
Effects of Vanadyl Complexes with Acetylacetonate Derivatives on Non-Tumor and Tumor Cell Lines

 , ,
, ,  , , ,
, , ,
Abstract
:1. Introduction
2. Results
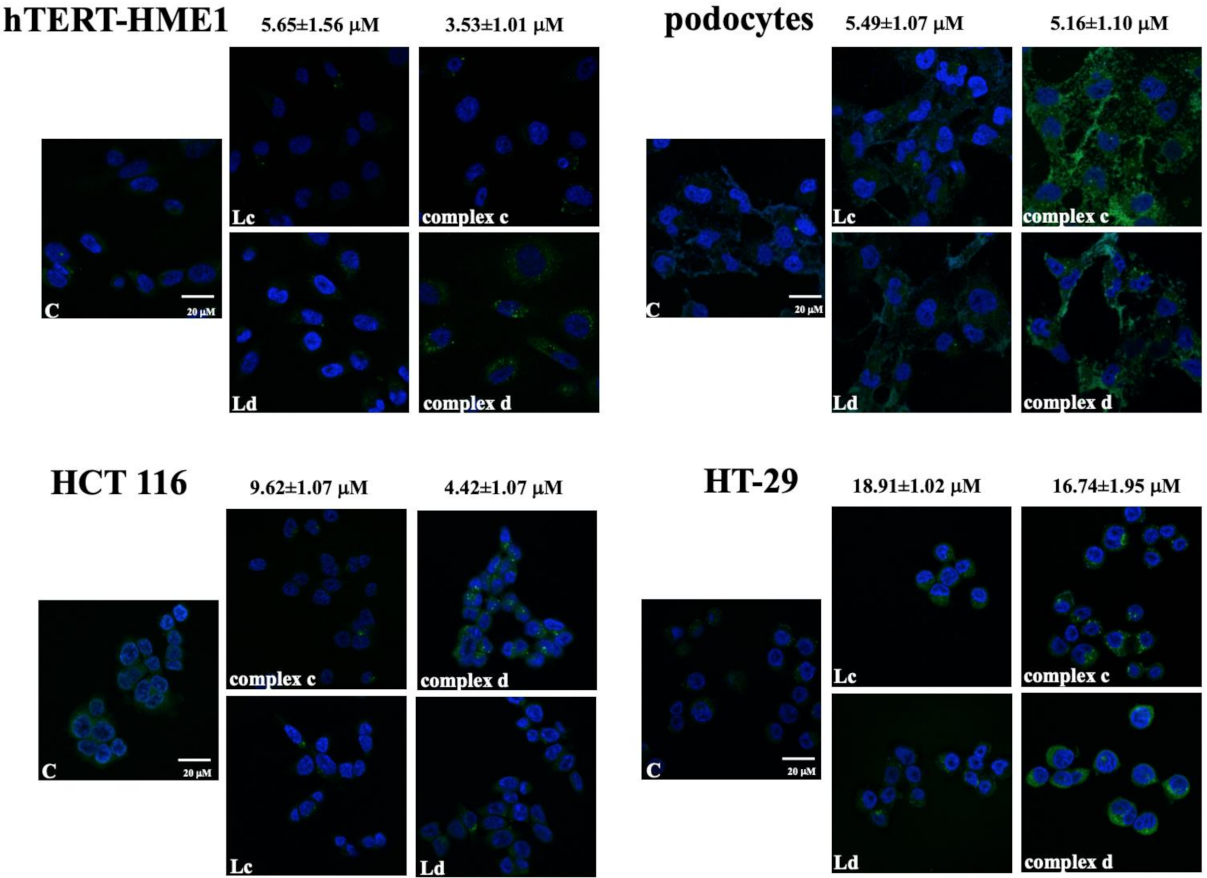
2.1. Intracellular Localization of Vanadyl Complexes
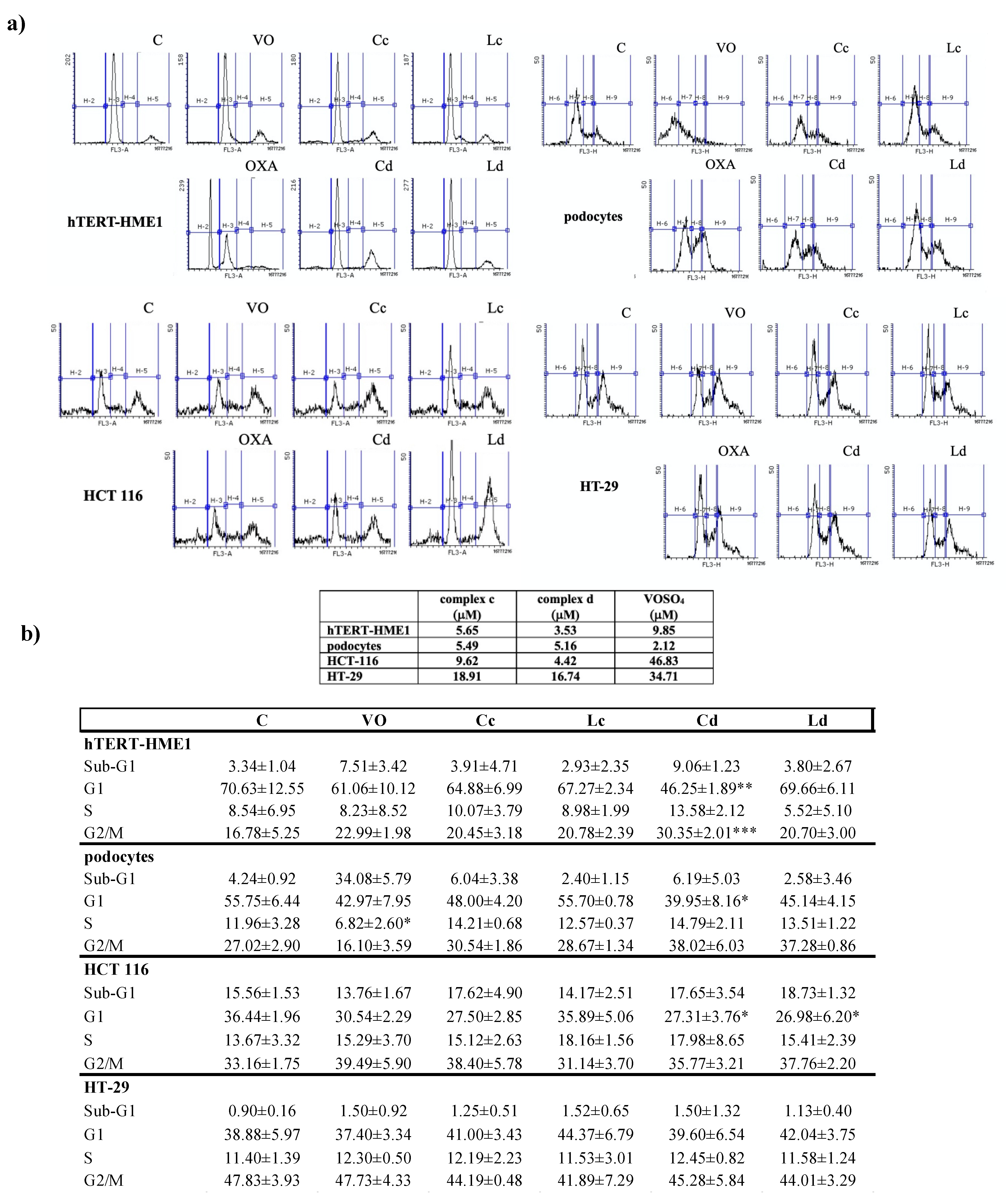
2.2. Effect of Vanadyl Complexes on Cell Cycle
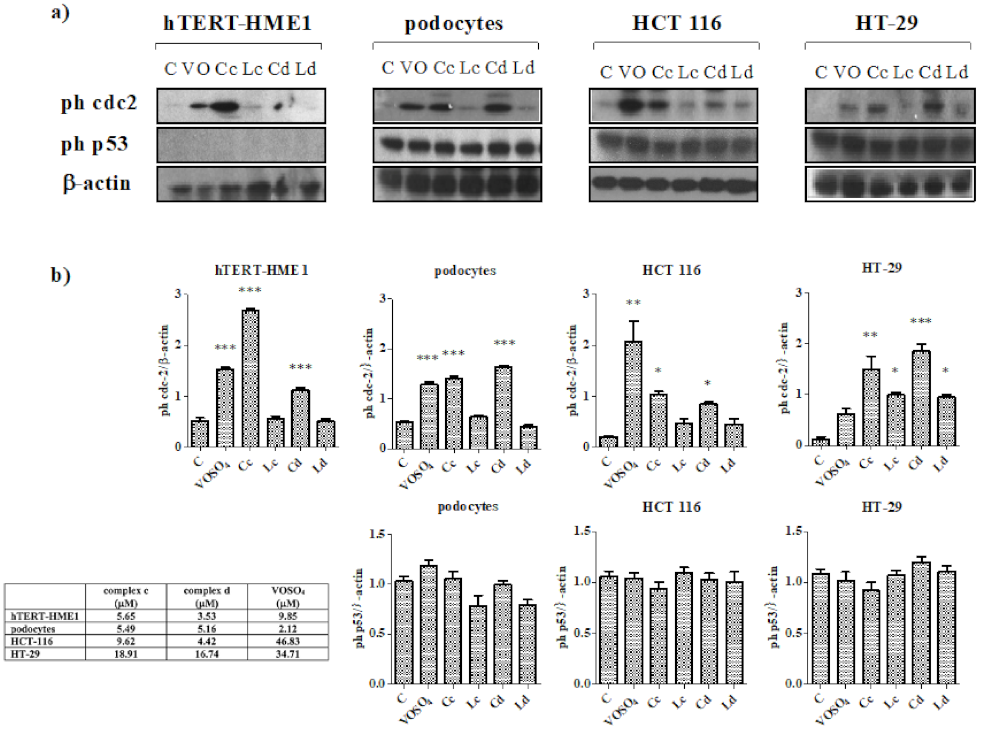
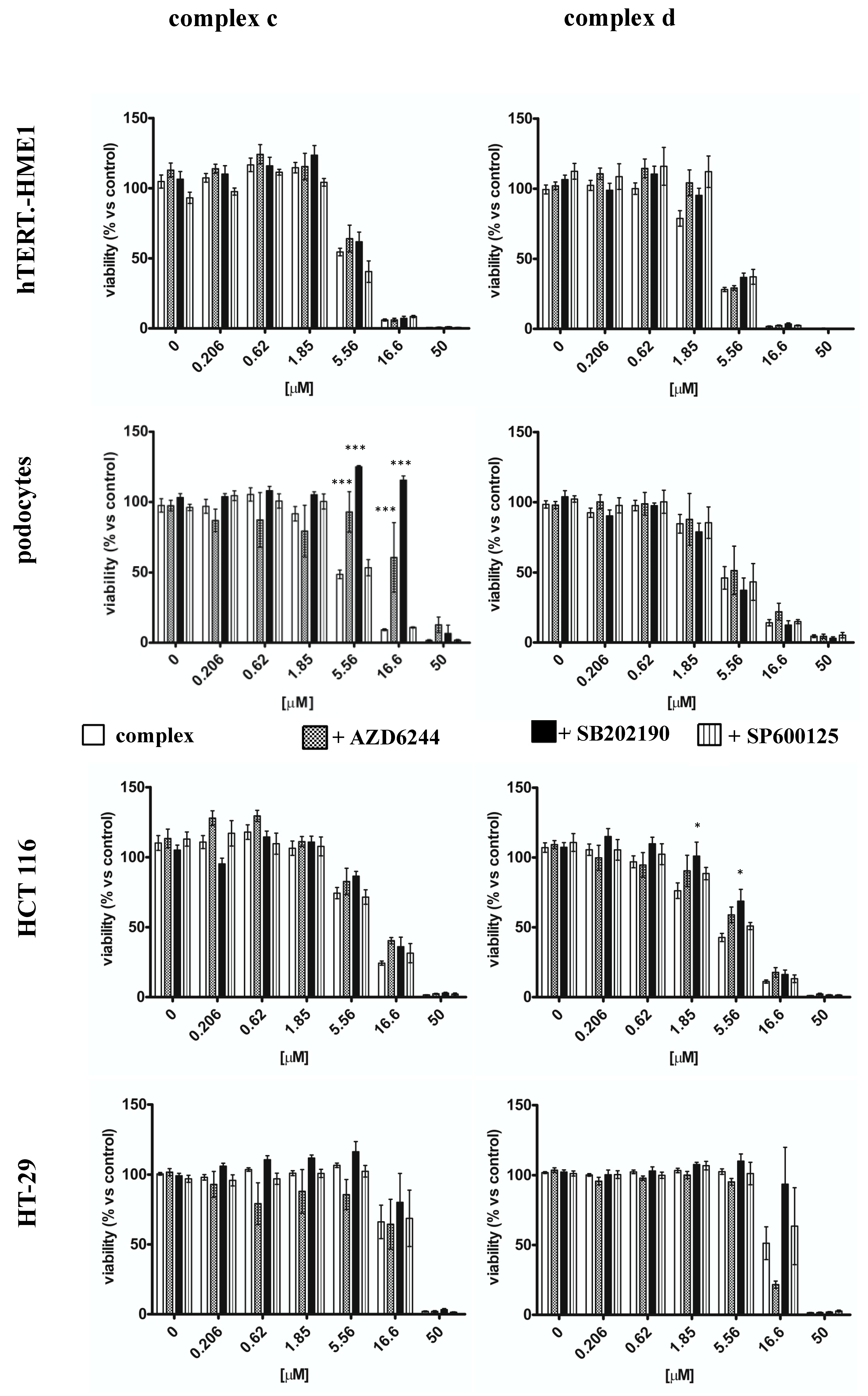
2.3. Effect of Vanadyl Complexes on MAPK
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Synthesis of Ligands and Complexes
4.3. Cell Culture
4.4. Confocal Microscopy
4.5. Cell Cycle
4.6. Western Blot Analysis
4.7. Cell Viability
4.8. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Rehder, D. The role of vanadium in biology. Metallomics 2015, 7, 730–742. [Google Scholar] [CrossRef] [Green Version]
- Evangelou, A.M. Vanadium in cancer treatment. Crit. Rev. Oncol. Hematol. 2002, 42, 249–265. [Google Scholar] [CrossRef]
- Kieler, J.; Gromek, A.; Nissen, N.I. Studies on the antineoplastic effect of vanadium salts. Acta Chir. Scand Suppl. 1965, 343, 154–164. [Google Scholar]
- English, L.H.; Macara, I.G.; Cantley, L.C. Vanadium stimulates the (Na+,K+) pump in friend erythroleukemia cells and blocks erythropoiesis. J. Cell Biol. 1983, 97, 1299–1302. [Google Scholar] [CrossRef] [Green Version]
- Thompson, H.J.; Chasteen, N.D.; Meeker, L.D. Dietary vanadyl(IV) sulfate inhibits chemically-induced mammary carcinogenesis. Carcinogenesis 1984, 5, 849–851. [Google Scholar] [CrossRef]
- Goldwaser, I.; Gefel, D.; Gershonov, E.; Fridkin, M.; Shechter, Y. Insulin-like effects of vanadium: Basic and clinical implications. J. Inorg. Biochem. 2000, 80, 21–25. [Google Scholar] [CrossRef]
- Brichard, S.M.; Henquin, J.C. The role of vanadium in the management of diabetes. Trends Pharmacol. Sci. 1995, 16, 265–270. [Google Scholar] [CrossRef]
- Faure, R.; Vincent, M.; Dufour, M.; Shaver, A.; Posner, B.I. Arrest at the G2/M transition of the cell cycle by protein-tyrosine phosphatase inhibition: Studies on a neuronal and a glial cell line. J. Cell Biochem. 1995, 59, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Krady, M.M.; Freyermuth, S.; Rogue, P.; Malviya, A.N. Pervanadate elicits proliferation and mediates activation of mitogen-activated protein (MAP) kinase in the nucleus. FEBS Lett. 1997, 412, 420–424. [Google Scholar] [CrossRef] [Green Version]
- Hanauske, U.; Hanauske, A.R.; Marshall, M.H.; Muggia, V.A.; Von Hoff, D.D. Biphasic effect of vanadium salts on in vitro tumor colony growth. Int. J. Cell Cloning 1987, 5, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Morinville, A.; Maysinger, D.; Shaver, A. From Vanadis to Atropos: Vanadium compounds as pharmacological tools in cell death signalling. Trends Pharmacol. Sci. 1998, 19, 452–460. [Google Scholar] [CrossRef]
- Jaspers, I.; Samet, J.M.; Erzurum, S.; Reed, W. Vanadium-induced kappaB-dependent transcription depends upon peroxide-induced activation of the p38 mitogen-activated protein kinase. Am. J. Respir. Cell Mol. Biol. 2000, 23, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.K.; Chiasson, J.L.; Srivastava, A.K. Vanadium salts stimulate mitogen-activated protein (MAP) kinases and ribosomal S6 kinases. Mol. Cell Biochem. 1995, 153, 69–78. [Google Scholar] [CrossRef]
- Samet, J.M.; Graves, L.M.; Quay, J.; Dailey, L.A.; Devlin, R.B.; Ghio, A.J.; Wu, W.; Bromberg, P.A.; Reed, W. Activation of MAPKs in human bronchial epithelial cells exposed to metals. Am. J. Physiol. 1998, 275, L551–L558. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Tan, Z.; Diltz, C.D.; You, M.; Fischer, E.H. Activation of mitogen-activated protein (MAP) kinase pathway by pervanadate, a potent inhibitor of tyrosine phosphatases. J. Biol. Chem. 1996, 271, 22251–22255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burkle, A. Poly(ADP-ribosyl)ation, genomic instability, and longevity. Ann. N. Y. Acad. Sci. 2000, 908, 126–132. [Google Scholar] [CrossRef]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef]
- Sgarbossa, S.; Diana, E.; Marabello, D.; Deagostino, A.; Cadamuro, S.; Barge, A.; Laurenti, E.; Gallicchio, M.; Boscaro, V.; Ghibaudi, E. Synthesis, characterization and cell viability test of six vanadyl complexes with acetylacetonate derivatives. J. Inorg. Biochem. 2013, 128, 26–37. [Google Scholar] [CrossRef]
- Lovisari, M.; Volpi, G.; Marabello, D.; Cadamuro, S.; Deagostino, A.; Diana, E.; Barge, A.; Gallicchio, M.; Boscaro, V.; Ghibaudi, E. EPR and photophysical characterization of six bioactive oxidovanadium(IV) complexes in the conditions of in vitro cell tests. J. Inorg. Biochem. 2017, 170, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Tournigand, C.; Andre, T.; Achille, E.; Lledo, G.; Flesh, M.; Mery-Mignard, D.; Quinaux, E.; Couteau, C.; Buyse, M.; Ganem, G.; et al. FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced colorectal cancer: A randomized GERCOR study. J. Clin. Oncol. 2004, 22, 229–237. [Google Scholar] [CrossRef] [Green Version]
- Ciombor, K.K.; Bekaii-Saab, T. A Comprehensive Review of Sequencing and Combination Strategies of Targeted Agents in Metastatic Colorectal Cancer. Oncologist 2018, 23, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Al-Hazmi, G.A.; Abou-Melha, K.S.; El-Metwaly, N.M.; Saleh, K.A. Synthesis of Novel VO(II)-Perimidine Complexes: Spectral, Computational, and Antitumor Studies. Bioinorg. Chem. Appl. 2018, 2018, 7176040. [Google Scholar] [CrossRef] [PubMed]
- Zecchin, D.; Arena, S.; Martini, M.; Sassi, F.; Pisacane, A.; Di Nicolantonio, F.; Bardelli, A. Modeling tumor progression by the sequential introduction of genetic alterations into the genome of human normal cells. Hum. Mutat. 2013, 34, 330–337. [Google Scholar] [CrossRef] [Green Version]
- Kowalski, S.; Wyrzykowski, D.; Inkielewicz-Stepniak, I. Molecular and Cellular Mechanisms of Cytotoxic Activity of Vanadium Compounds against Cancer Cells. Molecules 2020, 25, 1757. [Google Scholar] [CrossRef]
- Wells, N.J.; Watanabe, N.; Tokusumi, T.; Jiang, W.; Verdecia, M.A.; Hunter, T. The C-terminal domain of the Cdc2 inhibitory kinase Myt1 interacts with Cdc2 complexes and is required for inhibition of G(2)/M progression. J. Cell Sci. 1999, 112 Pt 19, 3361–3371. [Google Scholar] [CrossRef]
- Liu, T.T.; Liu, Y.J.; Wang, Q.; Yang, X.G.; Wang, K. Reactive-oxygen-species-mediated Cdc25C degradation results in differential antiproliferative activities of vanadate, tungstate, and molybdate in the PC-3 human prostate cancer cell line. J. Biol. Inorg. Chem. 2012, 17, 311–320. [Google Scholar] [CrossRef]
- Medico, E.; Russo, M.; Picco, G.; Cancelliere, C.; Valtorta, E.; Corti, G.; Buscarino, M.; Isella, C.; Lamba, S.; Martinoglio, B.; et al. The molecular landscape of colorectal cancer cell lines unveils clinically actionable kinase targets. Nat. Commun. 2015, 6, 7002. [Google Scholar] [CrossRef] [PubMed]
- Buecher, B.; Bouancheau, D.; Broquet, A.; Bezieau, S.; Denis, M.G.; Bonnet, C.; Heymann, M.F.; Jarry, A.; Galmiche, J.P.; Blottiere, H.M. Growth inhibitory effect of celecoxib and rofecoxib on human colorectal carcinoma cell lines. Anticancer Res. 2005, 25, 225–233. [Google Scholar]
- Zhang, Z.; Leonard, S.S.; Huang, C.; Vallyathan, V.; Castranova, V.; Shi, X. Role of reactive oxygen species and MAPKs in vanadate-induced G(2)/M phase arrest. Free Radic. Biol. Med. 2003, 34, 1333–1342. [Google Scholar] [CrossRef]
- Clark, O.; Park, I.; Di Florio, A.; Cichon, A.C.; Rustin, S.; Jugov, R.; Maeshima, R.; Stoker, A.W. Oxovanadium-based inhibitors can drive redox-sensitive cytotoxicity in neuroblastoma cells and synergise strongly with buthionine sulfoximine. Cancer Lett. 2015, 357, 316–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horniblow, R.D.; Bedford, M.; Hollingworth, R.; Evans, S.; Sutton, E.; Lal, N.; Beggs, A.; Iqbal, T.H.; Tselepis, C. BRAF mutations are associated with increased iron regulatory protein-2 expression in colorectal tumorigenesis. Cancer Sci. 2017, 108, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Rai, N.K.; Mathur, S.; Singh, S.K.; Tiwari, M.; Singh, V.K.; Haque, R.; Tiwari, S.; Kumar Sharma, L. Differential regulation of mitochondrial complex I and oxidative stress based on metastatic potential of colorectal cancer cells. Oncol. Lett. 2020, 20, 313. [Google Scholar] [CrossRef]
- Balaji, B.; Balakrishnan, B.; Perumalla, S.; Karande, A.A.; Chakravarty, A.R. Photoactivated cytotoxicity of ferrocenyl-terpyridine oxovanadium(IV) complexes of curcuminoids. Eur. J. Med. Chem. 2014, 85, 458–467. [Google Scholar] [CrossRef]
- Dubouchaud, H.; Walter, L.; Rigoulet, M.; Batandier, C. Mitochondrial NADH redox potential impacts the reactive oxygen species production of reverse Electron transfer through complex I. J. Bioenerg. Biomembr. 2018, 50, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Doublier, S.; Musante, L.; Lupia, E.; Candiano, G.; Spatola, T.; Caridi, G.; Zennaro, C.; Carraro, M.; Ghiggeri, G.M.; Camussi, G. Direct effect of plasma permeability factors from patients with idiopatic FSGS on nephrin and podocin expression in human podocytes. Int. J. Mol. Med. 2005, 16, 49–58. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
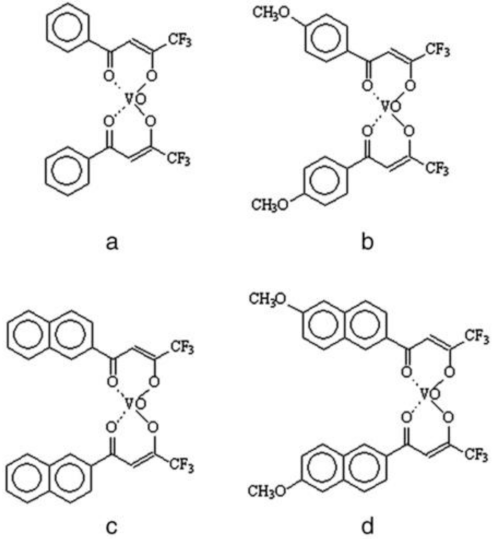


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex c | + AZD6244 | + SB202190 | + SP600125 | |
| hTERT-HME1 | 5.65 ± 1.56 | 6.58 ± 1.12 | 6.58 ± 1.12 | 5.20 ± 1.14 |
| Podocytes | 5.49 ± 1.07 | 20.16 ± 1.33 *** | 44.29 ± 1.54 *** | 6.03 ± 1.04 |
| HCT 116 | 9.62 ± 1.07 | 13.31 ± 1.15 | 13.06 ± 1.09 | 10.34 ± 1.13 |
| HT-29 | 18.91 ± 1.02 | 19.87 ± 1.02 | 22.64 ± 1.23 | 19.53 ± 1.31 |
| Complex d | + AZD6244 | + SB202190 | + SP600125 | |
| hTERT-HME1 | 3.53 ± 1.01 | 5.21 ± 1.89 | 4.71 ± 1.06 | 5.33 ± 1.59 |
| Podocytes | 5.16 ± 1.10 | 6.25 ± 1.24 | 4.14 ± 1.11 | 5.03 ± 1.06 |
| HCT 116 | 4.42 ± 1.07 | 6.81 ± 1.14 * | 7.99 ± 1.12 ** | 5.74 ± 1.09 |
| HT-29 | 16.74 ± 1.95 | 11.98 ± 1.05 | 25.99 ± 1.45 | 19.20 ± 1.20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boscaro, V.; Barge, A.; Deagostino, A.; Ghibaudi, E.; Laurenti, E.; Marabello, D.; Diana, E.; Gallicchio, M. Effects of Vanadyl Complexes with Acetylacetonate Derivatives on Non-Tumor and Tumor Cell Lines. Molecules 2021, 26, 5534. https://doi.org/10.3390/molecules26185534
Boscaro V, Barge A, Deagostino A, Ghibaudi E, Laurenti E, Marabello D, Diana E, Gallicchio M. Effects of Vanadyl Complexes with Acetylacetonate Derivatives on Non-Tumor and Tumor Cell Lines. Molecules. 2021; 26(18):5534. https://doi.org/10.3390/molecules26185534
Chicago/Turabian StyleBoscaro, Valentina, Alessandro Barge, Annamaria Deagostino, Elena Ghibaudi, Enzo Laurenti, Domenica Marabello, Eliano Diana, and Margherita Gallicchio. 2021. "Effects of Vanadyl Complexes with Acetylacetonate Derivatives on Non-Tumor and Tumor Cell Lines" Molecules 26, no. 18: 5534. https://doi.org/10.3390/molecules26185534