
Adsorption/Desorption Behaviors and SERS Chemical Enhancement of 6-Mercaptopurine on a Nanostructured Gold Surface: The Au20 Cluster Model



Abstract
:
1. Introduction
2. Computational Methods
3. Results and Discussion
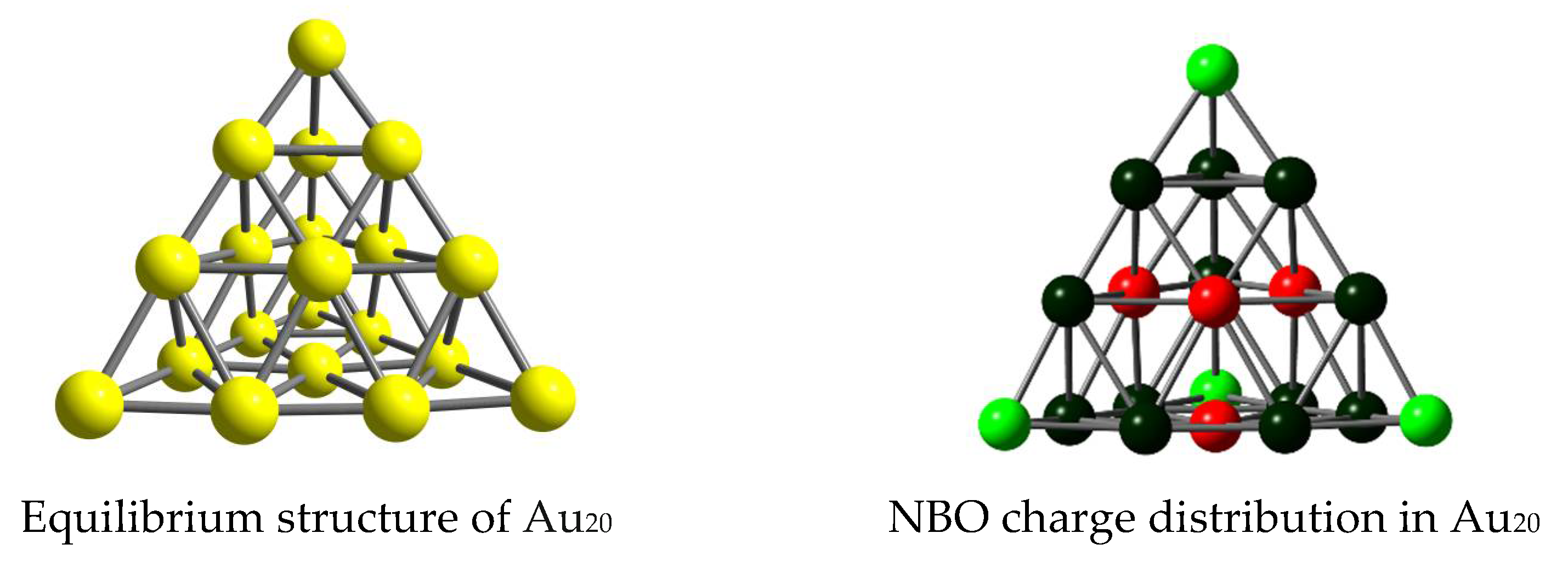
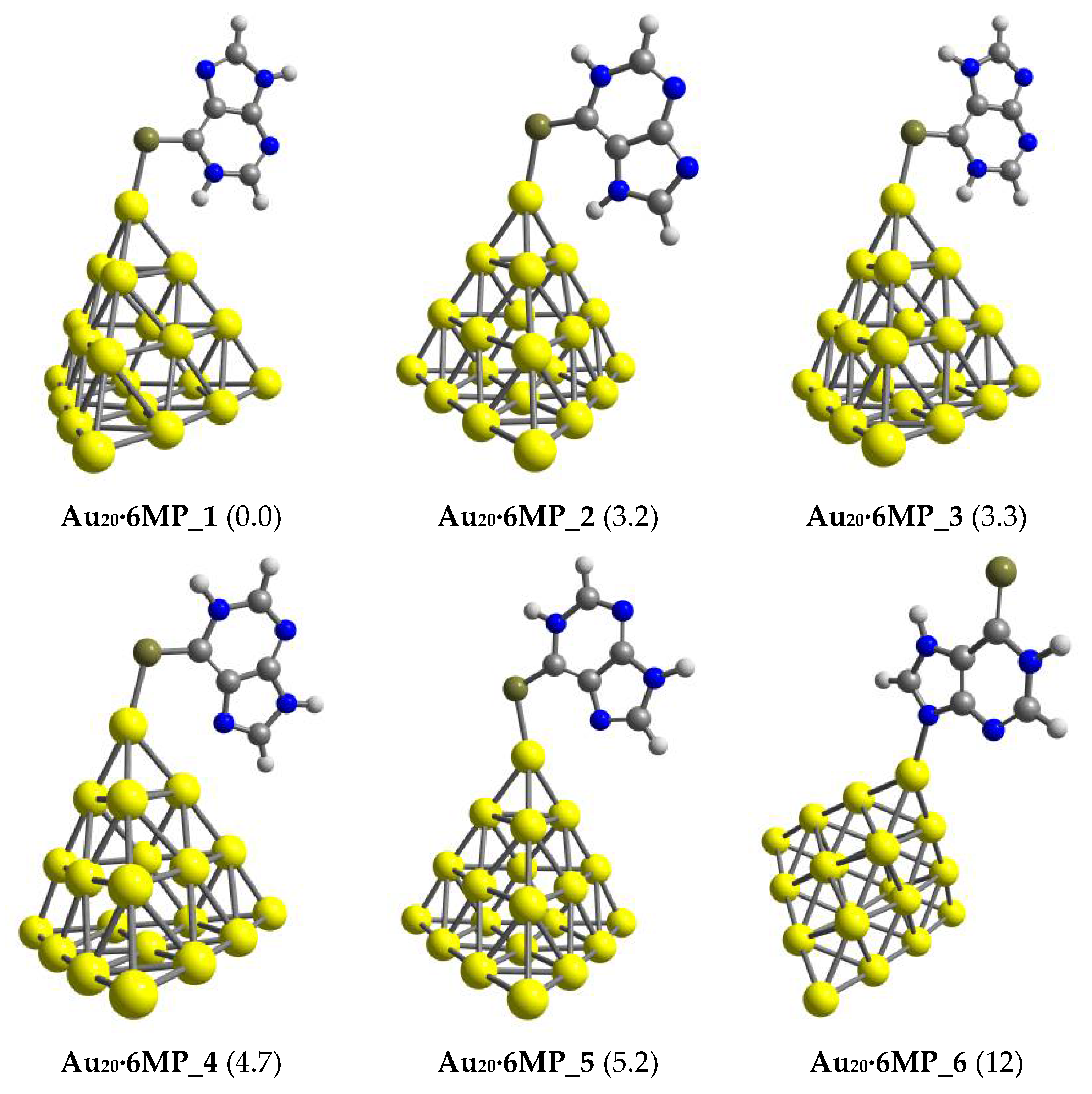
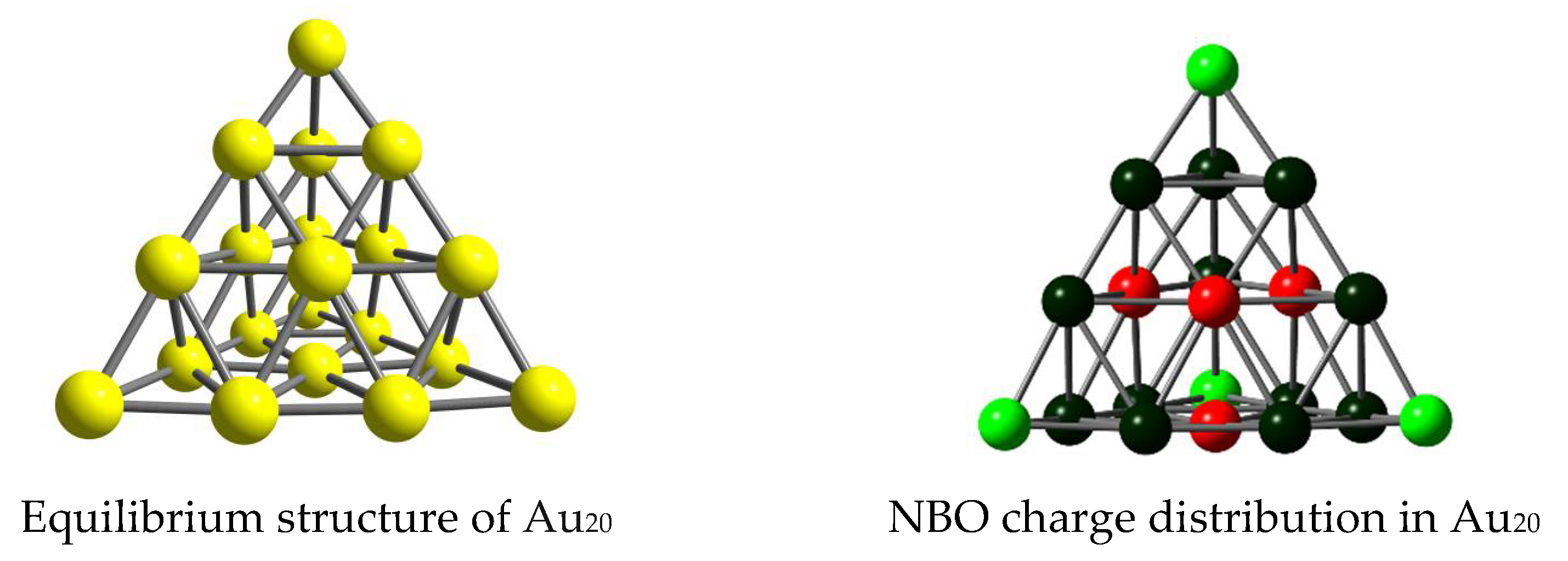
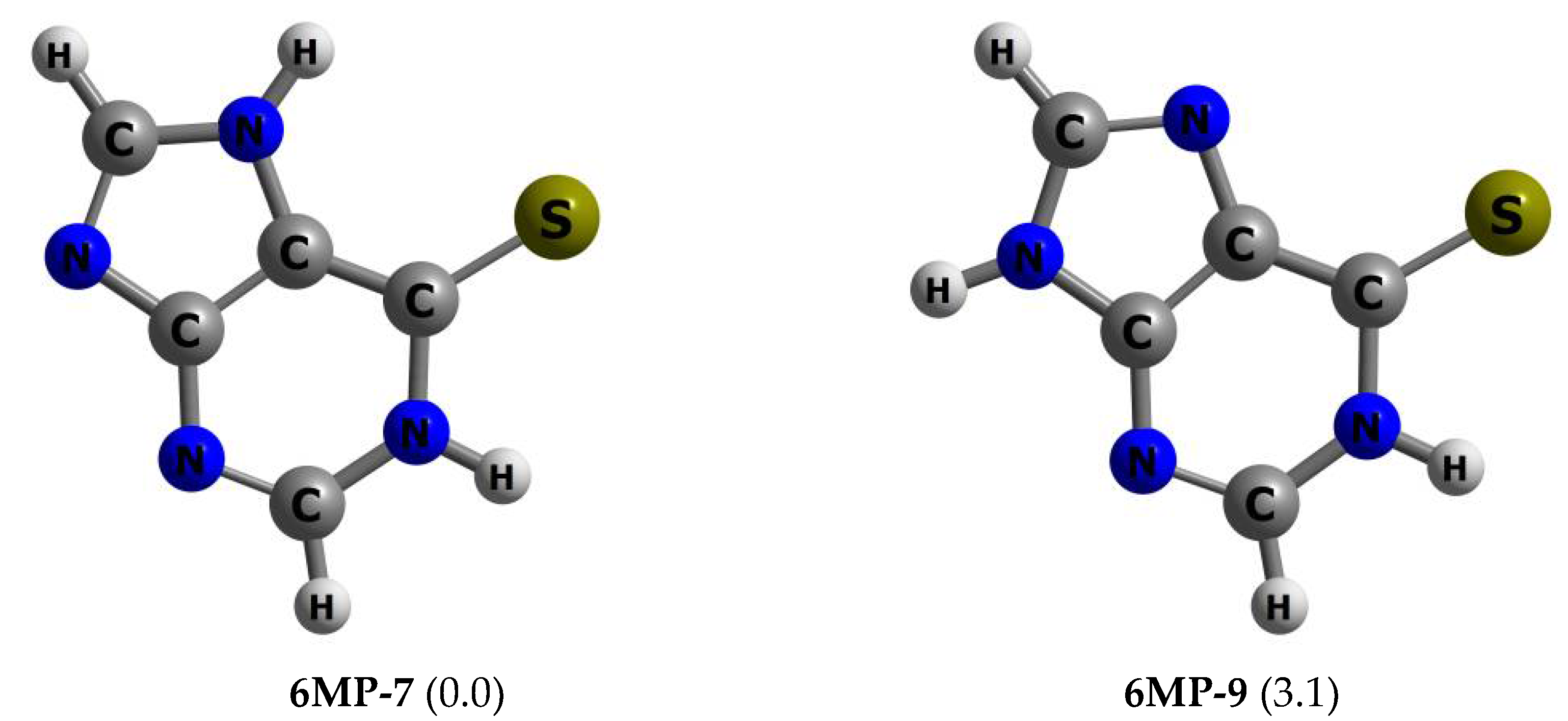
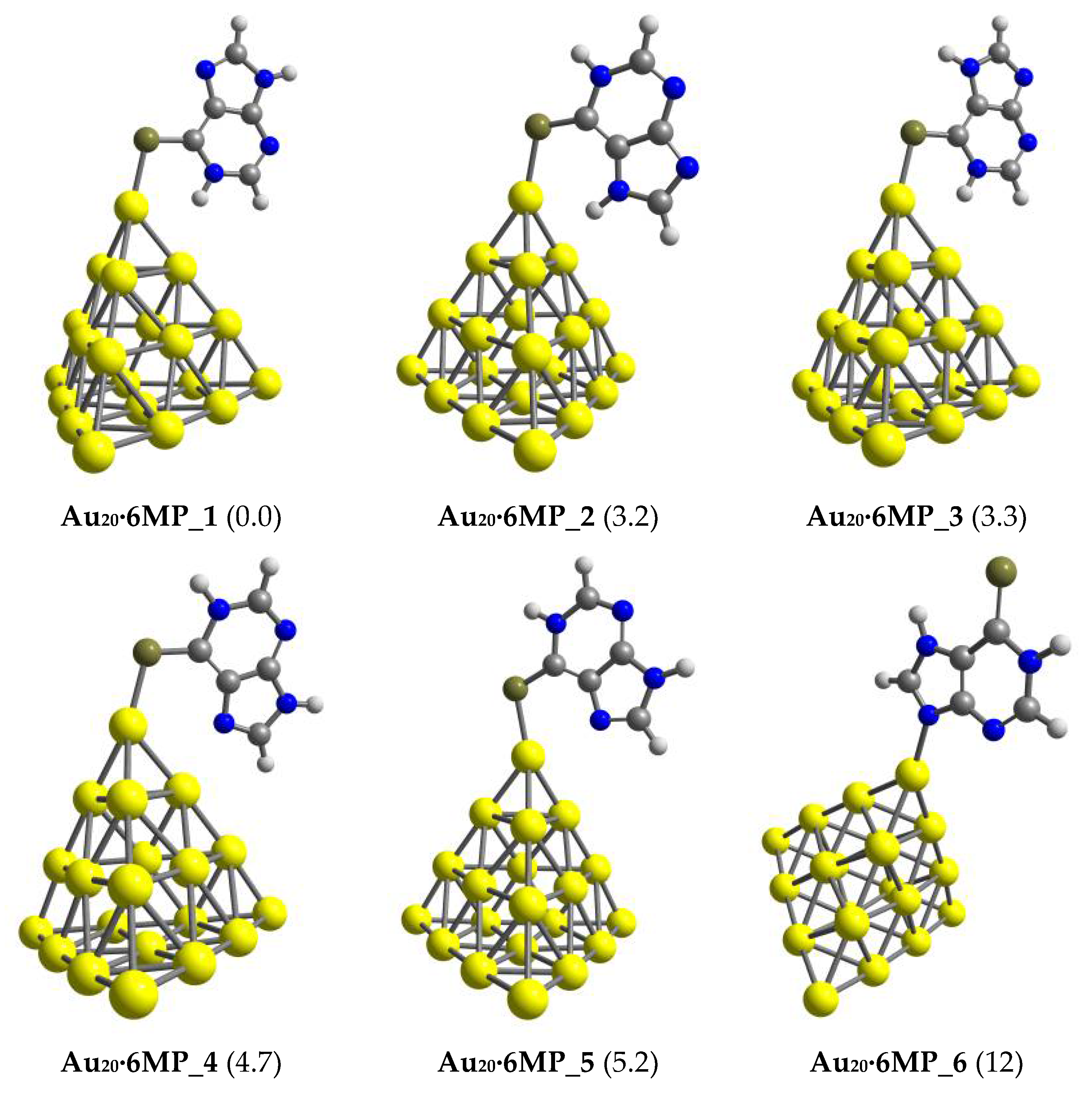
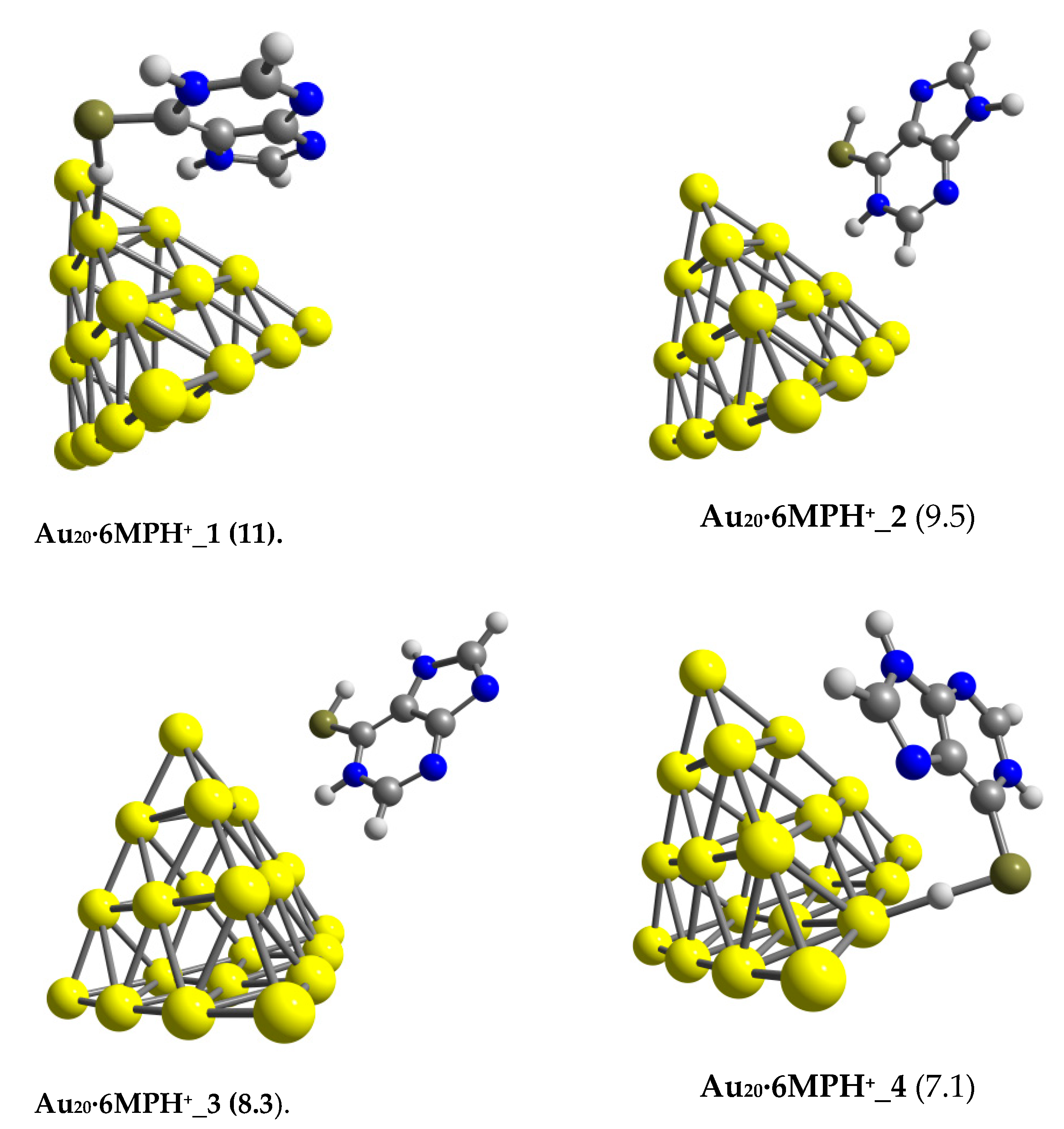
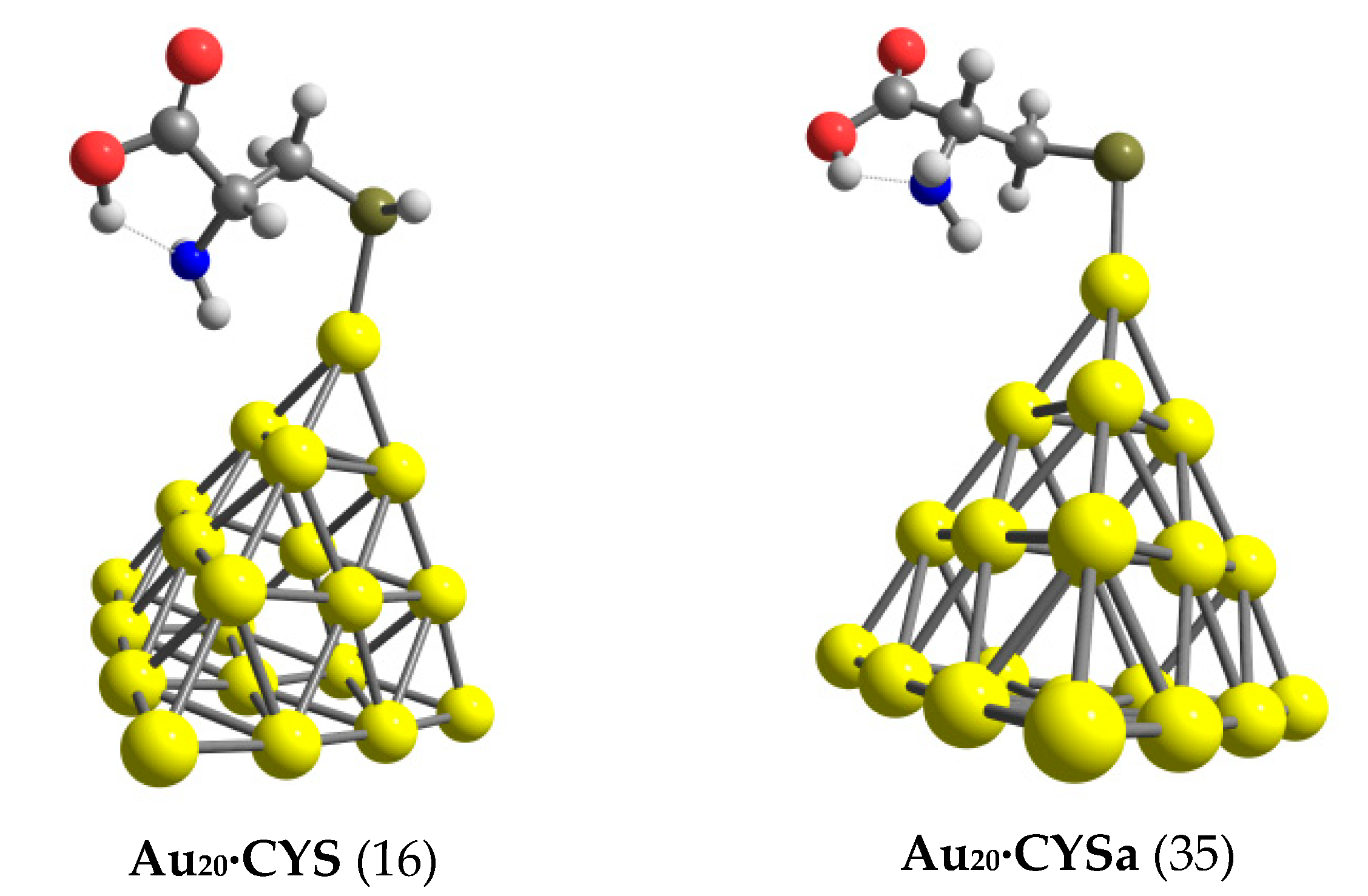
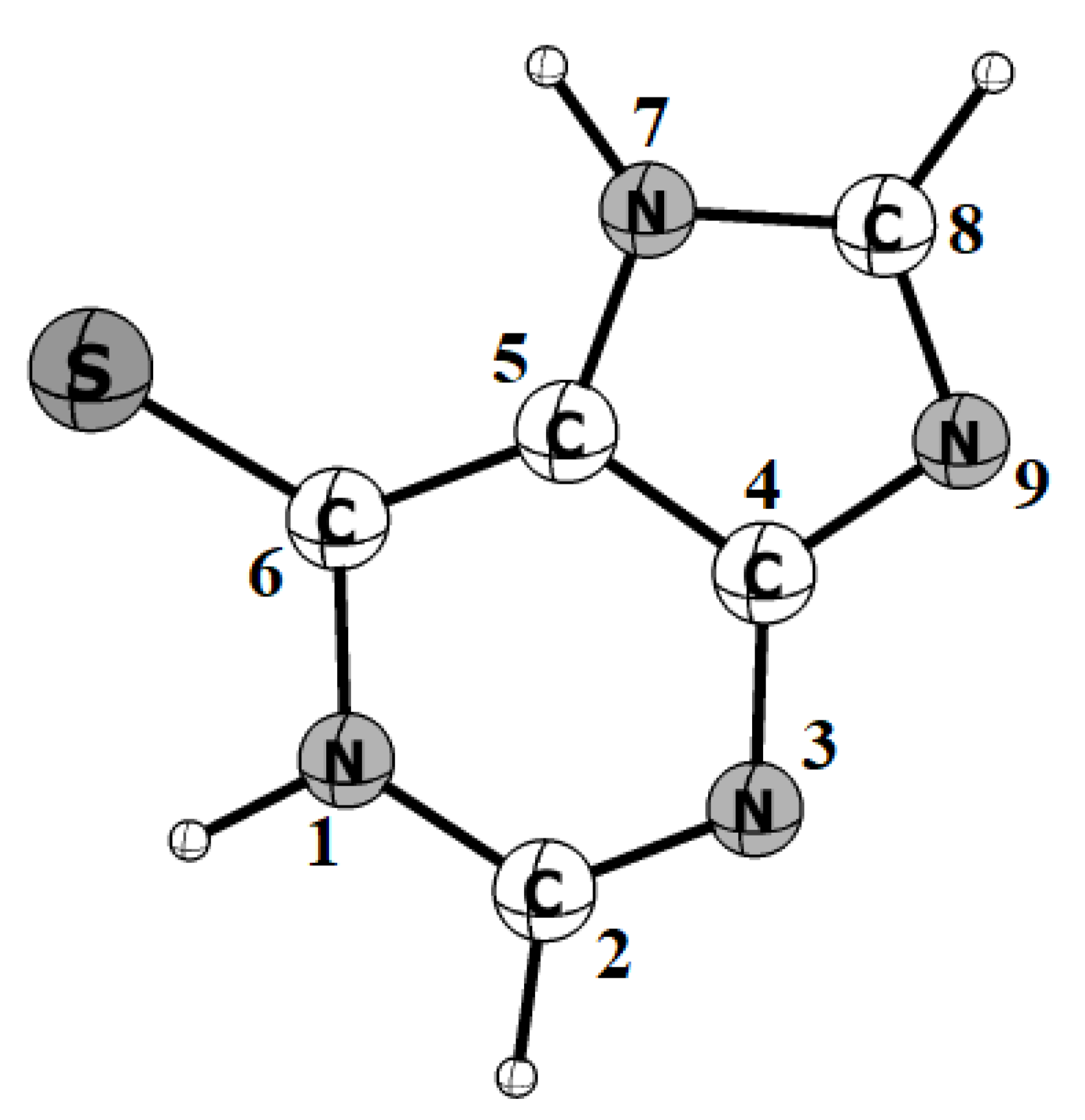
3.1. Structures and Energetics
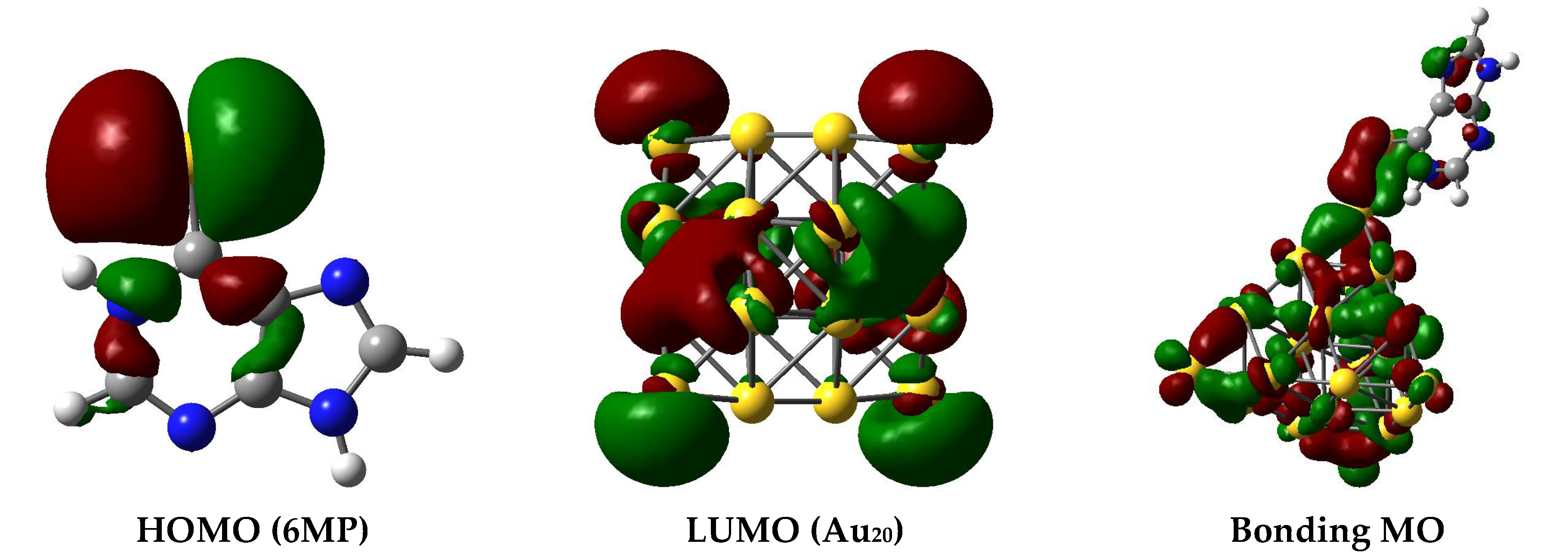
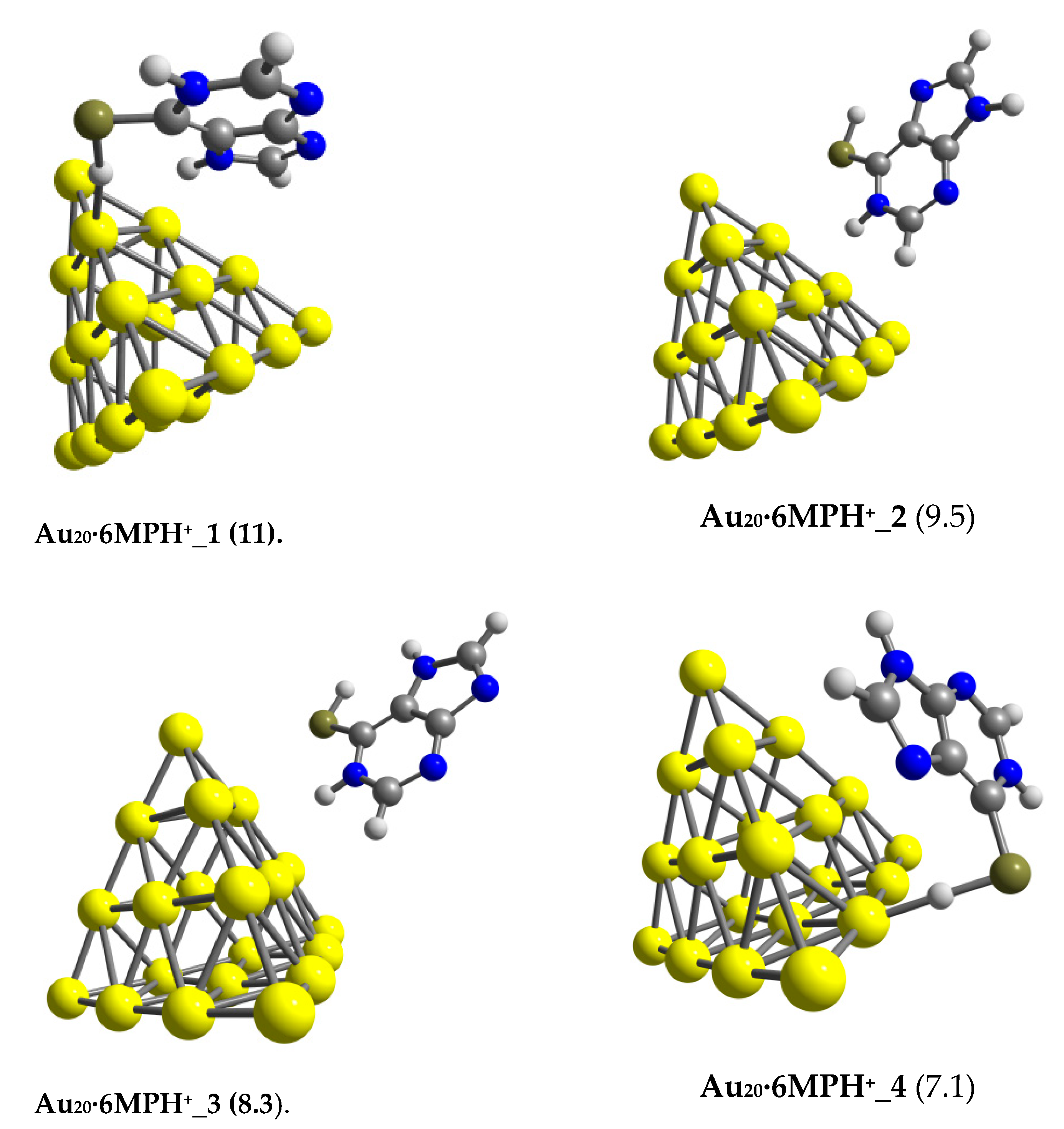
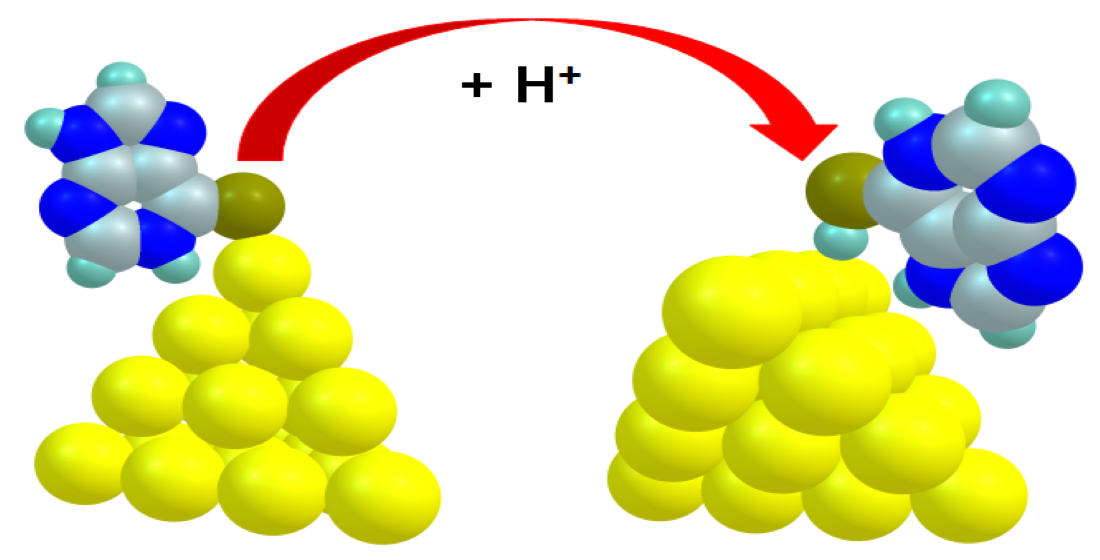
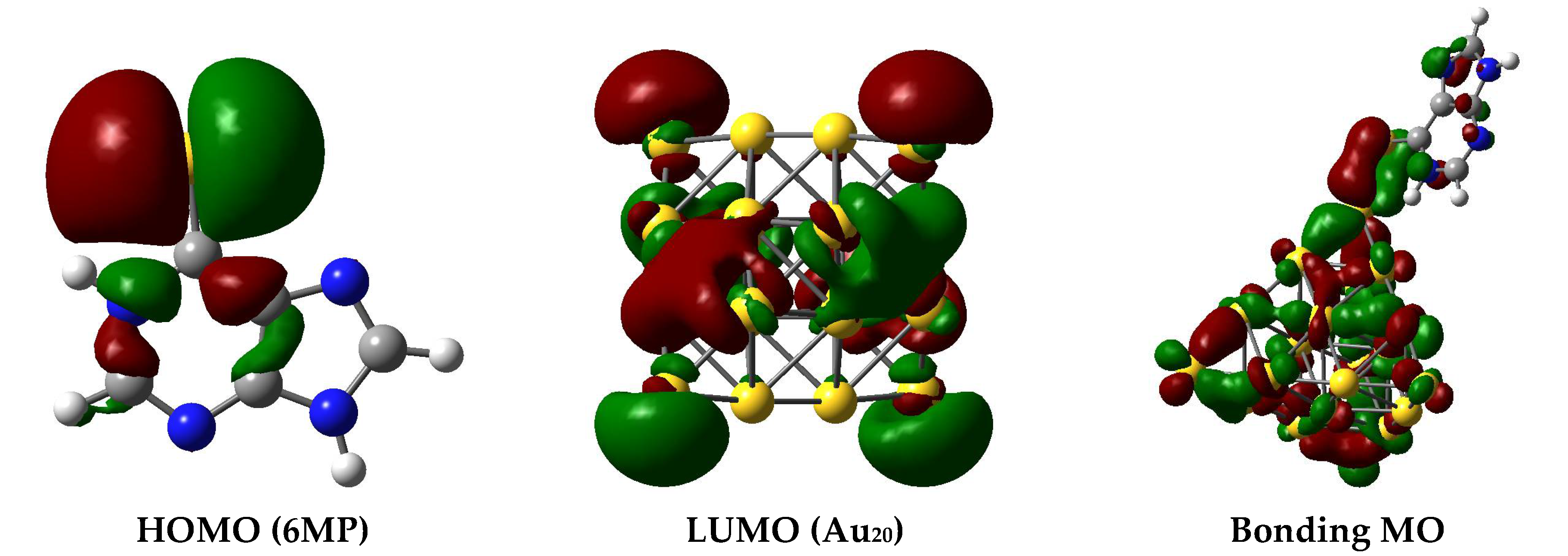
3.2. The Binding Mechanism and Drug Releasing
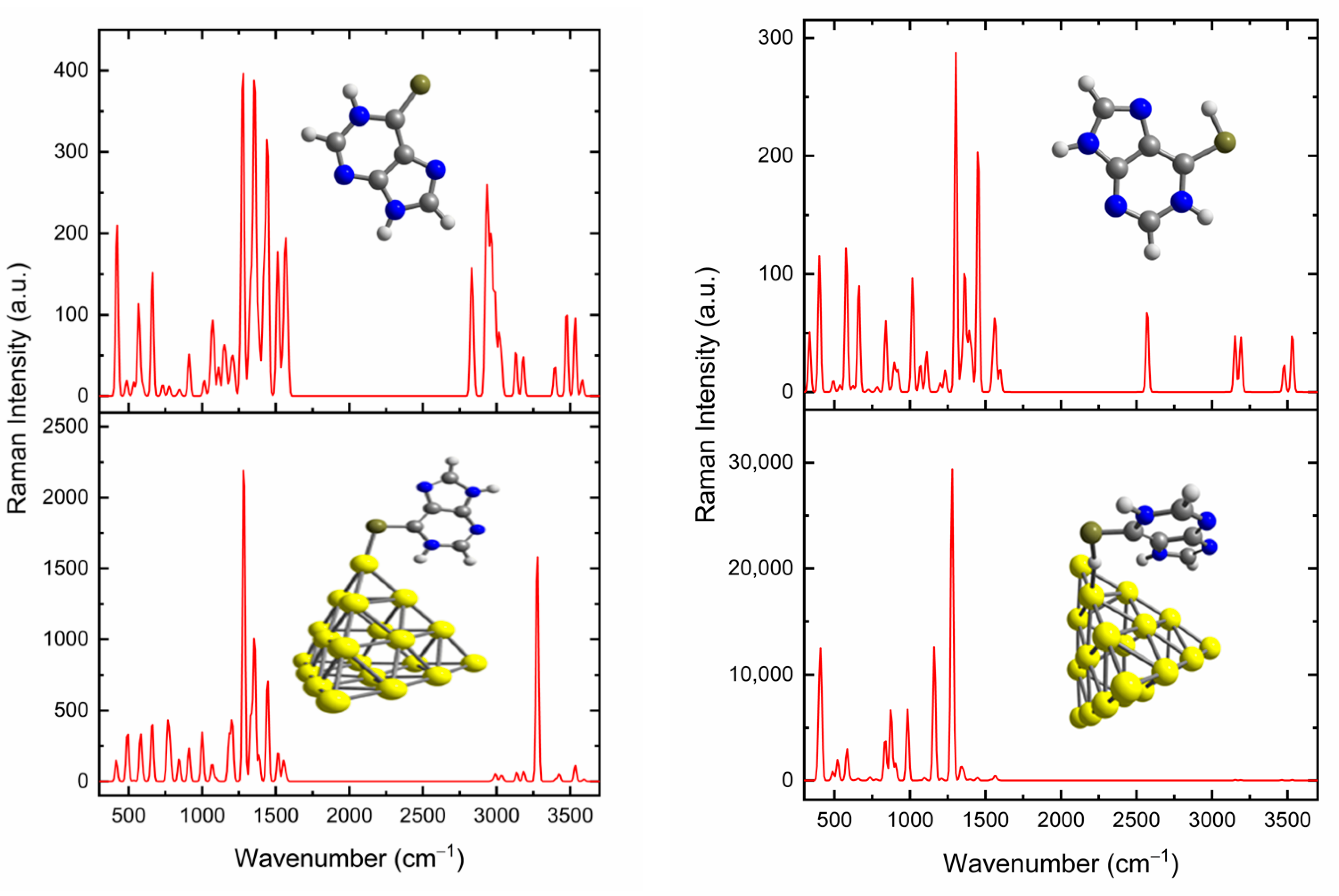
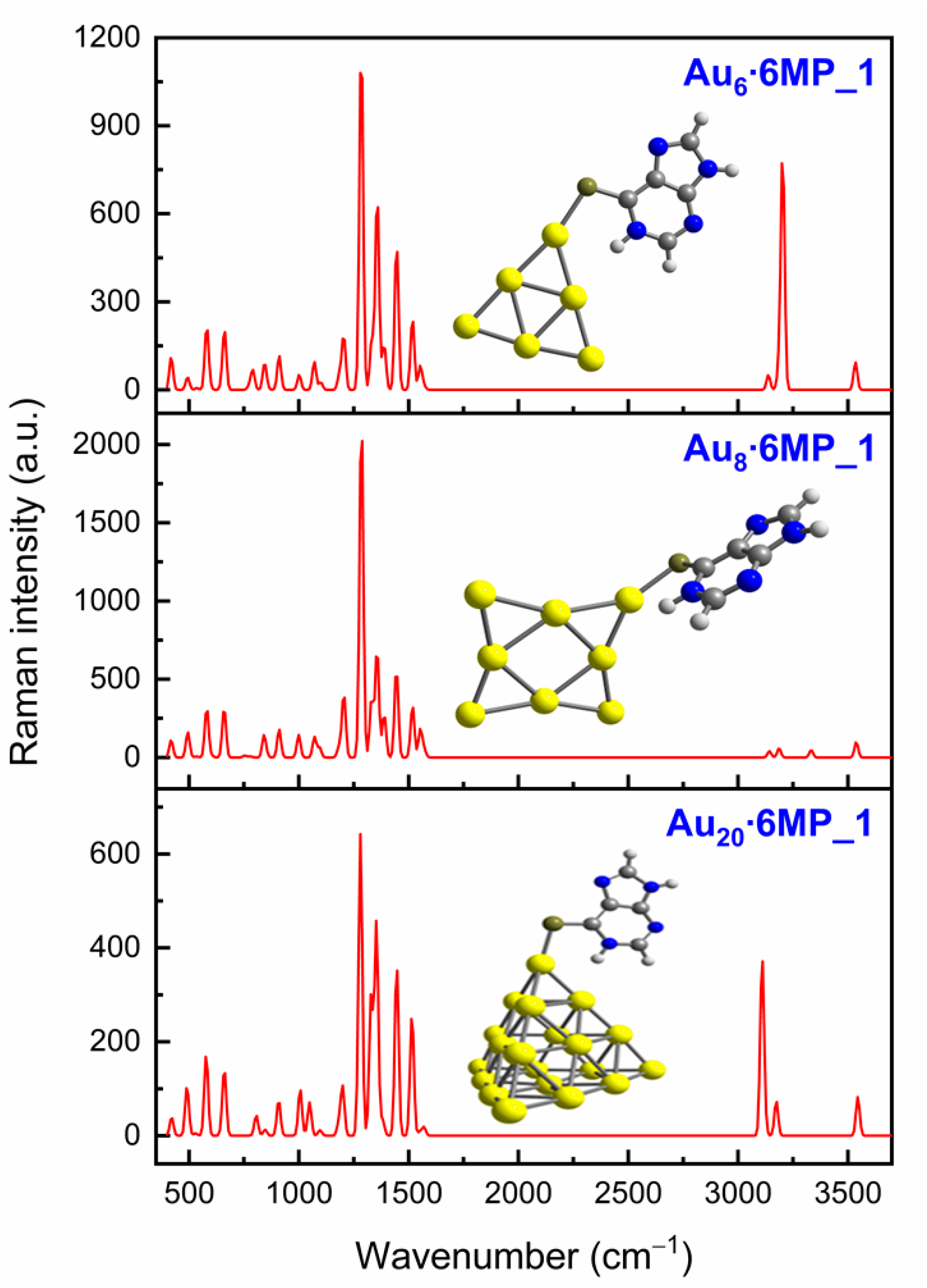
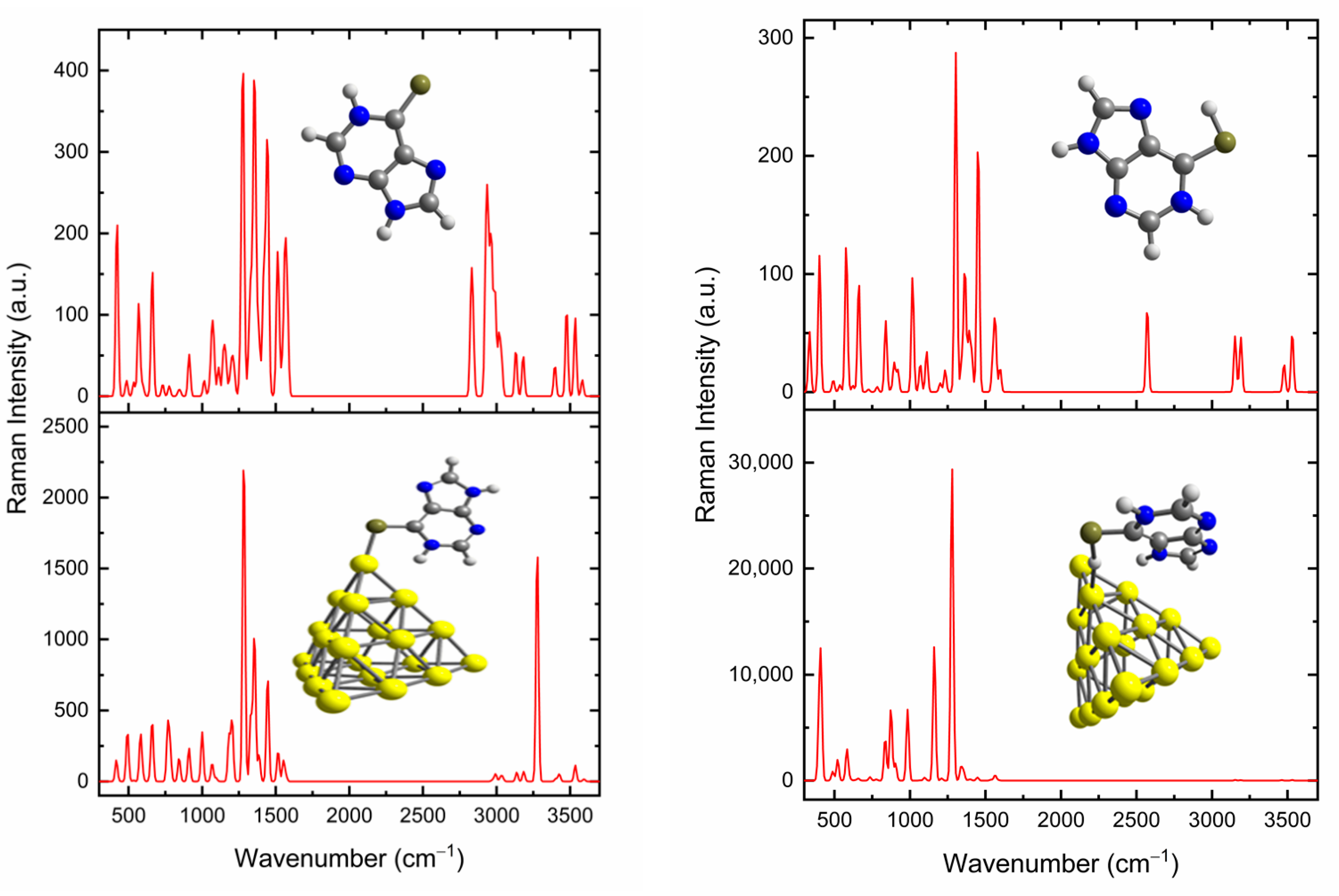
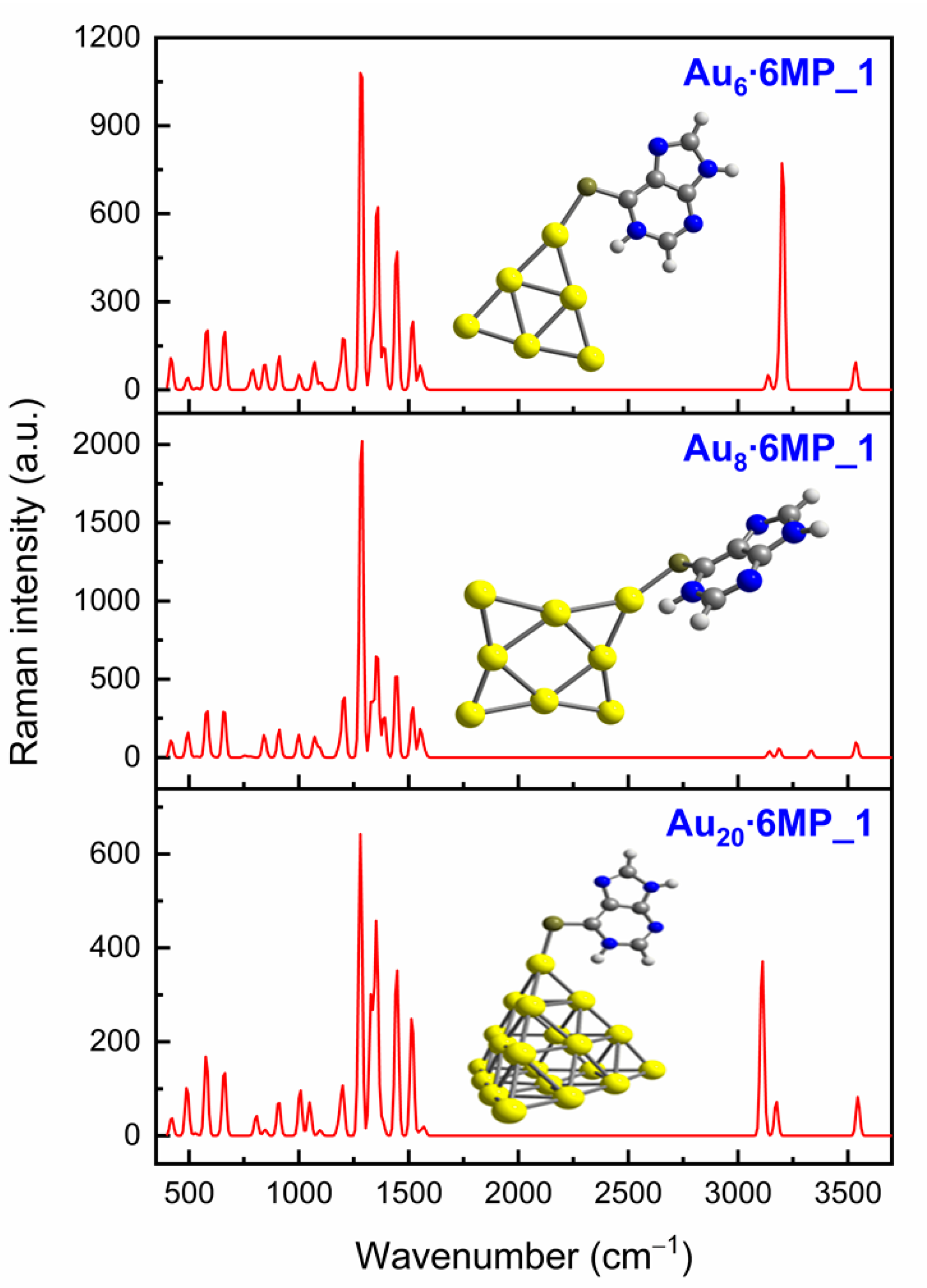
3.3. SERS Spectra of 6MP on Gold Surfaces
4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Scott, K.A.; Njardarson, J.T. Analysis of US FDA-approved drugs containing sulfur atoms. Topics Curr. Chem. 2018, 376, 5. [Google Scholar] [CrossRef]
- Parker, W.B. Enzymology of purine and pyrimidine antimetabolites used in the treatment of cancer. Chem. Rev. 2009, 109, 2880. [Google Scholar] [CrossRef] [Green Version]
- Sahasranaman, S.; Howard, D.; Roy, S. Clinical pharmacology and pharmacogenetics of thiopurines. Eur. J. Clin. Pharmacol. 2008, 64, 753. [Google Scholar] [CrossRef] [PubMed]
- Ensafi, A.A.; Karimi-Maleh, H. Determination of 6-mercaptopurine in the presence of uric acid using modified multiwall carbon nanotubes-TiO2 as a voltammetric sensor. Drug Test. Anal. 2012, 4, 970. [Google Scholar] [CrossRef]
- Yang, J.J.; Landier, W.; Yang, W.; Liu, C.; Hageman, L.; Cheng, C.; Pei, D.; Chen, Y.; Crews, K.R.; Kornegay, N. Inherited NUDT15 variant is a genetic determinant of mercaptopurine intolerance in children with acute lymphoblastic leukemia. Int. J. Clin. Oncol. 2015, 33, 1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javarsineh, S.; Vessally, E.; Bekhradnia, A.; Hosseinian, A.; Ahmadi, S. A computational study on the purinethol drug adsorption on the AlN nanocone and nanocluster. J. Clust. Sci. 2018, 29, 767. [Google Scholar] [CrossRef]
- Akhter, S.; Ahmad, I.; Ahmad, M.Z.; Ramazani, F.; Singh, A.; Rahman, Z.; Ahmad, F.J.; Storm, G.; Kok, R.J. Nanomedicines as cancer therapeutics: Current status. Curr. Cancer Drug Tar. 2013, 13, 362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lah, N.A.C.; Samykano, M.; Trigueros, S. Nanoscale metal particles as nanocarriers in targeted drug delivery system. J. Nanomed. Res. 2016, 4, 00086. [Google Scholar]
- Svenson, S.; Tomalia, D.A. Dendrimers in biomedical applications—reflections on the field. Adv. Drug Deliv. Rev. 2012, 64, 102–115. [Google Scholar] [CrossRef]
- Kalomiraki, M.; Thermos, K.; Chaniotakis, N.A. Dendrimers as tunable vectors of drug delivery systems and biomedical and ocular applications. Int. J. Nanomed. 2016, 11, 1. [Google Scholar]
- Duncan, R. The dawning era of polymer therapeutics. Nat. Rev. Drug Discov. 2003, 2, 347–360. [Google Scholar] [CrossRef]
- Vogus, D.R.; Krishnan, V.; Mitragotri, S. A review on engineering polymer drug conjugates to improve combination chemotherapy. Curr. Opin. Colloid Interface Sci. 2017, 31, 75–85. [Google Scholar] [CrossRef]
- Allen, T.M.; Cullis, P.R. Liposomal drug delivery systems: From concept to clinical applications. Adv. Drug Deliv. Rev. 2013, 65, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Pattni, B.S.; Chupin, V.V.; Torchilin, V.P. New developments in liposomal drug delivery. Chem. Rev. 2015, 115, 10938–10966. [Google Scholar] [CrossRef]
- Daraee, H.; Etemadi, A.; Kouhi, M.; Alimirzalu, S.; Akbarzadeh, A. Application of liposomes in medicine and drug delivery. Artif. Cells Nanomed. Biotechnol. 2016, 44, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Lu, J.; Xu, H.; Patel, A.; Chen, Z.-S.; Chen, G. Silver nanoparticles: Synthesis, properties, and therapeutic applications. Drug Discov. Today 2015, 20, 595–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hola, K.; Markova, Z.; Zoppellaro, G.; Tucek, J.; Zboril, R. Tailored functionalization of iron oxide nanoparticles for MRI, drug delivery, magnetic separation and immobilization of biosubstances. Biotechnol. Adv. 2015, 33, 1162–1176. [Google Scholar] [CrossRef]
- Kim, C.S.; Tonga, G.Y.; Solfiell, D.; Rotello, V.M. Inorganic nanosystems for therapeutic delivery: Status and prospects. Adv. Drug Deliv. Rev. 2013, 65, 93–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, S.; Singh, R.K.; Perez, R.A.; Abou Neel, E.A.; Kim, H.-W.; Chrzanowski, W. Silica-based mesoporous nanoparticles for controlled drug delivery. J. Tissue Eng. 2013, 4, 2041731413503357. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Feng, Z.; Hou, T.; Li, Y. Computational simulation of inorganic nanoparticle drug delivery systems at the molecular level. In Computational Pharmaceutics; Ouyang, D., Smith, S.C., Eds.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2015; pp. 149–167. [Google Scholar]
- Khan, A.; Rashid, R.; Murtaza, G.; Zahra, A. Gold nanoparticles: Synthesis and applications in drug delivery. Trop. J. Pharm. Res. 2014, 13, 1169–1177. [Google Scholar] [CrossRef]
- Bhatia, S. Nanoparticles types, classification, characterization, fabrication methods and drug delivery applications. In Natural Polymer Drug Delivery Systems; Springer: Berlin/Heidelberg, Germany, 2016; pp. 33–93. [Google Scholar]
- Bahadar, H.; Maqbool, F.; Niaz, K.; Abdollahi, M. Toxicity of nanoparticles and an overview of current experimental models. Iran. Biomed. J. 2016, 20, 1. [Google Scholar]
- Her, S.; Jaffray, D.A.; Allen, C. Gold nanoparticles for applications in cancer radiotherapy: Mechanisms and recent advancements. Adv. Drug Deliv. Rev. 2017, 109, 84–101. [Google Scholar] [CrossRef]
- Khodashenas, B.; Ardjmand, M.; Baei, M.S.; Rad, A.S.; Akbarzadeh, A. Conjugation of pectin biopolymer with Au-nanoparticles as a drug delivery system: Experimental and DFT studies. Appl. Organomet. Chem. 2020, 34, e5609. [Google Scholar] [CrossRef]
- Demurtas, M.; Perry, C.C. Facile one-pot synthesis of amoxicillin-coated gold nanoparticles and their antimicrobial activity. Gold Bull. 2014, 47, 103. [Google Scholar] [CrossRef] [Green Version]
- Austin, L.A.; Mackey, M.A.; Dreaden, E.C.; El-Sayed, M.A. The optical, photothermal, and facile surface chemical properties of gold and silver nanoparticles in biodiagnostics, therapy, and drug delivery. Arch. Toxicol. 2014, 88, 1391–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torchilin, V.P. Multifunctional, stimuli-sensitive nanoparticulate systems for drug delivery. Nat. Rev. Drug Discov. 2014, 13, 813. [Google Scholar] [CrossRef] [Green Version]
- Hainfeld, J.F.; Slatkin, D.N.; Focella, T.M.; Smilowitz, H.M. Gold nanoparticles: A new X-ray contrast agent. Br. J. Radiol. 2005, 79, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Ataka, K.; Kottke, T.; Heberle, J. Thinner, smaller, faster: IR techniques to probe the functionality of biological and biomimetic systems. Angew. Chem., Int. Ed. 2010, 49, 5416–5424. [Google Scholar] [CrossRef]
- Ma, L.-N.; Zhang, J.; Chen, H.-T.; Zhou, J.-H.; Ding, Y.-Z.; Liu, Y.-S. An overview on ELISA techniques for FMD. Virol. J. 2011, 8, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malou, N.; Raoult, D. Immuno-PCR: A promising ultrasensitive diagnostic method to detect antigens and antibodies. Trends Microbiol. 2011, 19, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Seibel, J.; König, S.; Göhler, A.; Doose, S.; Memmel, E.; Bertleff, N.; Sauer, M. Investigating infection processes with a workflow from organic chemistry to biophysics: The combination of metabolic glycoengineering, super-resolution fluorescence imaging and proteomics. Expert Rev. Proteom. 2013, 10, 25–31. [Google Scholar] [CrossRef]
- Petryayeva, E.; Algar, W.R.; Medintz, I.L. Quantum dots in bioanalysis: A review of applications across various platforms for fluorescence spectroscopy and imaging. Appl. Spectrosc. 2013, 67, 215–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brauchle, E.; Schenke-Layland, K. Raman spectroscopy in biomedicine–non-invasive in vitro analysis of cells and extracellular matrix components in tissues. Biotechnol. J. 2013, 8, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Bantz, K.C.; Meyer, A.F.; Wittenberg, N.J.; Im, H.; Kurtuluş, Ö.; Lee, S.H.; Lindquist, N.C.; Oh, S.-H.; Haynes, C.L. Recent progress in SERS biosensing. Phys. Chem. Chem. Phys. 2011, 13, 11551–11567. [Google Scholar] [CrossRef]
- Culha, M. Surface-enhanced raman scattering: An emerging label-free detection and identification technique for proteins. Appl. Spectrosc. 2013, 67, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Cialla, D.; Pollok, S.; Steinbrücker, C.; Weber, K.; Popp, J. SERS-based detection of biomolecules. Nanophotonics 2014, 3, 383–411. [Google Scholar] [CrossRef]
- Bauman, S.J.; Brawley, Z.T.; Darweesh, A.A.; Herzog, J.B. Substrate oxide layer thickness optimization for a dual-width plasmonic grating for surface-enhanced Raman spectroscopy (SERS) biosensor applications. Sensors 2017, 17, 1530. [Google Scholar] [CrossRef] [Green Version]
- Dies, H.; Siampani, M.; Escobedo, C.; Docoslis, A. Direct detection of toxic contaminants in minimally processed food products using dendritic surface-enhanced Raman scattering substrates. Sensors 2018, 18, 2726. [Google Scholar] [CrossRef] [Green Version]
- Aikens, C.M.; Schatz, G.C. TDDFT studies of absorption and SERS spectra of pyridine interacting with Au20. J. Phys. Chem. A 2006, 110, 13317–13324. [Google Scholar] [CrossRef]
- Sharma, B.; Frontiera, R.R.; Henry, A.-I.; Ringe, E.; Van Duyne, R.P. SERS: Materials, applications, and the future. Mater. Today 2012, 15, 16–25. [Google Scholar] [CrossRef]
- Häkkinen, H. The gold–sulfur interface at the nanoscale. Nat. Chem. 2012, 4, 443–455. [Google Scholar] [CrossRef] [PubMed]
- Maza, F.L.; Grumelli, D.; Carro, P.; Vericat, C.; Kern, K.; Salvarezza, R.C. The role of the crystalline face in the ordering of 6-mercaptopurine self-assembled monolayers on gold. Nanoscale 2016, 8, 17231–17240. [Google Scholar] [CrossRef] [PubMed]
- Fernández, C.C.; Pensa, E.; Carro, P.; Salvarezza, R.; Williams, F.J. Electronic structure of a self-assembled monolayer with two surface anchors: 6-mercaptopurine on Au (111). Langmuir 2018, 34, 5696–5702. [Google Scholar] [CrossRef] [Green Version]
- Ren, H.; Cao, X.; Zhang, Y.; Chehelamirani, M.; Salahub, D.R. Theoretical investigation of 6-mercaptopurine isomers’ adsorption on the Au (001) surface: Revealing the fate of different isomers. ACS Omega 2019, 5, 610–618. [Google Scholar] [CrossRef] [Green Version]
- Rashidpour, N.; Kashid, V.; Shah, V. Effect of tautomerism on Au-6-mercaptopurine nanocluster stability. AIP Conf. Proc. 2013, 1512, 390–391. [Google Scholar]
- Ren, H.; Chen, F.; Li, X.; He, Y. A new insight of structures, bonding and electronic properties for 6-mercaptopurine and Ag 8 clusters configurations: A theoretical perspective. BMC Chem. 2019, 13, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Nhat, P.V.; Si, N.T.; Tram, N.T.T.; Duong, L.V.; Nguyen, M.T. Elucidating the binding mechanism of thione-containing mercaptopurine and thioguanine drugs to small gold clusters. J. Comput. Chem. 2020, 41, 1748–1758. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16; Revision B.01; Software For Theoretical Calculation: Wallingford, CT, USA, 2016; Revision B.01. [Google Scholar]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [Green Version]
- Peterson, K.A. Systematically convergent basis sets with relativistic pseudo-potentials. I. Correlation consistent basis sets for the post-d group 13–15 elements. J. Chem. Phys. 2003, 119, 11099. [Google Scholar] [CrossRef] [Green Version]
- Nhat, P.V.; Nguyen, P.T.N.; Si, N.T. A computational study of thiol-containing cysteine amino acid binding to Au6 and Au8 gold clusters. J. Mol. Model. 2020, 26, 58. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999. [Google Scholar] [CrossRef]
- Skyner, R.; McDonagh, J.; Groom, C.; Van Mourik, T.; Mitchell, J. A review of methods for the calculation of solution free energies and the modelling of systems in solution. Phys. Chem. Chem. Phys. 2015, 17, 6174–6191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadipour, N.L.; Ahmadi Peyghan, A.; Soleymanabadi, H. Theoretical study on the Al-doped ZnO nanoclusters for CO chemical sensors. J. Phys. Chem. C 2015, 119, 6398. [Google Scholar] [CrossRef]
- Yang, Y.; Ostadhosseini, N. A theoretical investigation on the mercaptopurine drug interaction with boron nitride nanocage: Solvent and density functional effect. Physica E Low-Dimens. Syst. Nanostruct. 2021, 125, 114337. [Google Scholar] [CrossRef]
- Baikie, I.D.; Mackenzie, S.; Estrup, P.J.Z.; Meyer, J.A. Noise and the Kelvin method. Rev. Sci. Instrum. 1991, 62, 1326–1332. [Google Scholar] [CrossRef]
- Korotcenkov, G. Sensing layers in work-function-type gas sensors. In Handbook of Gas Sensor Materials; Springer: Berlin/Heidelberg, Germany, 2013; pp. 377–388. [Google Scholar]
- Nhat, P.V.; Si, N.T.; Leszczynski, J.; Nguyen, M.T. Another look at structure of gold clusters Aun from perspective of phenomenological shell model. Chem. Phys. 2017, 493, 140–148. [Google Scholar] [CrossRef]
- Zhao, H.-Y.; Ning, H.; Wang, J.; Su, X.-J.; Guo, X.-G.; Liu, Y. Structural Evolution of Aun (n = 20–32) Clusters: Lowest-Lying Structures and Relativistic Effects. Phys. Lett. A 2010, 374, 1033–1038. [Google Scholar] [CrossRef]
- Schwerdtfeger, P. Gold goes nano-from small clusters to low-dimensional assemblies. Angew. Chem. Int. Ed. 2003, 42, 1892–1895. [Google Scholar] [CrossRef] [PubMed]
- Gruene, P.; Rayner, D.M.; Redlich, B.; van der Meer, A.F.; Lyon, J.T.; Meijer, G.; Fielicke, A. Structures of neutral Au7, Au19, and Au20 clusters in the gas phase. Science 2008, 321, 674–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao-Zong, L. DFT calculations on 6-thiopurine tautomers. Acta Chim. Sin. 2004, 62, 1075–1079. [Google Scholar]
- Suresh Kumar, S.; Athimoolam, S.; Sridhar, B. XRD, vibrational spectra and quantum chemical studies of an anticancer drug: 6-Mercaptopurine. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2015, 146, 204–213. [Google Scholar] [CrossRef]
- Pazderski, L.; Łakomska, I.; Wojtczak, A.; Szłyk, E.; Sitkowski, J.; Kozerski, L.; Kamieński, B.; Koźmiński, W.; Tousek, J.; Marek, R. The studies of tautomerism in 6-mercaptopurine derivatives by 1H-13C, 1H-15N NMR and 13C, 15N CPMAS-experimental and quantum chemical approach. J. Mol. Struct. 2006, 785, 205–215. [Google Scholar] [CrossRef]
- Pakiari, A.H.; Jamshidi, Z. Interaction of amino acids with gold and silver clusters. J. Phys. Chem. A 2007, 111, 4391. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Liu, Y.; Liu, Z.; Yang, Y.; Jiang, J.; Zhang, Z.; Shen, G.; Yu, R. Raman Mapping and In Situ SERS Spectroelectrochemical Studies of 6-Mercaptopurine SAMs on the Gold Electrode. J. Phys. Chem. B 2005, 109, 2739–2744. [Google Scholar] [CrossRef] [PubMed]
- Pearson, R.G. Hard and Soft Acids and Bases. J. Am. Chem. Soc. 1963, 85, 3533. [Google Scholar] [CrossRef]
- James, A.M.; Lord, M.P. Macmillan’s Chemical and Physical Data; Macmillan: Basingstoke, UK, 1992. [Google Scholar]
- Ahmadi Peyghan, A.; Hadipour, N.L.; Bagheri, Z. Effects of Al doping and double-antisite defect on the adsorption of HCN on a BC2N nanotube: Density functional theory studies. J. Phys. Chem. C 2013, 117, 2427. [Google Scholar] [CrossRef]
- Ahmadi, A.; Hadipour, N.L.; Kamfiroozi, M.; Bagheri, Z. Theoretical study of aluminum nitride nanotubes for chemical sensing of formaldehyde. Sensor. Actuat. B-Chem. 2012, 161, 1025. [Google Scholar] [CrossRef]
- Kim, C.; Kim, B.; Lee, S.M.; Jo, C.; Lee, Y.H. Electronic structures of capped carbon nanotubes under electric fields. Phys. Rev. B 2002, 65, 165418. [Google Scholar] [CrossRef] [Green Version]
- Rajsky, T.; Urban, M. Aun (n = 1, 11) clusters interacting with lone-pair ligands. J. Phys. Chem. A 2016, 120, 3938–3949. [Google Scholar] [CrossRef]
- Si, N.T.; Nhung, N.T.A.; Bui, T.Q.; Nguyen, M.T.; Nhat, P.V. Gold nanoclusters as prospective carriers and detectors of pramipexole. RSC Adv. 2021, 11, 16619–16632. [Google Scholar] [CrossRef]
- Ghosh, P.; Han, G.; De, M.; Kim, C.K.; Rotello, V.M. Gold nanoparticles in delivery applications. Adv. Drug Deliv. Rev. 2008, 60, 1307. [Google Scholar] [CrossRef]
- Swietach, P.; Vaughan-Jones, R.D.; Harris, A.L.; Hulikova, A. The chemistry, physiology and pathology of pH in cancer. Philos. Trans. R. Soc. B 2014, 369, 20130099. [Google Scholar] [CrossRef] [Green Version]
- Le Guével, X.; Hoötzer, B.; Jung, G.; Hollemeyer, K.; Trouillet, V.; Schneider, M. Formation of fluorescent metal (Au, Ag) nanoclusters capped in bovine serum albumin followed by fluorescence and spectroscopy. J. Phys. Chem. C 2011, 115, 10955–10963. [Google Scholar] [CrossRef]
- Eckhardt, S.; Brunetto, P.S.; Gagnon, J.; Priebe, M.; Giese, B.; Fromm, K.M. Nanobio silver: Its interactions with peptides and bacteria, and its uses in medicine. Chem. Rev. 2013, 113, 4708–4754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neil, M.J. The Merck index: An Encyclopedia of Chemicals, Drugs, and Biologicals; RSC Publishing: London, UK, 2013. [Google Scholar]
- Peng, S.; Cho, K.; Qi, P.; Dai, H. Ab initio study of CNT NO2 gas sensor. Chem. Phys. Lett. 2004, 387, 271–276. [Google Scholar] [CrossRef]
- Le Ru, E.; Etchegoin, P. Principles of Surface-Enhanced Raman Spectroscopy: And Related Plasmonic Effects; Elsevier: Amsterdam, The Netherlands, 2008. [Google Scholar]
- Etchegoin, P.; Cohen, L.; Hartigan, H.; Brown, R.; Milton, M.; Gallop, J. Electromagnetic contribution to surface enhanced Raman scattering revisited. J. Chem. Phys. 2003, 119, 5281–5289. [Google Scholar] [CrossRef]
- Pannico, M.; Musto, P. SERS spectroscopy for the therapeutic drug monitoring of the anticancer drug 6-Mercaptopurine: Molecular and kinetic studies. Appl. Surf. Sci. 2021, 539, 148225. [Google Scholar] [CrossRef]
- Itoh, T.; Yoshida, K.; Biju, V.; Kikkawa, Y.; Ishikawa, M.; Ozaki, Y. Second enhancement in surface-enhanced resonance Raman scattering revealed by an analysis of anti-Stokes and Stokes Raman spectra. Phys. Rev. B 2007, 76, 129901. [Google Scholar] [CrossRef]
- Jensen, L.; Aikens, C.M.; Schatz, G.C. Electronic structure methods for studying surface-enhanced Raman scattering. Chem. Soc. Rev. 2008, 37, 1061–1073. [Google Scholar] [CrossRef]
- Yao, G.; Huang, Q. DFT and SERS study of L-Cysteine adsorption on the surface of gold nanoparticles. J. Phys. Chem. C 2018, 122, 15241–15251. [Google Scholar] [CrossRef]
- Jeanmaire, D.L.; Van Duyne, R.P. Surface Raman spectroelectrochemistry: Part, I. Heterocyclic, aromatic, and aliphatic amines adsorbed on the anodized silver electrode. J. Electroanal. Chem. Interfacial Electrochem. 1977, 84, 1–20. [Google Scholar] [CrossRef]
- Ngo, T.C.; Trinh, Q.T.; Thi Thai An, N.; Tri, N.N.; Trung, N.T.; Truong, D.H.; Huy, B.T.; Nguyen, M.T.; Dao, D.Q. Sers spectra of the pesticide chlorpyrifos adsorbed on silver nanosurface: The Ag20 cluster model. J. Phys. Chem. C 2020, 124, 21702–21716. [Google Scholar] [CrossRef]
- Vivoni, A.; Chen, S.P.; Ejeh, D.; Hosten, C.M. Normal-mode analysis of the Raman-active modes of the anti-tumor agent 6-mercaptopurine. J. Raman Spectrosc. 2001, 32, 1–8. [Google Scholar] [CrossRef]
- Bazylewski, P.; Divigalpitiya, R.; Fanchini, G. In situ Raman spectroscopy distinguishes between reversible and irreversible thiol modifications in l-cysteine. RSC Adv. 2017, 7, 2964–2970. [Google Scholar] [CrossRef] [Green Version]
- Ishida, T.; Murayama, T.; Taketoshi, A.; Haruta, M. Importance of size and contact structure of gold nanoparticles for the genesis of unique catalytic processes. Chem. Rev. 2020, 120, 464–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | RE | ||||||
|---|---|---|---|---|---|---|---|
| In a Vacuum | In Water | In a Vacuum | In Water | In a Vacuum | In Water | ||
| Au20∙6MP_1 | 0.0 | 0.0 | 29.4 | 22.4 | −19.6 | −10.5 | 2.41 |
| Au20∙6MP_2 | 3.2 | 1.0 | 26.2 | 21.5 | −16.9 | −7.82 | 2.41 |
| Au20∙6MP_3 | 3.3 | 1.0 | 26.2 | 21.4 | −16.6 | −11.5 | 2.42 |
| Au20∙6MP_4 | 4.7 | 9.9 | 24.7 | 12.6 | −14.7 | −1.91 | 2.41 |
| Au20∙6MP_5 | 5.2 | 1.4 | 24.2 | 21.0 | −15.3 | −9.64 | 2.44 |
| Au20∙6MP_6 | 12 | 7.8 | 17.3 | 14.7 | −9.08 | −3.32 | 2.21 |
| Species | In a Vacuum | In an Aqueous Solution | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HOMO | LUMO | HOMO | LUMO | ||||||||||
| 6MP-7 | −5.37 | −3.96 | −2.55 | 2.81 | - | 3.96 | −5.52 | −4.05 | −2.58 | 2.94 | - | 4.05 | |
| 6MP-9 | −4.93 | −3.58 | −2.23 | 2.70 | - | 3.58 | −5.39 | −3.91 | −2.42 | 2.98 | - | 3.91 | |
| Au20 | −5.73 | −4.84 | −3.95 | 1.78 | - | 4.84 | −5.07 | −4.13 | −3.18 | 1.90 | - | 4.13 | |
| Au20∙MP_1 | −5.15 | −4.42 | −3.69 | 1.47 | −17.4 | 4.42 | −4.81 | −4.01 | −3.20 | 1.60 | −15.8 | 4.01 | |
| Au20∙MP_2 | −5.28 | −4.52 | −3.76 | 1.52 | −14.6 | 4.52 | −4.84 | −4.03 | −3.22 | 1.62 | −14.7 | 4.03 | |
| Au20∙MP_3 | −5.25 | −4.50 | −3.75 | 1.49 | −16.3 | 4.50 | −4.82 | −4.02 | −3.22 | 1.60 | −15.8 | 4.02 | |
| Au20∙MP_4 | −4.86 | −4.30 | −3.73 | 1.13 | −36.5 | 4.30 | −4.74 | −3.98 | −3.22 | 1.52 | −20.0 | 3.98 | |
| Au20∙MP_5 | −4.87 | −4.26 | −3.65 | 1.23 | −30.9 | 4.26 | −4.77 | −3.98 | −3.18 | 1.60 | −15.8 | 3.98 | |
| Au20∙MP_6 | −5.10 | −4.35 | −3.59 | 1.51 | −15.2 | 4.35 | −4.85 | −4.01 | −3.16 | 1.68 | −11.6 | 4.01 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hang, N.T.N.; Si, N.T.; Nguyen, M.T.; Nhat, P.V. Adsorption/Desorption Behaviors and SERS Chemical Enhancement of 6-Mercaptopurine on a Nanostructured Gold Surface: The Au20 Cluster Model. Molecules 2021, 26, 5422. https://doi.org/10.3390/molecules26175422
Hang NTN, Si NT, Nguyen MT, Nhat PV. Adsorption/Desorption Behaviors and SERS Chemical Enhancement of 6-Mercaptopurine on a Nanostructured Gold Surface: The Au20 Cluster Model. Molecules. 2021; 26(17):5422. https://doi.org/10.3390/molecules26175422
Chicago/Turabian StyleHang, Nguyen Thi Nhat, Nguyen Thanh Si, Minh Tho Nguyen, and Pham Vu Nhat. 2021. "Adsorption/Desorption Behaviors and SERS Chemical Enhancement of 6-Mercaptopurine on a Nanostructured Gold Surface: The Au20 Cluster Model" Molecules 26, no. 17: 5422. https://doi.org/10.3390/molecules26175422
APA StyleHang, N. T. N., Si, N. T., Nguyen, M. T., & Nhat, P. V. (2021). Adsorption/Desorption Behaviors and SERS Chemical Enhancement of 6-Mercaptopurine on a Nanostructured Gold Surface: The Au20 Cluster Model. Molecules, 26(17), 5422. https://doi.org/10.3390/molecules26175422