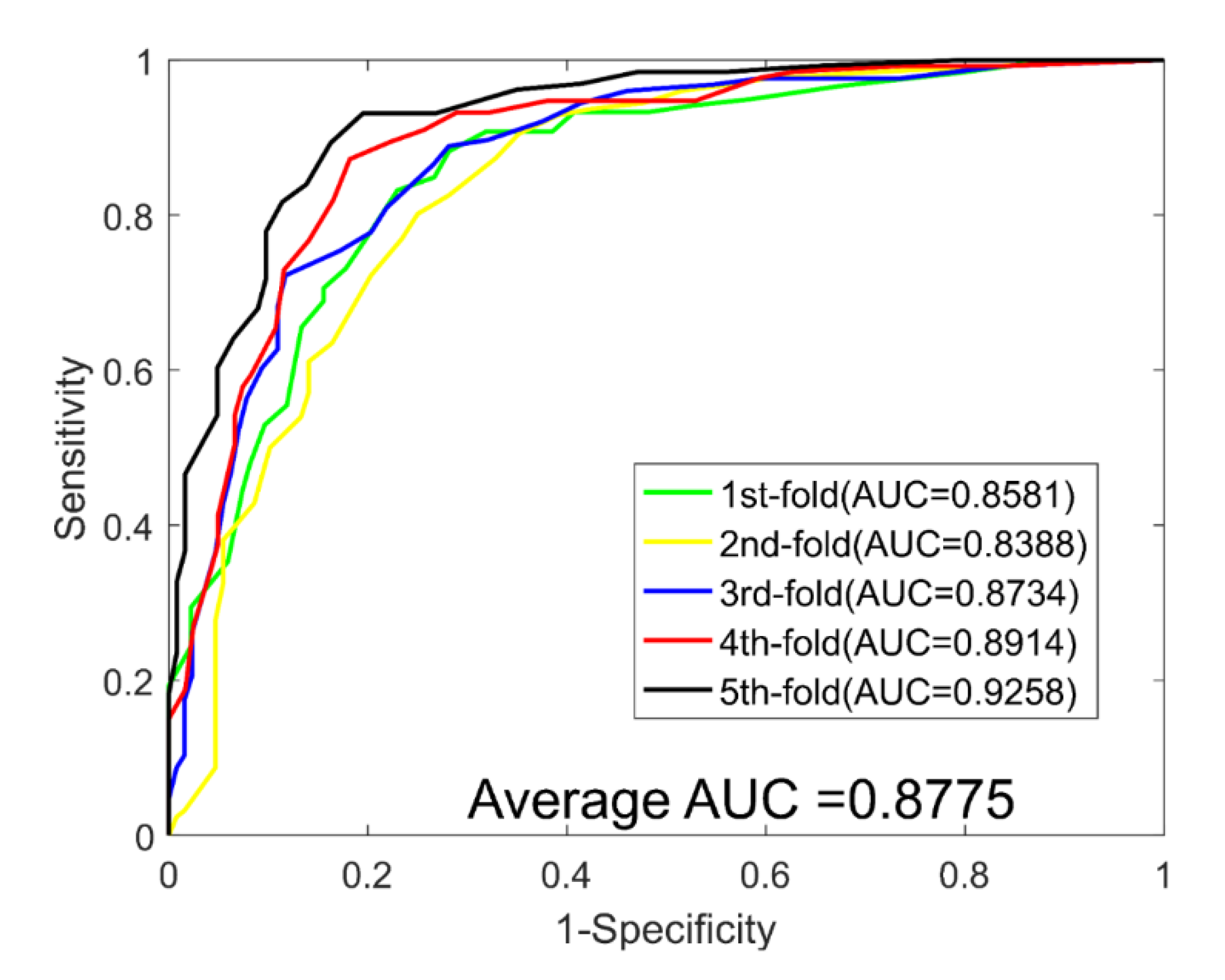
| 1 | 79.13 | 81.06 | 79.26 | 66.88 | 0.8581 |
| 2 | 76.77 | 77.60 | 75.78 | 64.33 | 0.8388 |
| 3 | 79.53 | 80.16 | 78.91 | 67.44 | 0.8734 |
| 4 | 83.46 | 84.35 | 80.17 | 72.24 | 0.8914 |
| 5 | 86.22 | 84.38 | 87.80 | 76.23 | 0.9258 |
| Average | 81.02 ± 3.77 | 81.51 ± 2.90 | 80.38 ± 4.47 | 69.42 ± 4.76 | 0.8775 ± 0.0332 |
| Test Set | ACC. (%) | PR. (%) | Sen. (%) | MCC (%) | AUC |
|---|---|---|---|---|---|
| 1 | 72.22 | 60.00 | 59.23 | 57.67 | 0.7642 |
| 2 | 77.78 | 72.73 | 88.89 | 64.58 | 0.7963 |
| 3 | 80.56 | 73.69 | 87.50 | 68.43 | 0.8109 |
| 4 | 66.67 | 78.95 | 65.22 | 53.74 | 0.7609 |
| 5 | 75.00 | 76.19 | 80.00 | 61.73 | 0.7453 |
| Average | 74.44 ± 5.34 | 72.31 ± 7.29 | 78.17 ± 10.64 | 61.23 ± 5.75 | 0.7755 ± 0.0271 |
| Negative Samples | ACC. (%) | PR. (%) | Sen. (%) | MCC (%) | AUC |
|---|---|---|---|---|---|
| Sample 1 | 81.73 ± 2.18 | 82.47 ± 2.48 | 80.65 ± 3.93 | 70.03 ± 2.94 | 0.8882 ± 0.0126 |
| Sample 2 | 81.13 ± 1.80 | 83.04 ± 2.49 | 80.59 ± 4.64 | 70.51 ± 2.36 | 0.8904 ± 0.0167 |
| Sample 3 | 81.97 ± 1.06 | 82.19 ± 1.79 | 81.59 ± 3.39 | 70.29 ± 1.48 | 0.8876 ± 0.0046 |
| Sample 4 | 81.10 ± 1.18 | 82.04 ± 1.61 | 79.53 ± 4.17 | 69.18 ± 1.65 | 0.8881 ± 0.0162 |
| Sample 5 | 81.57 ± 1.53 | 82.79 ± 2.66 | 81.23 ± 4.59 | 69.76 ± 2.06 | 0.8900 ± 0.0124 |
| Dataset | Descriptor | ACC. (%) | PR. (%) | Sen. (%) | MCC (%) | AUC |
|---|---|---|---|---|---|---|
| Enzyme | LPQ | 87.45 ± 3.87 | 88.62 ± 5.32 | 86.20 ± 2.02 | 78.24 ± 5.49 | 0.9329 ± 0.0358 |
| DTCWT | 89.21 ± 0.69 | 90.77 ± 1.40 | 87.30 ± 1.62 | 80.73 ± 1.09 | 0.9498 ± 0.0059 | |
| Ion Channel | LPQ | 84.07 ± 3.59 | 83.10 ± 3.98 | 85.49 ± 4.85 | 73.43 ± 5.03 | 0.9008 ± 0.0404 |
| DTCWT | 85.49 ± 1.18 | 85.58 ± 1.32 | 85.40 ± 3.54 | 75.12 ± 1.73 | 0.9270 ± 0.0113 | |
| GPCRs | LPQ | 79.61 ± 4.26 | 78.69 ± 6.04 | 81.07 ± 3.76 | 67.71 ± 4.69 | 0.8474 ± 0.0402 |
| DTCWT | 81.02 ± 3.77 | 81.51 ± 2.90 | 80.38 ± 4.47 | 69.42 ± 4.76 | 0.8775 ± 0.0332 | |
| NRs | LPQ | 71.11 ± 3.17 | 70.03 ± 10.36 | 75.55 ± 13.97 | 56.53 ± 3.54 | 0.7403 ± 0.0667 |
| DTCWT | 74.44 ± 5.34 | 72.31 ± 7.29 | 78.17 ± 10.64 | 61.23 ± 5.75 | 0.7755 ± 0.0271 |
| Dataset | Model | ACC. (%) | PR. (%) | Sen. (%) | MCC (%) | AUC |
|---|---|---|---|---|---|---|
| Enzyme | SVM | 77.25 ± 0.73 | 79.84 ± 1.31 | 72.86 ± 2.45 | 64.67 ± 0.89 | 0.8012 ± 0.0191 |
| KNN | 80.53 ± 1.85 | 77.42 ± 2.20 | 86.06 ± 3.17 | 68.46 ± 2.17 | 0.8050 ± 0.0168 | |
| RoF | 89.21 ± 0.69 | 90.77 ± 1.40 | 87.30 ± 1.62 | 80.73 ± 1.09 | 0.9498 ± 0.0059 | |
| Ion Channel | SVM | 71.08 ± 1.93 | 70.28 ± 1.56 | 73.21 ± 4.58 | 58.77 ± 1.61 | 0.7683 ± 0.0224 |
| KNN | 79.02 ± 2.28 | 75.11 ± 3.60 | 86.72 ± 3.89 | 66.41 ± 2.68 | 0.7901 ± 0.0204 | |
| RoF | 85.49 ± 1.18 | 85.58 ± 1.32 | 85.40 ± 3.54 | 75.12 ± 1.73 | 0.9270 ± 0.0113 | |
| GPCRs | SVM | 61.65 ± 2.44 | 62.26 ± 4.33 | 59.53 ± 1.50 | 52.72 ± 1.09 | 0.6652 ± 0.0252 |
| KNN | 63.86 ± 3.83 | 61.47 ± 3.05 | 74.65 ± 2.21 | 52.86 ± 2.61 | 0.6379 ± 0.0408 | |
| RoF | 81.02 ± 3.77 | 81.51 ± 2.90 | 80.38 ± 4.47 | 69.42 ± 4.76 | 0.8775 ± 0.0332 | |
| NRs | SVM | 62.22 ± 5.41 | 61.99 ± 11.70 | 60.90 ± 9.47 | 52.22 ± 3.51 | 0.6220 ± 0.0611 |
| KNN | 48.33 ± 6.97 | 48.65 ± 9.71 | 46.68 ± 2.26 | 49.58 ± 0.77 | 0.4775 ± 0.0728 | |
| RoF | 74.44 ± 5.34 | 72.31 ± 7.29 | 78.17 ± 10.64 | 61.23 ± 5.75 | 0.7755 ± 0.0271 |
| Method | Enzyme | GPCRs | Ion Channel | NRs |
|---|---|---|---|---|
| Yamanishi et al. | 0.845 | 0.812 | 0.731 | 0.830 |
| KBMF2K | 0.832 | 0.857 | 0.799 | 0.824 |
| MLCLE | 0.842 | 0.850 | 0.795 | 0.790 |
| AM-PSSM | 0.843 | 0.839 | 0.722 | 0.767 |
| SIMCOMP | 0.863 | 0.867 | 0.776 | 0.856 |
| DBSI | 0.8075 | 0.8022 | 0.8029 | 0.7578 |
| NETCBP | 0.8251 | 0.8235 | 0.8034 | 0.8394 |
| Our Method | 0.9498 | 0.8775 | 0.9270 | 0.7755 |
| Dataset | Drug | Target Protein | Interactions |
|---|---|---|---|
| Enzyme | 445 | 664 | 2926 |
| Ion Channel | 210 | 204 | 1476 |
| NRs | 54 | 26 | 90 |
| GPCRs | 223 | 95 | 635 |
| Drugbank-approved | 1555 | 1591 | 5831 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, J.; Li, L.-P.; You, Z.-H.; Yu, C.-Q.; Ren, Z.-H.; Chen, Y. Prediction of Drug–Target Interactions by Combining Dual-Tree Complex Wavelet Transform with Ensemble Learning Method. Molecules 2021, 26, 5359. https://doi.org/10.3390/molecules26175359
Pan J, Li L-P, You Z-H, Yu C-Q, Ren Z-H, Chen Y. Prediction of Drug–Target Interactions by Combining Dual-Tree Complex Wavelet Transform with Ensemble Learning Method. Molecules. 2021; 26(17):5359. https://doi.org/10.3390/molecules26175359
Chicago/Turabian StylePan, Jie, Li-Ping Li, Zhu-Hong You, Chang-Qing Yu, Zhong-Hao Ren, and Yao Chen. 2021. "Prediction of Drug–Target Interactions by Combining Dual-Tree Complex Wavelet Transform with Ensemble Learning Method" Molecules 26, no. 17: 5359. https://doi.org/10.3390/molecules26175359