
Sensitization to Drug Treatment in Precursor B-Cell Acute Lymphoblastic Leukemia Is Not Achieved by Stromal NF-κB Inhibition of Cell Adhesion but by Stromal PKC-Dependent Inhibition of ABC Transporters Activity

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
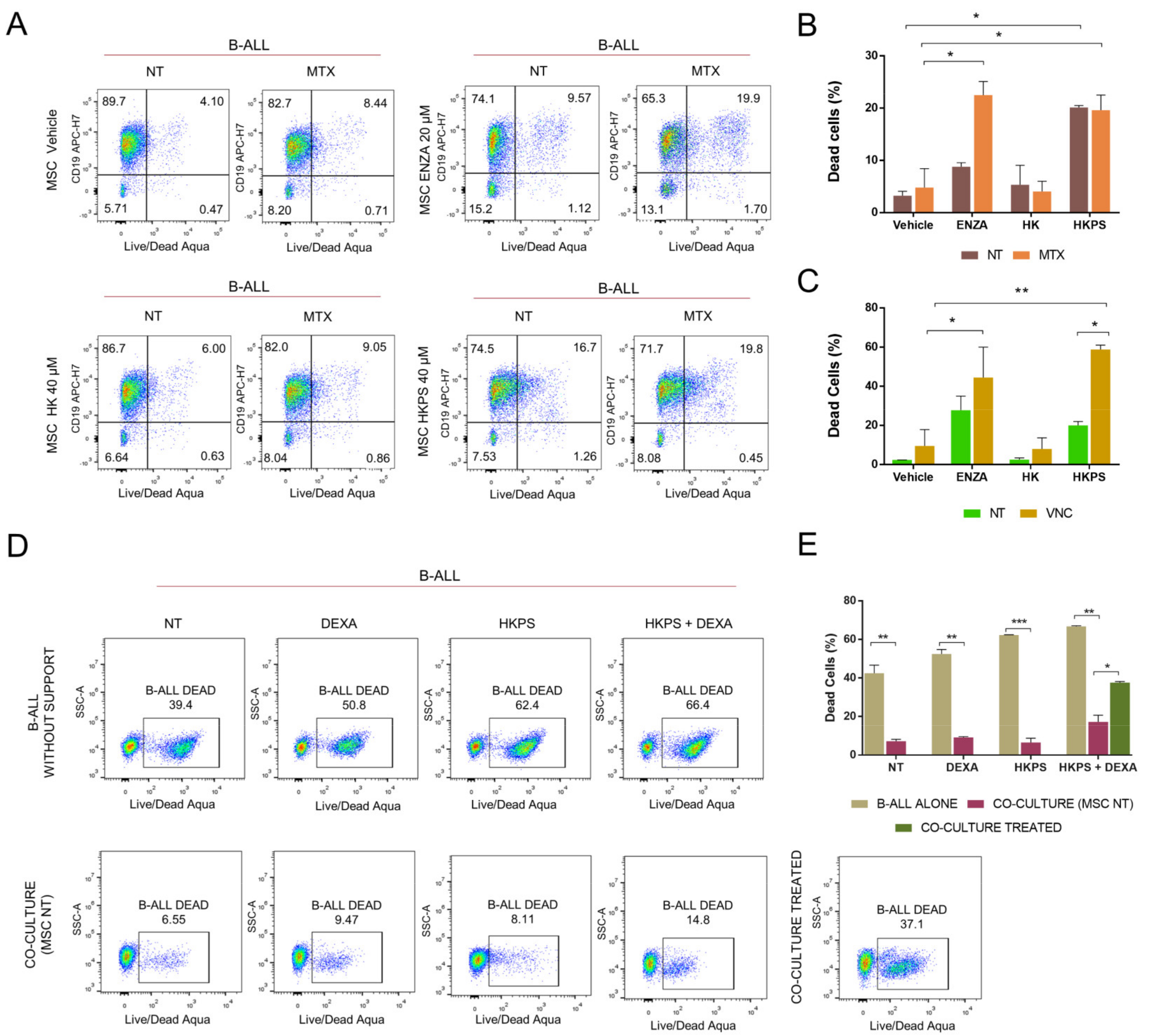
2.1. Increased Susceptibility to MTX and VNC with Inhibitors of Stromal PKC
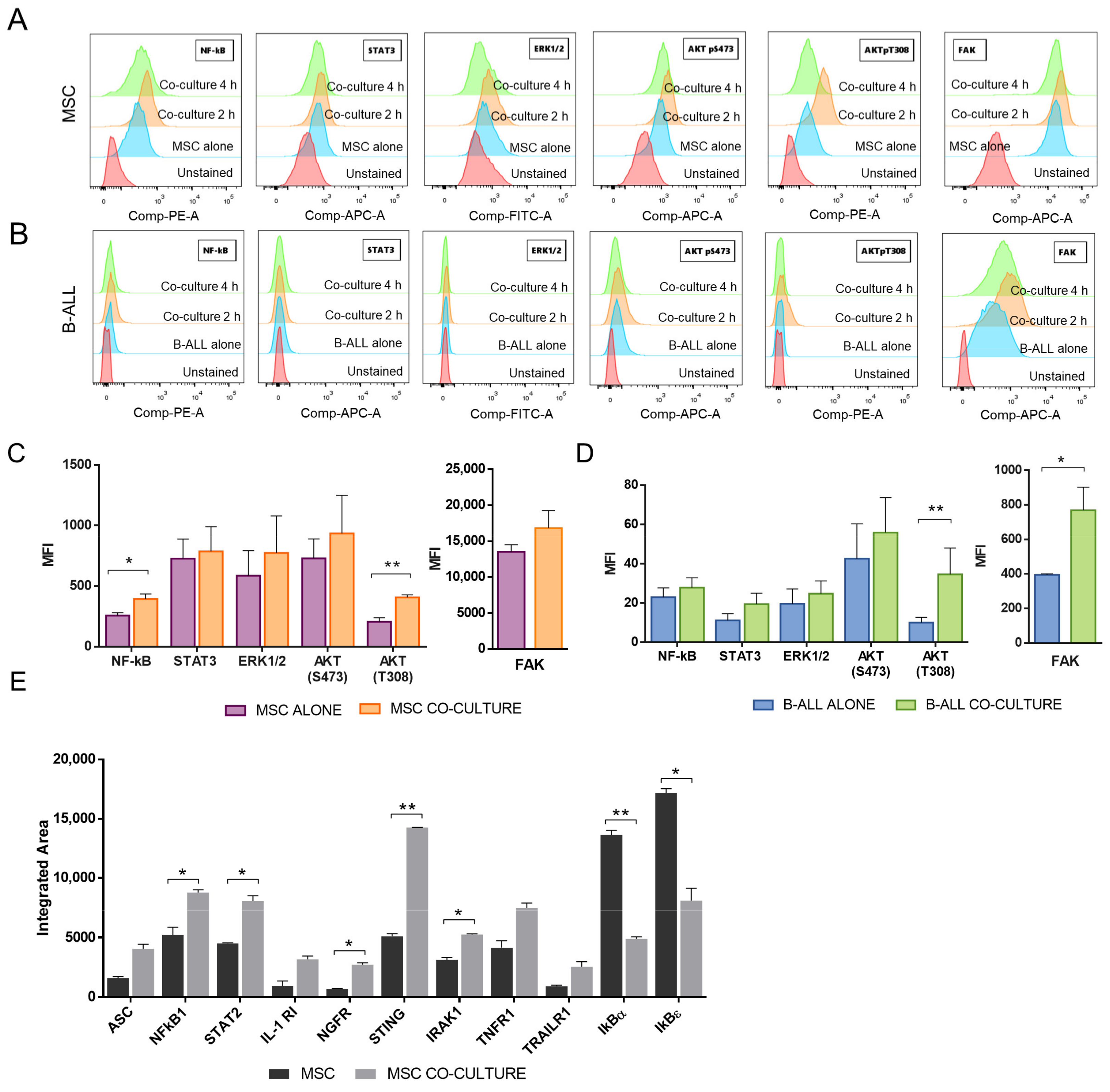
2.2. NF-κB and Akt Signalling Are Increased in MSC after the Co-Culture with B-ALL Cells
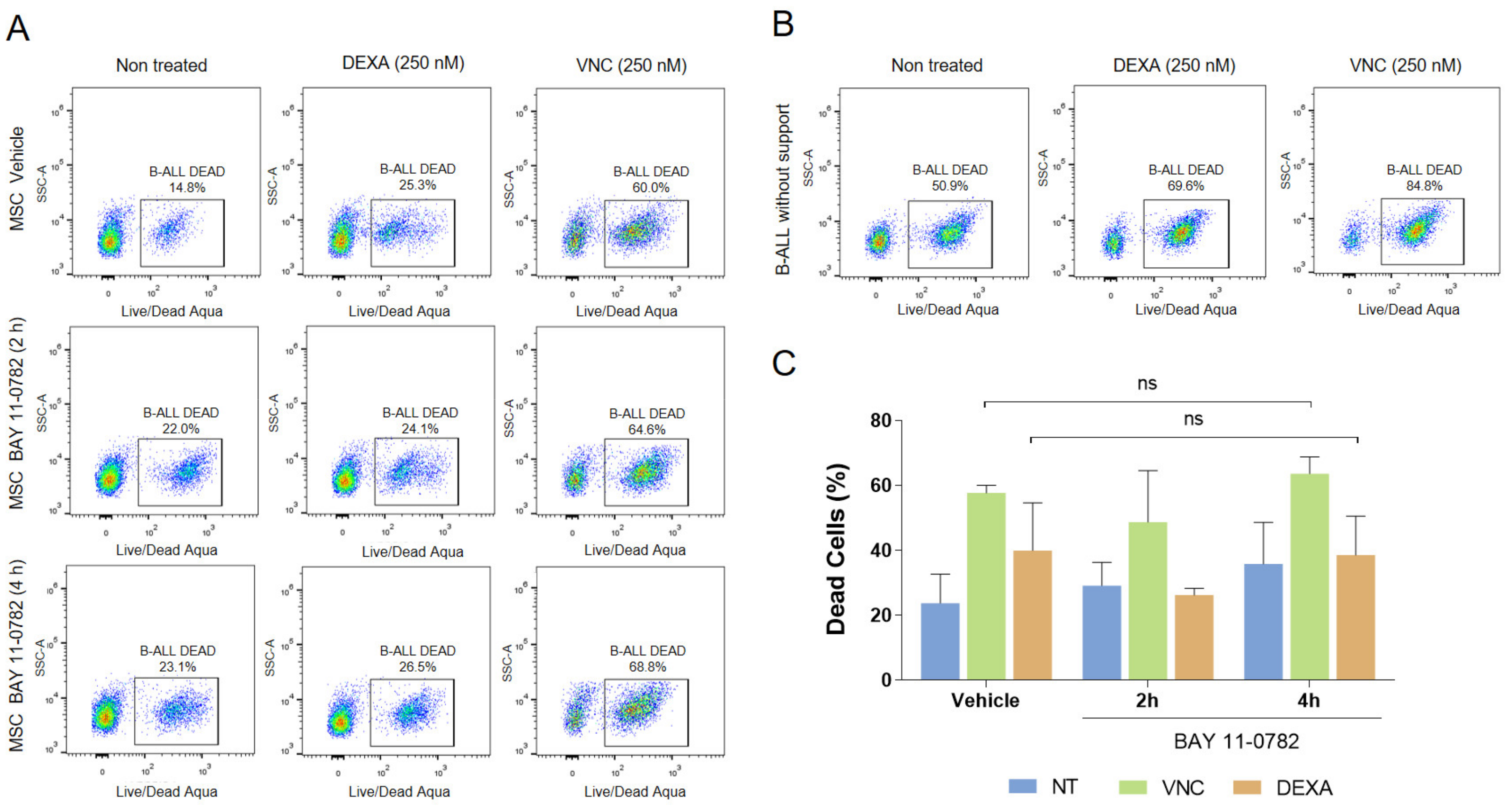
2.3. Blocking of NF-κB, but Not Akt, Inhibited Adhesion of B-ALL Cells to MSC without Increasing Susceptibility to DEXA or VNC
2.4. Increased Sensitization to Drug Treatment Could also Be Dissociated from NF-κB and Akt Signalling
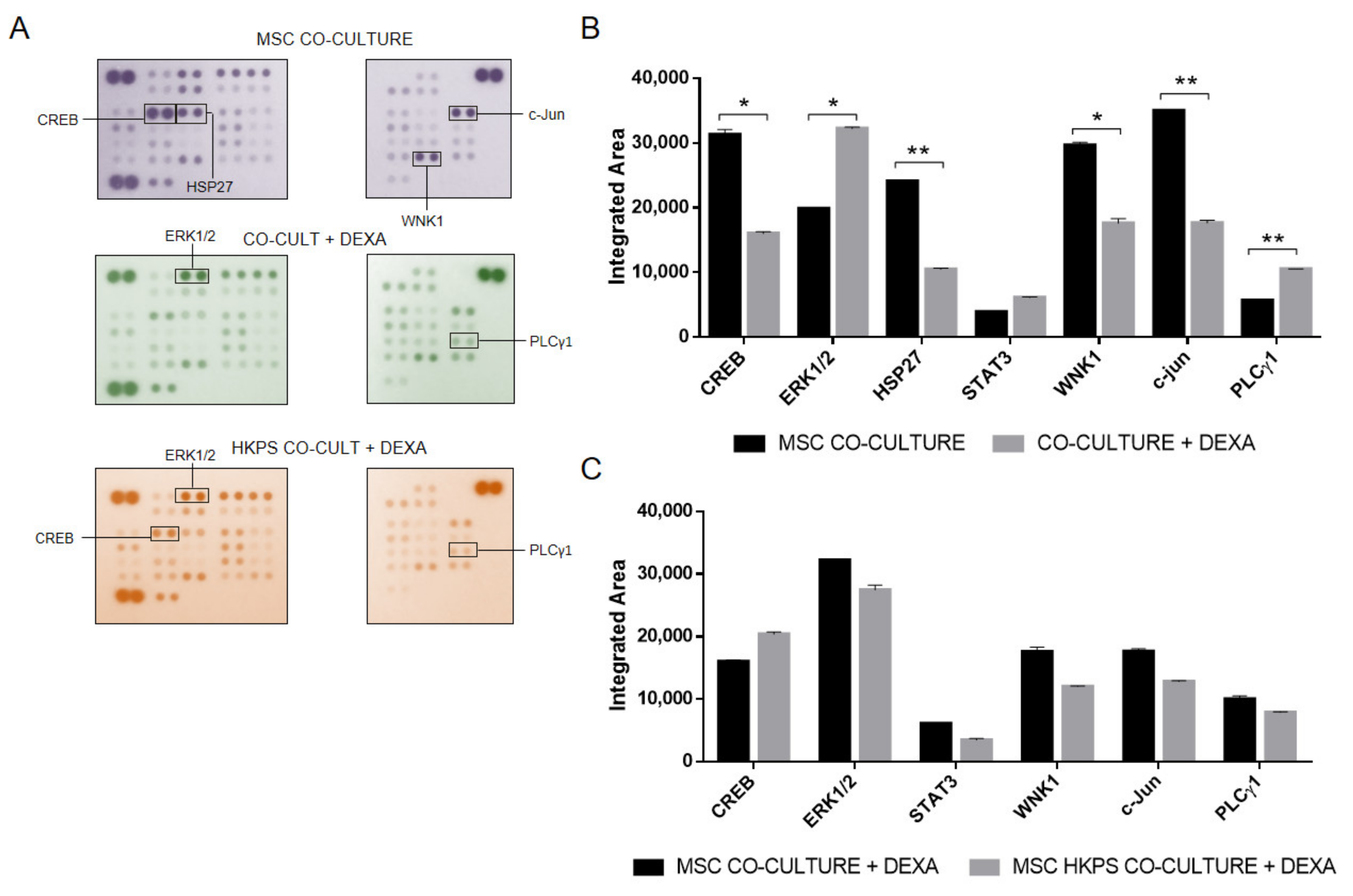
2.5. Evaluation of Other Kinases That Could Be Involved in Drug Protection
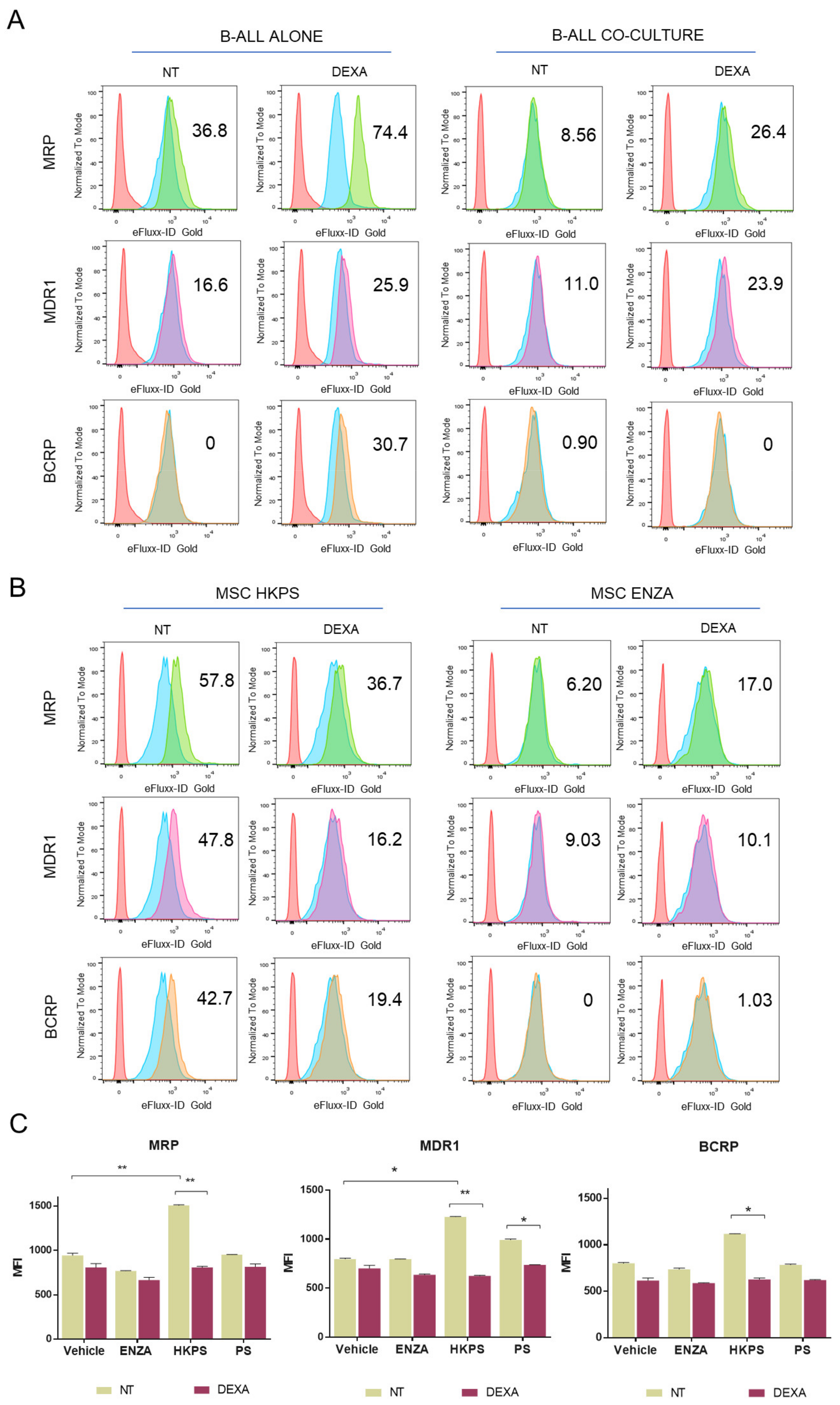
2.6. B-ALL ABC Transporters Are Indirectly Affected by the MSC Support
3. Discussion
4. Materials and Methods
4.1. B-ALL Cell Samples and MSC Isolation and Characterization
4.2. Inhibitors of Signalling Molecules Used
4.3. Cell Adhesion Assays
4.4. Phosphorylation Evaluation by Flow Cytometry
4.5. ABC Transporters Activity
4.6. Phospho Kinase Activity and NF-κB Pathway
4.7. Drug Sensitivity in B-ALL Cells
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Hunger, S.P.; Mullighan, C.G. Redefining ALL classification: Toward detecting high-risk ALL and implementing precision medicine. Blood 2015, 125, 3977–3987. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, K.; Devidas, M.; Cheng, S.C.; La, M.; Raetz, E.A.; Carroll, W.L.; Winick, N.J.; Hunger, S.P.; Gaynon, P.S.; Loh, M.L. Factors influencing survival after relapse from acute lymphoblastic leukemia: A Children’s Oncology Group study. Leukemia 2008, 22, 2142–2150. [Google Scholar] [CrossRef] [Green Version]
- Van Delft, F.W.; Horsley, S.; Colman, S.; Anderson, K.; Bateman, C.; Kempski, H.; Zuna, J.; Eckert, C.; Saha, V.; Kearney, L.; et al. Clonal origins of relapse in ETV6-RUNX1 acute lymphoblastic leukemia. Blood 2011, 117, 6247–6254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebinger, S.; Özdemir, E.Z.; Ziegenhain, C.; Tiedt, S.; Castro Alves, C.; Grunert, M.; Dworzak, M.; Lutz, C.; Turati, V.A.; Enver, T.; et al. Characterization of Rare, Dormant, and Therapy-Resistant Cells in Acute Lymphoblastic Leukemia. Cancer Cell 2016, 30, 849–862. [Google Scholar] [CrossRef] [Green Version]
- Iwamoto, S.; Mihara, K.; Downing, J.R.; Pui, C.-H.; Campana, D. Mesenchymal cells regulate the response of acute lymphoblastic leukemia cells to asparaginase. J. Clin. Investig. 2007, 117, 1049. [Google Scholar] [CrossRef] [PubMed]
- Mangolini, M.; Ringshausen, I. Bone marrow stromal cells drive key hallmarks of B cell malignancies. Int. J. Mol. Sci. 2020, 21, 1466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colmone, A.; Amorim, M.; Pontier, A.L.; Wang, S.; Jablonski, E.; Sipkins, D.A. Leukemic cells create bone marrow niches that disrupt the behavior of normal hematopoietic progenitor cells. Science 2008, 322, 1861–1865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, C.W.; Shi, J.; Chen, J.; Wang, B.; Yu, Y.H.; Qin, X.; Zhou, X.C.; Cai, Y.J.; Li, Z.Q.; Zhang, F.; et al. Leukemia Propagating Cells Rebuild an Evolving Niche in Response to Therapy. Cancer Cell 2014, 25, 778–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schepers, K.; Pietras, E.M.; Reynaud, D.; Flach, J.; Binnewies, M.; Garg, T.; Wagers, A.J.; Hsiao, E.C.; Passegué, E. Myeloproliferative Neoplasia Remodels the Endosteal Bone Marrow Niche into a Self-Reinforcing Leukemic Niche. Cell Stem Cell 2013, 13, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Ho, Y.W.; Huang, Q.; Maeda, T.; Lin, A.; Lee, S.U.; Hair, A.; Holyoake, T.L.; Huettner, C.; Bhatia, R. Altered Microenvironmental Regulation of Leukemic and Normal Stem Cells in Chronic Myelogenous Leukemia. Cancer Cell 2012, 21, 577–592. [Google Scholar] [CrossRef] [Green Version]
- Manabe, A.; Coustan-Smith, E.; Behm, F.G.; Raimondi, S.C.; Campana, D. Bone marrow-derived stromal cells prevent apoptotic cell death in B- lineage acute lymphoblastic leukemia. Blood 1992, 79, 2370–2377. [Google Scholar] [CrossRef] [Green Version]
- Bendall, L.J.; Daniel, A.; Kortlepel, K.; Gottlieb, D.J. Bone marrow adherent layers inhibit apoptosis of acute myeloid leukemia cells. Exp. Hematol. 1994, 22, 1252–1260. [Google Scholar]
- Panayiotidis, P.; Jones, D.; Ganeshaguru, K.; Foroni, L.; Hoffbrand, A.V. Human bone marrow stromal cells prevent apoptosis and support the survival of chronic lymphocytic leukaemia cells in vitro. Br. J. Haematol. 1996, 92, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Kurtova, A.V.; Balakrishnan, K.; Chen, R.; Ding, W.; Schnabl, S.; Quiroga, M.P.; Sivina, M.; Wierda, W.G.; Estrov, Z.; Keating, M.J.; et al. Diverse marrow stromal cells protect CLL cells from spontaneous and drug-induced apoptosis: Development of a reliable and reproducible system to assess stromal cell adhesion-mediated drug resistance. Blood 2009, 114, 4441–4450. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, E.; Azab, A.K.; Manley, P.W.; Kung, A.L.; Christie, A.L.; Bronson, R.; Ghobrial, I.M.; Griffin, J.D. Inhibition of CXCR4 in CML cells disrupts their interaction with the bone marrow microenvironment and sensitizes them to nilotinib. Leukemia 2012, 26, 985–990. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Aparicio, P.F.; Vanegas, N.-D.P.; Uribe, G.I.; Ortiz-Montero, P.; Cadavid-Cortés, C.; Lagos, J.; Flechas-Afanador, J.; Linares-Ballesteros, A.; Vernot, J.-P. Dual Targeting of Stromal Cell Support and Leukemic Cell Growth by a Peptidic PKC Inhibitor Shows Effectiveness against B-ALL. Int. J. Mol. Sci. 2020, 21, 3705. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Chen, J.; Moore, A.; Mangolini, M.; Santoro, A.; Boyd, J.R.; Schjerven, H.; Ecker, V.; Buchner, M.; Williamson, J.C.; et al. Stromal cell protein kinase C-β inhibition enhances chemosensitivity in B cell malignancies and overcomes drug resistance. Sci. Transl. Med. 2020, 12, eaax9340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, L.C.; Lange, B.J.; Rheingold, S.R.; Bunin, N.J. Bone-marrow relapse in paediatric acute lymphoblastic leukaemia. Lancet Oncol. 2008, 9, 873–883. [Google Scholar] [CrossRef]
- Hawkins, E.D.; Duarte, D.; Akinduro, O.; Khorshed, R.A.; Passaro, D.; Nowicka, M.; Straszkowski, L.; Scott, M.K.; Rothery, S.; Ruivo, N.; et al. T-cell acute leukaemia exhibits dynamic interactions with bone marrow microenvironments. Nature 2016, 538, 518–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Zimmerman, G.; Huang, X.; Yu, S.; Myers, J.; Wang, Y.; Moreton, S.; Nthale, J.; Awadallah, A.; Beck, R.; et al. Aberrant notch signaling in the bone marrow microenvironment of acute lymphoid leukemia suppresses osteoblast-mediated support of hematopoietic niche function. Cancer Res. 2016, 76, 1641–1652. [Google Scholar] [CrossRef] [Green Version]
- Nishii, K.; Katayama, N.; Miwa, H.; Shikami, M.; Masuya, M.; Shiku, H.; Kita, K. Survival of human leukaemic B-cell precursors is supported by stromal cells and cytokines: Association with the expression of bcl-2 protein. Br. J. Haematol. 1999, 105, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Mudry, M.E.; Fortney, J.E.; York, T.; Hall, B.M.; Gibson, L.F. Stromal cells regulate survival of B-lineage leukemic cells during chemotherapy. Blood 2000, 96, 1926–1932. [Google Scholar] [CrossRef] [PubMed]
- Schneider, P.; Vasse, M.; Al Bayati, A.; Lenormand, B.; Vannier, J.P. Is high expression of the chemokine receptor CXCR-4 of predictive value for early relapse in childhood acute lymphoblastic leukaemia? Br. J. Haematol. 2002, 119, 579–580. [Google Scholar] [CrossRef]
- Hsieh, Y.T.; Gang, E.J.; Geng, H.; Park, E.; Huantes, S.; Chudziak, D.; Dauber, K.; Schaefer, P.; Scharman, C.; Shimada, H.; et al. Integrin alpha4 blockade sensitizes drug resistant pre-B acute lymphoblastic leukemia to chemotherapy. Blood 2013, 121, 1814–1818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fei, F.; Joo, E.J.; Tarighat, S.S.; Schiffer, I.; Paz, H.; Fabbri, M.; Abdel-Azim, H.; Groffen, J.; Heisterkamp, N. B-cell precursor acute lymphoblastic leukemia and stromal cells communicate through Galectin-3. Oncotarget 2015, 6, 11378–11394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, I.; Yoshida, T.; Jena, N.; Qi, X.; Zhang, J.; Van Etten, R.A.; Georgopoulos, K. Loss of Ikaros DNA-binding function confers integrin-dependent survival on pre-B cells and progression to acute lymphoblastic leukemia. Nat. Immunol. 2014, 15, 294–304. [Google Scholar] [CrossRef]
- Gang, E.J.; Kim, H.N.; Hsieh, Y.-T.; Ruan, Y.; Ogana, H.A.; Lee, S.; Pham, J.; Geng, H.; Park, E.; Klemm, L.; et al. Integrin α6 mediates the drug resistance of acute lymphoblastic B-cell leukemia. Blood 2020, 136, 210–223. [Google Scholar] [CrossRef]
- Tabe, Y.; Jin, L.; Tsutsumi-Ishii, Y.; Xu, Y.; McQueen, T.; Priebe, W.; Mills, G.B.; Ohsaka, A.; Nagaoka, I.; Andreeff, M.; et al. Activation of Integrin-Linked Kinase Is a Critical Prosurvival Pathway Induced in Leukemic Cells by Bone Marrow-Derived Stromal Cells. Cancer Res. 2007, 67, 684–694. [Google Scholar] [CrossRef] [Green Version]
- Jones, C.L.; Gearheart, C.M.; Fosmire, S.; Delgado-Martin, C.; Evensen, N.A.; Bride, K.; Waanders, A.J.; Pais, F.; Wang, J.; Bhatla, T.; et al. MAPK signaling cascades mediate distinct glucocorticoid resistance mechanisms in pediatric leukemia. Blood 2015, 126, 2202–2212. [Google Scholar] [CrossRef] [Green Version]
- Uy, G.L.; Rettig, M.P.; Motabi, I.H.; McFarland, K.; Trinkaus, K.M.; Hladnik, L.M.; Kulkarni, S.; Abboud, C.N.; Cashen, A.F.; Stockerl-Goldstein, K.E.; et al. A phase 1/2 study of chemosensitization with the CXCR4 antagonist plerixafor in relapsed or refractory acute myeloid leukemia. Blood 2012, 119, 3917–3924. [Google Scholar] [CrossRef]
- Bajaj, J.; Konuma, T.; Lytle, N.K.; Kwon, H.Y.; Ablack, J.N.; Cantor, J.M.; Rizzieri, D.; Chuah, C.; Oehler, V.G.; Broome, E.H.; et al. CD98-Mediated Adhesive Signaling Enables the Establishment and Propagation of Acute Myelogenous Leukemia. Cancer Cell 2016, 30, 792–805. [Google Scholar] [CrossRef] [Green Version]
- Matsunaga, T.; Takemoto, N.; Sato, T.; Takimoto, R.; Tanaka, I.; Fujimi, A.; Akiyama, T.; Kuroda, H.; Kawano, Y.; Kobune, M.; et al. Interaction between leukemic-cell VLA-4 and stromal fibronectin is a decisive factor for minimal residual disease of acute myelogenous leukemia. Nat. Med. 2003, 9, 1158–1165. [Google Scholar] [CrossRef]
- Amigo-Jiménez, I.; Bailón, E.; Aguilera-Montilla, N.; Terol, M.J.; García-Marco, J.A.; García-Pardo, A. Bone marrow stroma-induced resistance of chronic lymphocytic leukemia cells to arsenic trioxide involves Mcl-1 upregulation and is overcome by inhibiting the PI3Kδ or PKCβ signaling pathways. Oncotarget 2015, 6, 44832–44848. [Google Scholar] [CrossRef]
- Niedermeier, M.; Hennessy, B.T.; Knight, Z.A.; Henneberg, M.; Hu, J.; Kurtova, A.V.; Wierda, W.G.; Keating, M.J.; Shokat, K.M.; Burger, J.A. Isoform-selective phosphoinositide 3′-kinase inhibitors inhibit CXCR4 signaling and overcome stromal cell-mediated drug resistance in chronic lymphocytic leukemia: A novel therapeutic approach. Blood 2009, 113, 5549–5557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evangelisti, C.; Cappellini, A.; Oliveira, M.; Fragoso, R.; Barata, J.T.; Bertaina, A.; Locatelli, F.; Simioni, C.; Neri, L.M.; Chiarini, F.; et al. Phosphatidylinositol 3-kinase inhibition potentiates glucocorticoid response in B-cell acute lymphoblastic leukemia. J. Cell Physiol. 2018, 233, 1796–1811. [Google Scholar] [CrossRef]
- Feldhahn, N.; Arutyunyan, A.; Stoddart, S.; Zhang, B.; Schmidhuber, S.; Yi, S.-J.; Kim, Y.; Groffen, J.; Heisterkamp, N. Environment-mediated drug resistance in Bcr/Abl-positive acute lymphoblastic leukemia. Oncoimmunology 2012, 1, 618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, C.; Witkowski, M.T.; Harris, J.; Dolgalev, I.; Sreeram, S.; Qian, W.; Tong, J.; Chen, X.; Aifantis, I.; Chen, W. Leukemia-on-a-chip: Dissecting the chemoresistance mechanisms in B cell acute lymphoblastic leukemia bone marrow niche. Sci. Adv. 2020, 6, eaba5536. [Google Scholar] [CrossRef] [PubMed]
- Lutzny, G.; Kocher, T.; Schmidt-Supprian, M.; Rudelius, M.; Klein-Hitpass, L.; Finch, A.J.; Dürig, J.; Wagner, M.; Haferlach, C.; Kohlmann, A.; et al. Protein Kinase C-β-Dependent Activation of NF-κB in Stromal Cells Is Indispensable for the Survival of Chronic Lymphocytic Leukemia B Cells In Vivo. Cancer Cell 2013, 23, 77–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacamo, R.; Chen, Y.; Wang, Z.; Wencai, M.; Zhang, M.; Spaeth, E.L.; Wang, Y.; Battula, V.L.; Mak, P.Y.; Schallmoser, K.; et al. Reciprocal leukemia-stroma VCAM-1/VLA-4-dependent activation of NF-κB mediates chemoresistance. Blood 2014, 123, 2691–2702. [Google Scholar] [CrossRef] [PubMed]
- Polak, R.; De Rooij, B.; Pieters, R.; Den Boer, M.L. B-cell precursor acute lymphoblastic leukemia cells use tunneling nanotubes to orchestrate their microenvironment. Blood 2015, 126, 2404–2414. [Google Scholar] [CrossRef] [Green Version]
- Burt, R.; Dey, A.; Aref, S.; Aguiar, M.; Akarca, A.; Bailey, K.; Day, W.; Hooper, S.; Kirkwood, A.; Kirschner, K.; et al. Activated stromal cells transfer mitochondria to rescue acute lymphoblastic leukemia cells from oxidative stress. Blood 2019, 134, 1415–1429. [Google Scholar] [CrossRef]
- Dander, E.; Palmi, C.; D’amico, G.; Cazzaniga, G. The bone marrow niche in b-cell acute lymphoblastic leukemia: The role of microenvironment from pre-leukemia to overt leukemia. Int. J. Mol. Sci. 2021, 22, 4426. [Google Scholar] [CrossRef]
- Bonilla, X.; Vanegas, N.D.P.; Vernot, J.P. Acute leukemia induces senescence and impaired osteogenic differentiation in mesenchymal stem cells endowing leukemic cells with functional advantages. Stem Cells Int. 2019, 2019, 3864948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boutin, L.; Arnautou, P.; Trignol, A.; Ségot, A.; Farge, T.; Desterke, C.; Soave, S.; Clay, D.; Raffoux, E.; Sarry, J.E.; et al. Mesenchymal stromal cells confer chemoresistance to myeloid leukemia blasts through Side Population functionality and ABC transporter activation. Haematologica 2020, 105, 987–998. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, M.T.; Dolgalev, I.; Evensen, N.A.; Ma, C.; Chambers, T.; Roberts, K.G.; Sreeram, S.; Dai, Y.; Tikhonova, A.N.; Lasry, A.; et al. Extensive Remodeling of the Immune Microenvironment in B Cell Acute Lymphoblastic Leukemia. Cancer Cell 2020, 37, 867–882.e12. [Google Scholar] [CrossRef]
- Anderson, D.; Skut, P.; Hughes, A.M.; Ferrari, E.; Tickner, J.; Xu, J.; Mullin, B.H.; Tang, D.; Malinge, S.; Kees, U.R.; et al. The bone marrow microenvironment of pre-B acute lymphoblastic leukemia at single-cell resolution. Sci. Rep. 2020, 10, 19173. [Google Scholar] [CrossRef] [PubMed]
- Kamdje, A.H.N.; Mosna, F.; Bifari, F.; Lisi, V.; Bassi, G.; Malpeli, G.; Ricciardi, M.; Perbellini, O.; Scupoli, M.T.; Pizzolo, G.; et al. Notch-3 and Notch-4 signaling rescue from apoptosis human B-ALL cells in contact with human bone marrow-derived mesenchymal stromal cells. Blood 2011, 118, 380–389. [Google Scholar] [CrossRef]
- Hu, K.; Gu, Y.; Lou, L.; Liu, L.; Hu, Y.; Wang, B.; Luo, Y.; Shi, J.; Yu, X.; Huang, H. Galectin-3 mediates bone marrow microenvironment-induced drug resistance in acute leukemia cells via Wnt/β-catenin signaling pathway. J. Hematol. Oncol. 2015, 8, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, J.-H. Protein Kinase C (PKC) Isozymes and Cancer. New J. Sci. 2014, 2014, 231418. [Google Scholar] [CrossRef] [Green Version]
- Simon-Gabriel, C.P.; Foerster, K.; Saleem, S.; Bleckmann, D.; Benkisser-Petersen, M.; Thornton, N.; Umezawa, K.; Decker, S.; Burger, M.; Veelken, H.; et al. Microenvironmental stromal cells abrogate NF-κB inhibitor-induced apoptosis in chronic lymphocytic leukemia. Haematologica 2018, 103, 136–147. [Google Scholar] [CrossRef] [Green Version]
- Woyach, J.A.; Furman, R.R.; Liu, T.-M.; Ozer, H.G.; Zapatka, M.; Ruppert, A.S.; Xue, L.; Li, D.H.-H.; Steggerda, S.M.; Versele, M.; et al. Resistance Mechanisms for the Bruton’s Tyrosine Kinase Inhibitor Ibrutinib. N. Engl. J. Med. 2014, 370, 2286–2294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Krause, D.S.; Deans, R.J.; Keating, A.; Prockop, D.J.; Horwitz, E.M. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- World Medical Association declaration of Helsinki: Ethical principles for medical research involving human subjects. J. Am. Med. Assoc. 2013, 310, 2191–2194. [CrossRef] [PubMed] [Green Version]
- Perdomo-Arciniegas, A.M.; Patarroyo, M.E.; Vernot, J.-P. Novel Chimeric Peptide Inhibits Protein Kinase C and Induces Apoptosis in Human Immune Cells. Int. J. Pept. Res. Ther. 2008, 14, 64–74. [Google Scholar] [CrossRef]
- Amblard, M.; Fehrentz, J.A.; Martinez, J.; Subra, G. Methods and protocols of modern solid phase peptide synthesis. Mol. Biotechnol. 2006, 33, 239–254. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruiz-Aparicio, P.F.; Uribe, G.I.; Linares-Ballesteros, A.; Vernot, J.-P. Sensitization to Drug Treatment in Precursor B-Cell Acute Lymphoblastic Leukemia Is Not Achieved by Stromal NF-κB Inhibition of Cell Adhesion but by Stromal PKC-Dependent Inhibition of ABC Transporters Activity. Molecules 2021, 26, 5366. https://doi.org/10.3390/molecules26175366
Ruiz-Aparicio PF, Uribe GI, Linares-Ballesteros A, Vernot J-P. Sensitization to Drug Treatment in Precursor B-Cell Acute Lymphoblastic Leukemia Is Not Achieved by Stromal NF-κB Inhibition of Cell Adhesion but by Stromal PKC-Dependent Inhibition of ABC Transporters Activity. Molecules. 2021; 26(17):5366. https://doi.org/10.3390/molecules26175366
Chicago/Turabian StyleRuiz-Aparicio, Paola Fernanda, Gloria Inés Uribe, Adriana Linares-Ballesteros, and Jean-Paul Vernot. 2021. "Sensitization to Drug Treatment in Precursor B-Cell Acute Lymphoblastic Leukemia Is Not Achieved by Stromal NF-κB Inhibition of Cell Adhesion but by Stromal PKC-Dependent Inhibition of ABC Transporters Activity" Molecules 26, no. 17: 5366. https://doi.org/10.3390/molecules26175366