Abstract
The aim of the present work is the synthesis and characterization of new perfluorinated monomers bearing, similarly to Nafion®, acidic groups for proton transport for potential and future applications in proton exchange membrane (PEM) fuel cells. To this end, we focused our attention on the synthesis of various molecules with (i) sufficient volatility to be used in vacuum polymerization techniques (e.g., PECVD)), (ii) sulfonic, phosphonic, or carboxylic acid functionalities for proton transport capacity of the resulting membrane, (iii) both aliphatic and aromatic perfluorinated tags to diversify the membrane polarity with respect to Nafion®, and (iv) a double bond to facilitate the polymerization under vacuum giving a preferential way for the chain growth of the polymer. A retrosynthetic approach persuaded us to attempt three main synthetic strategies: (a) organometallic Heck-type cross-coupling, (b) nucleophilic displacement, and (c) Wittig–Horner reaction (carbanion approach). Preliminary results on the plasma deposition of a polymeric film are also presented. The variation of plasma conditions allowed us to point out that the film prepared in the mildest settings (20 W) shows the maximum monomer retention in its structure. In this condition, plasma polymerization likely occurs mainly by rupture of the π bond in the monomer molecule.
1. Introduction
Proton exchange membrane fuel cells (PEMFCs), also referred as solid polymer fuel cells, are the simplest type of fuel cells, with application spanning from portable power to automotive [1,2,3]. As solid polymer electrolytes, PEMFCs employ a membrane constituted by a perfluorinated polymer bearing acidic groups, working both as an electronic insulator (to keep separate the oxidative and the reductive half reactions) and as a proton conductor.
Among proton exchange membranes for PEM-type fuel cells, Nafion® is still the most used and studied electrolyte due to its high proton conductivity and moderate swelling in water [4,5]. Alongside its many advantages, the main drawback of Nafion® polymers is due to the membrane hydration, which still represents an operative limit for fuel cells, since the temperature must be maintained below 100 °C [6]. Another factor still limiting the commercial availability of PEMFCs is the cost associated to the active materials employed for their fabrication, i.e., Pt-based catalyst and the Nafion® membrane [6,7,8].
In the last few years, scientists have focused their attention on the development of new catalytic systems with the aim of reducing costs through the enhancement of the performance of the catalyst, by lowering the amount of noble metal (essentially Pt) [9] or by using less expensive metals [8,10,11,12]. Nevertheless, only a few efforts have been made in the direction of finding a valid alternative to Nafion® [5,13,14].
Most alternative proton exchange polymeric membranes are perfluorinated polymers bearing sulfonic or sulfonamidic groups. However, their performances are still not sufficient. Since the nature and chemical characteristics of the membranes are strictly connected to the water transport, it is necessary to increase the number of sulfonic groups per monomeric unit in order to achieve a much higher proton conductivity [15]. Fluorophosphonic acid monomers, which have higher chemical and thermal stabilities than sulfonic acid moieties, are still rarely investigated [16,17,18,19,20,21].
Furthermore, for portable applications, the miniaturization of the FC is crucial (micro-FC), and vacuum-based techniques such as plasma-enhanced chemical vapor deposition (PECVD) are highly desirable to produce both the catalyst layer and the proton exchange membrane in the form of thin films [13,22,23,24,25]. In order to be vacuum-compatible, the volatility of the starting monomer for the deposition of the thin film protonic membrane is mandatory.
In most of the works reported in the literature, these membranes are fabricated from the polymerization of trifluoromethane sulfonic acid, chlorosulfonic acid, or vinyl phosphonic acid with different fluorocarbons or vinylbenzenes [26]. However, most of the commercially available acidic precursors have scarce volatility, due to the hydrogen bond or acid/basic interactions, are highly corrosive, and therefore are not adapted for vacuum deposition [27]. Moreover, the presence of a π bond in the monomer structure can represent a preferential way for the chain growth of the polymer in a plasma environment, avoiding the degradation of other functional groups, such as the acidic one, during the polymerization process [27]. Therefore, tailored precursor synthesis is needed as the commercial ones do not meet all the requirements.
With this in mind, we planned several strategies for synthesizing monomers possessing the following features: (i) a sufficient volatility to be used in vacuum polymerization techniques (e.g., PECVD), (ii) sulfonic, phosphonic, or carboxylic acid functionalities to increase the proton transport capacity of the resulting membrane, (iii) both aliphatic and aromatic perfluorinated tags to diversify the membrane polarity with respect to Nafion®, and (iv) a double bond to facilitate the polymerization under vacuum, giving a preferential way for the chain growth of the polymer [28].
On these bases, we focused our attention on perfluorinated esters having the general formula depicted in Scheme 1. After polymerization, the free acidic functionalities responsible for the protonic conductivity can be restored by alkaline hydrolysis of the polymer.

Scheme 1.
General structure of unsaturated perfluorinated monomers.
We report here the synthesis and the characterization of new perfluorinated molecules bearing, similarly to Nafion®, acidic moieties for the protonic transport, potentially suitable as monomers or co-monomers in PEM fuel cells construction. Finally, we report some preliminary results on the plasma polymerization of one of the synthesized precursors. Since there is a lack of papers dedicated to the synthesis of this type of monomer, our work could represent a valid tool for subsequent applicative research in the preparation of membranes with potential applications in PEM fuel cells.
2. Results
2.1. Planning the Synthetic Strategy
Initial investigations were directed toward the synthesis of unsaturated perfluorinated sulfonic esters of general formula A (Scheme 2), which to the best of our knowledge has no precedent in the literature [29].

Scheme 2.
General structure of α,β-unsaturated perfluorinated sulfonic esters.
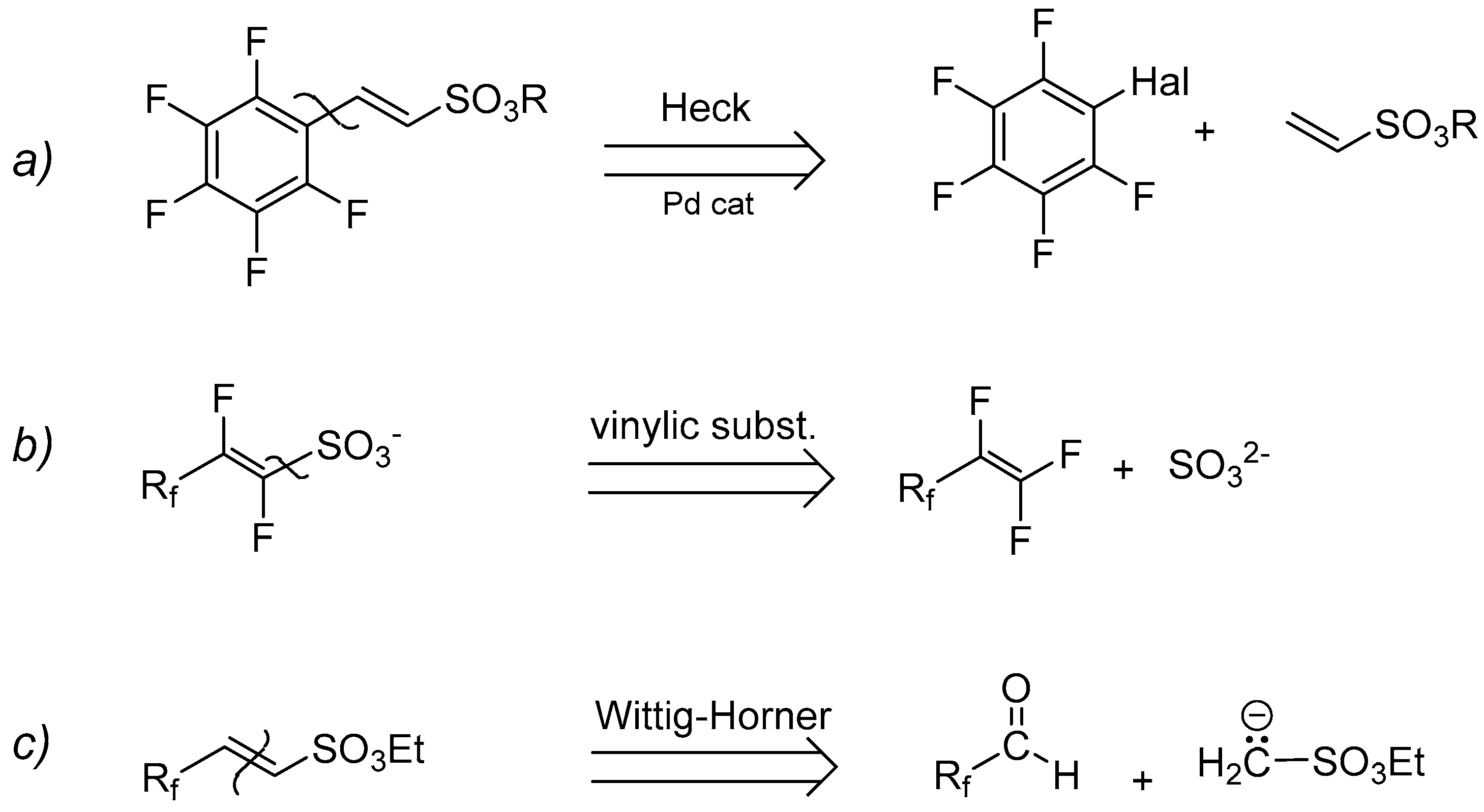
A retrosynthetic approach persuaded us to attempt three main synthetic strategies (Scheme 3): (a) organometallic Heck-type cross-coupling, (b) nucleophilic displacement, and (c) Wittig–Horner reaction (carbanion approach).
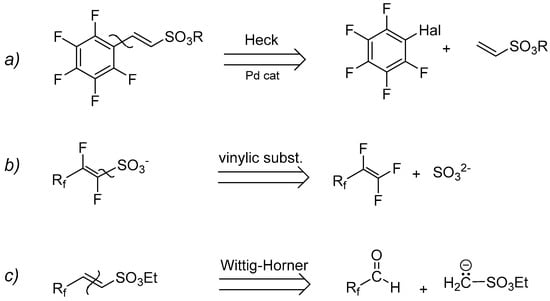
Scheme 3.
Retrosynthetic routes for the synthesis of perfluorinated sulfonyl monomers (Rf = perfluorinated tag).
2.1.1. The Heck-Type Coupling Reaction
In the last few decades, we studied several synthetic protocols involving palladium or copper-catalyzed Heck reactions utilizing greener or alternative reaction solvents such as water or ionic liquids [30,31,32]. These latter proved to be particularly effective in the activation of unreactive chloroarenes and hindered olefins [33].
In the present work, since an obvious disconnection analysis results in a direct coupling of a vinyl ester with the iodopentafluorobenzene, the palladium-catalyzed Heck coupling can be considered as an easy and direct way to obtain α,β-unsaturated fluoroaromatic esters. Unfortunately, the lack of papers dealing with the synthesis of these kinds of unsaturated compounds [34] can be considered clear evidence of a difficulty in the Heck coupling when perfluoroaromatic molecules are employed. Moreover, to the best of our knowledge, only two examples were described until now, in which the non-fluorinated analogous phenyl vinylsulfonate (and its sodium salt) have been directly coupled with iodobenzene [35,36].
During the screening, both well-established palladium–phosphine-based complexes and less common but more reactive palladium nanoparticles (PdNPs) were tested as catalysts [30]. In addition, in order to prevent undesirable side reactions such as desulfonation or polymerization processes, we also planned to modify the structure of the target molecule by inserting a spacer between the sulfonate moiety and the double bond. Heck couplings were carried out both in water and in organic solvents, depending on the nature of the starting vinyl substrate.
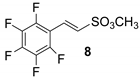
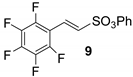
Obtained results are summarized in Table 1. Unfortunately, reactions performed in organic solvents were unproductive, even in the presence of Pd colloids (Table 1, runs 1–4). GC-MS analyses revealed only the presence of unreacted iodopentafluorobenzene and the disappearance of the vinyl substrate, which may be due to a plausible palladium-catalyzed degradation or a thermal desulfonation of the starting material.

Table 1.
Heck-type coupling of unsaturated sulfonates with perfluoroaryl halides a.
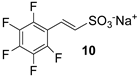
On the contrary, encouraging results emerged by using vinylsulfonate salt 3 instead of the corresponding ester, under typical phase transfer catalysis conditions. In fact, following a protocol of ours [37], Pd colloids were used in an aqueous solution of tetrabutylammonium hydroxide (TBAOH), affording a 50% of conversion into the coupled product 10, as revealed by the 1HNMR spectrum of the reaction mixture (Table 1, run 5 and Figure S2).
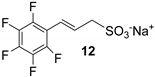
However, all attempts made to raise conversions were unsuccessful, and even the isolation of 10 from the reaction mixture led to very difficult results. Furthermore, no improvements were found even with the less substituted and more reactive 3,4,5-trifluoroiodobenzene (Table 1, run 6). Lower yields were also obtained in the presence of a methylene spacer between the sulfonic group and the double bond (Table 1, run 7).
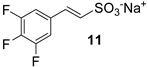
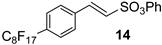
Then, few modifications were made, and the substitution of fluorine atoms on the aromatic ring, with a perfluorinated tag to the p-position, provided satisfactory results only in the case of phenyl sulfonate 14, which was isolated in a 65% of yield (Table 1, runs 9).
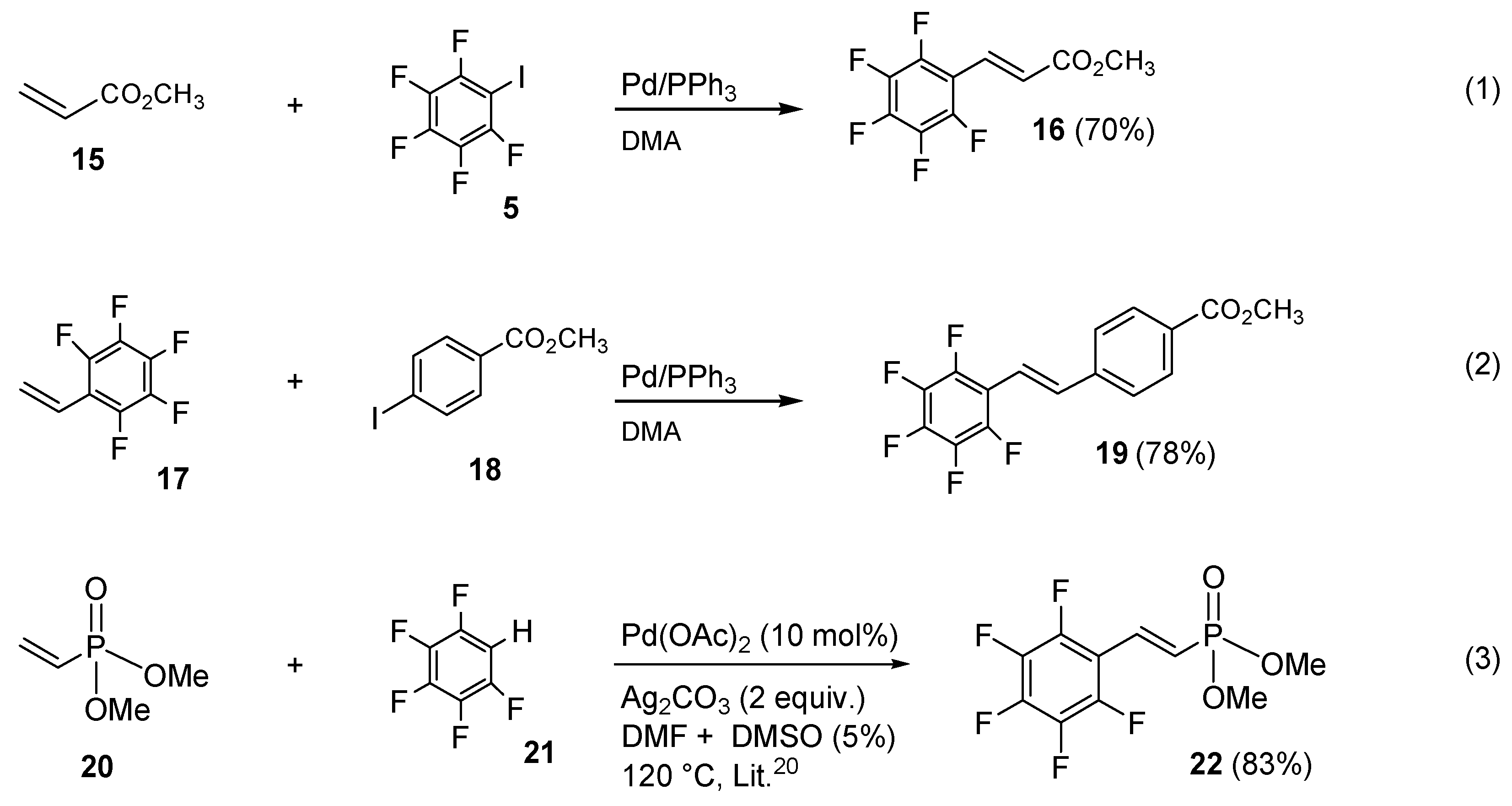
Much better results were obtained with carboxylic esters. Thus, the Heck coupling of methyl acrylate 15 with perfluorinated iodobenzene 5 afforded smoothly (E)-methyl-3-(perfluorophenyl)acrylate 16 in 70% of isolated yield (Scheme 4, Equation (1)). Similar satisfactory results were also reached by coupling pentafluorostyrene (17), which afforded methyl 4-(perfluorostyryl)benzoate 19 in 78% of yield (Scheme 4, Equation (2)).
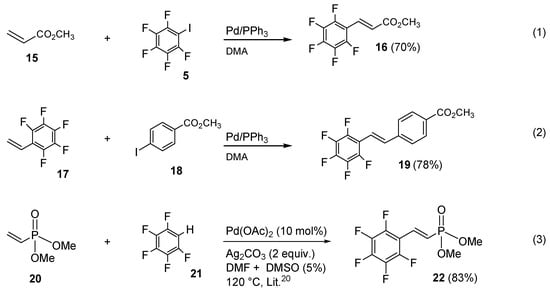
Scheme 4.
Heck strategy applied to other fluorinated vinyl substrates.
Unfortunately, under similar conditions, all the attempts made to synthesize aryl vinyl phosphonate were unproductive. Therefore, we paid attention to an alternative strategy based on the oxidative Heck coupling by adopting a known procedure of Caryl-H activation for the direct olefination of highly electron-deficient perfluoroarenes catalyzed by Pd(OAc)2 and silver carbonate as a re-oxidant [38]. By means of a slight modification of this method, commercially available vinyl diethyl phosphonate 20 and pentafluorobenzene 21 were coupled for the first time in good yield, affording (E)-dimethyl 2-(perfluorophenyl)vinylphosphonate 22 (Scheme 4, Equation (3)).
In summary, although the organometallic approach can be considered a short and quick way to synthesize olefinic substrates, under explored conditions, it proved to be poorly effective, affording low to moderate yields and showing some drawbacks that could limit the scale up (e.g., toxic solvents and expensive reagents). Studies are in progress to improve these results.
2.1.2. The Nucleophilic Displacement Approach
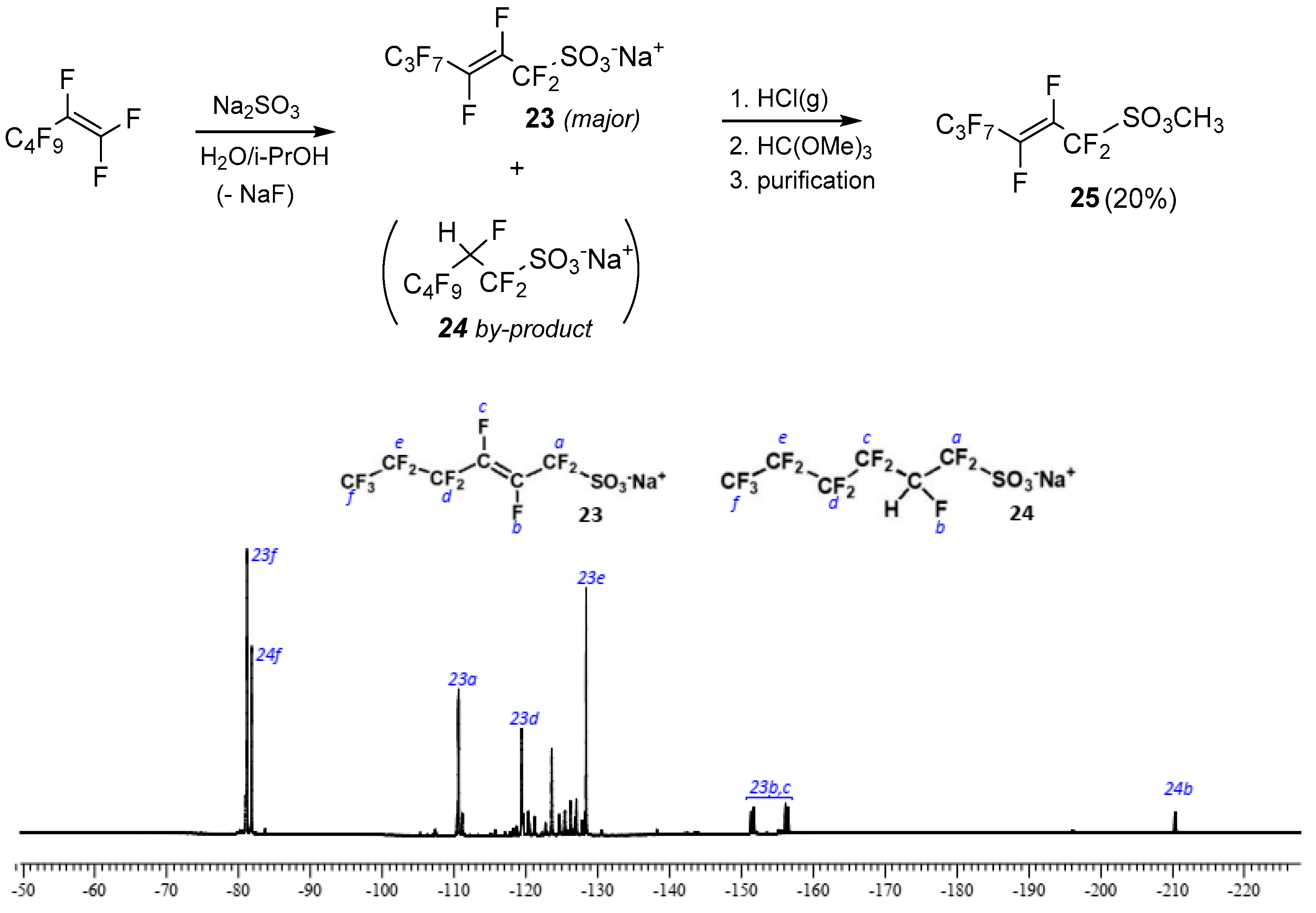
A different strategy was adopted involving the direct functionalization of terminal perfluoroalkenes with a sulfonic group through the nucleophilic displacement of a fluorine atom attached to the double bond. Based on results by Gross and Engler [39], such a vinylic substitution can be carried out smoothly with sulfite anion, but it occurs preeminently with the double bond rearrangement, affording the allyl sulfonate as a product with only trace amounts of the expected α,β-unsaturated compound. With this in mind, perfluoro-hex-2-ensulfonate 23 was initially prepared by reacting perfluorohex-1-ene with sodium sulfite in water/isopropanol (Figure 1). Unfortunately, this strategy also suffered from the drawback of giving high rates of addition to the double bond as a side reaction, which leads to minor amounts (25% ca.) of the saturated sulfonic salt 24, as revealed by 19F-NMR spectrum in Figure 1.
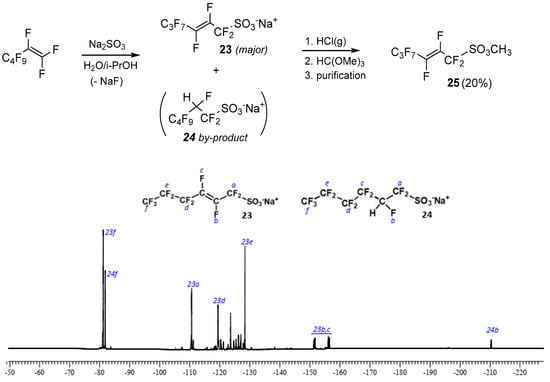
Figure 1.
Nucleophilic displacement strategy for sulfonic esters and sulfonate salts 23–24 distribution by 19F spectrum of the reaction mixture obtained by the treatment of perfluorohex-1-ene with SO32−. In the spectrum, the following assignments are possible according to the literature [39]: CF3- triplets (23f and 24f) at δ-81.22 ppm, intensity 1 (allylic product 23) and δ-81.86 ppm, intensity 0.5 (addition product 24); -CF = CF- at δ−153.83 ppm (23b,c), AB spin system, J(AB) 139.8 Hz; -CFH- at δ−210.44 ppm, 2JF,H 30.5 Hz (24a); -CF2-S- at δ-110.6 ppm (23a); -CF2- at δ−119.40 ppm and 128.44 ppm (23d and 23e, respectively). The CFH group of the addition product creates a magnetic inequivalence of neighbor CF2-groups (-CF2-CFH-CF2-SO3Na) and thus AB spin systems (δ from −108 to −127.5 ppm), which has not been specified in detail (signals attributable to fluorine atoms 24a,c,d,e).
To obtain highly volatile compounds, salts 23 and 24 were transformed into the corresponding methyl esters by treatment of the reaction mixture with gaseous HCl followed by the addition of trimethylortoformate [40]. After distillation, sulfonate derivative 25 was only obtained in 20% of overall yield (see Experimental section). It is worth mentioning that the addition of compound 24 was the sole by-product observed with this strategy; no compounds coming from the addition of HCl to the double bond were detected during the successive steps of esterification, which was most probably due to the presence of fluorine atoms on vinylic carbons [41].
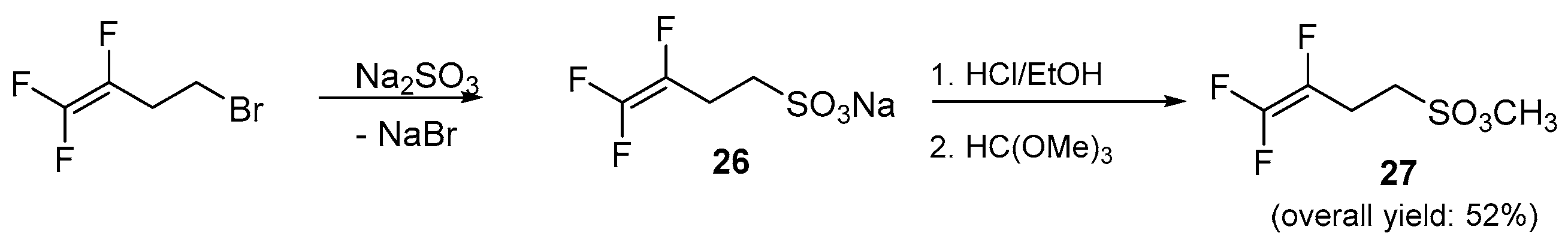
To overcome all these problems, the vinylic substitution approach was replaced with a simple nucleophilic displacement on a saturated carbon atom bearing a good leaving group such as a bromide. In addition, for a further increase in reactivity, fluorine atoms near the leaving group were replaced by hydrogen atoms starting from a homoallylic bromide fluorinated onto the double bond, namely 4-bromo-1,1,2-trifluorobut-1-ene, as shown in Scheme 5. More encouraging results were achieved by means of this new strategy and the nucleophilic displacement occurred smoothly enabling the synthesis of homoallylic fluorosulfonate ester 27 in a 52% of overall yield (Scheme 5).
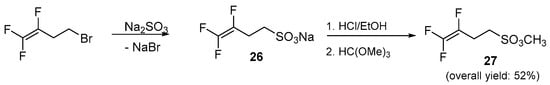
Scheme 5.
Synthetic strategy for homoallylic fluorosulfonic ester 27.
Due to the practical importance of this compound, which seems to attract quite a bit of interest in patents and has no precedent in the literature, the method was validated performing a scale-up experiment on a gram scale, obtaining 20 g of the desired product.
2.1.3. Carbanion Approach: The Wittig–Horner Reaction
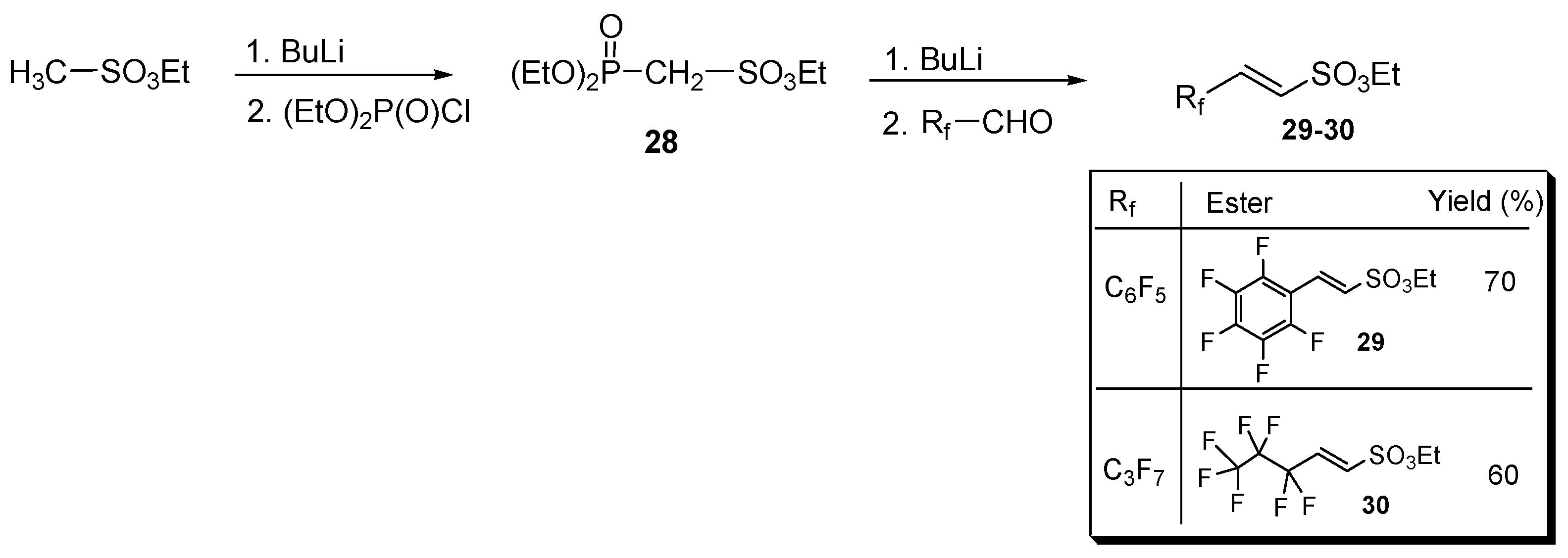
The previous strategies enabled the synthesis of perfluorinated aromatic, allylic, and homoallylic sulfonates but failed in the case of their α,β-unsaturated analogous. To overcome this limitation, a literature procedure [41] was adopted based on the Wittig–Horner condensation of ethyl diethylphosphoryl methanesulfonate 28 and a proper perfluorinated aldehyde Rf-CHO (Figure 2).
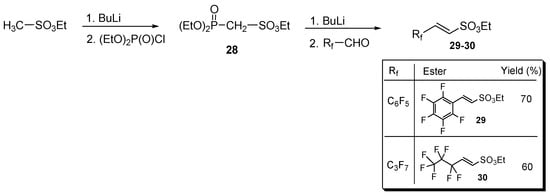
Figure 2.
The Wittig–Horner route for synthesizing both aliphatic and aromatic perfluorinated α,β-unsaturated sulfonic esters 29 and 30 (yields in the table are referred to the second step).
In particular, diester (28), generated from the commercially available ethyl methansulfonate, was condensed with pentafluorobenzaldehyde and with 2,2,3,3,4,4,4-heptafluorobutanal, affording the perfluoroaryl and perfluoroalkyl sulfonyl esters 29 and 30, respectively (Figure 2).
In turn, methanesulfonate 28 intermediate was prepared by reacting the carbanion to the alpha position of ethyl methanesulfonate with ethyl chlorophosphite (see for details Section S7 in the Supplementary Materials).
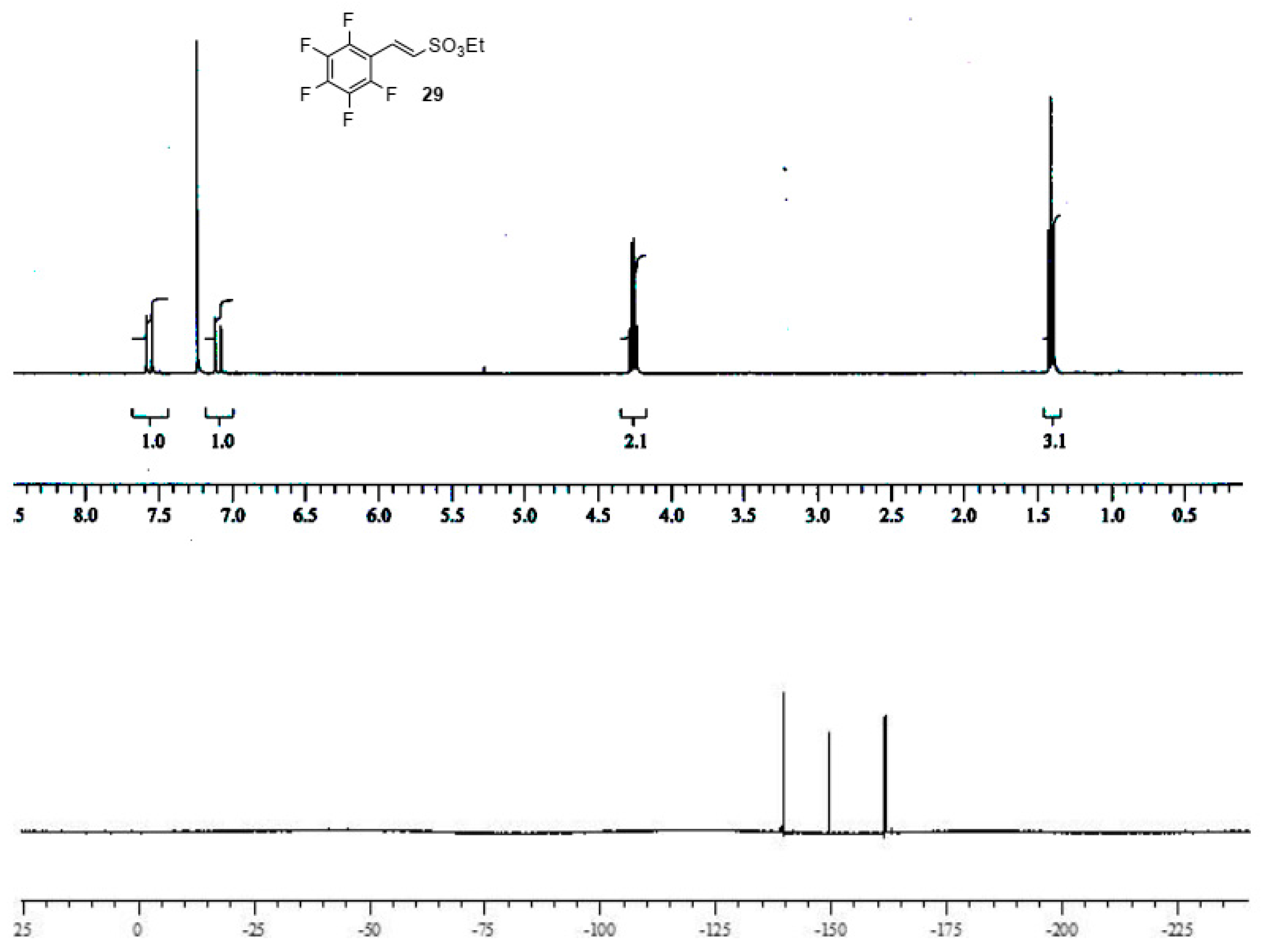
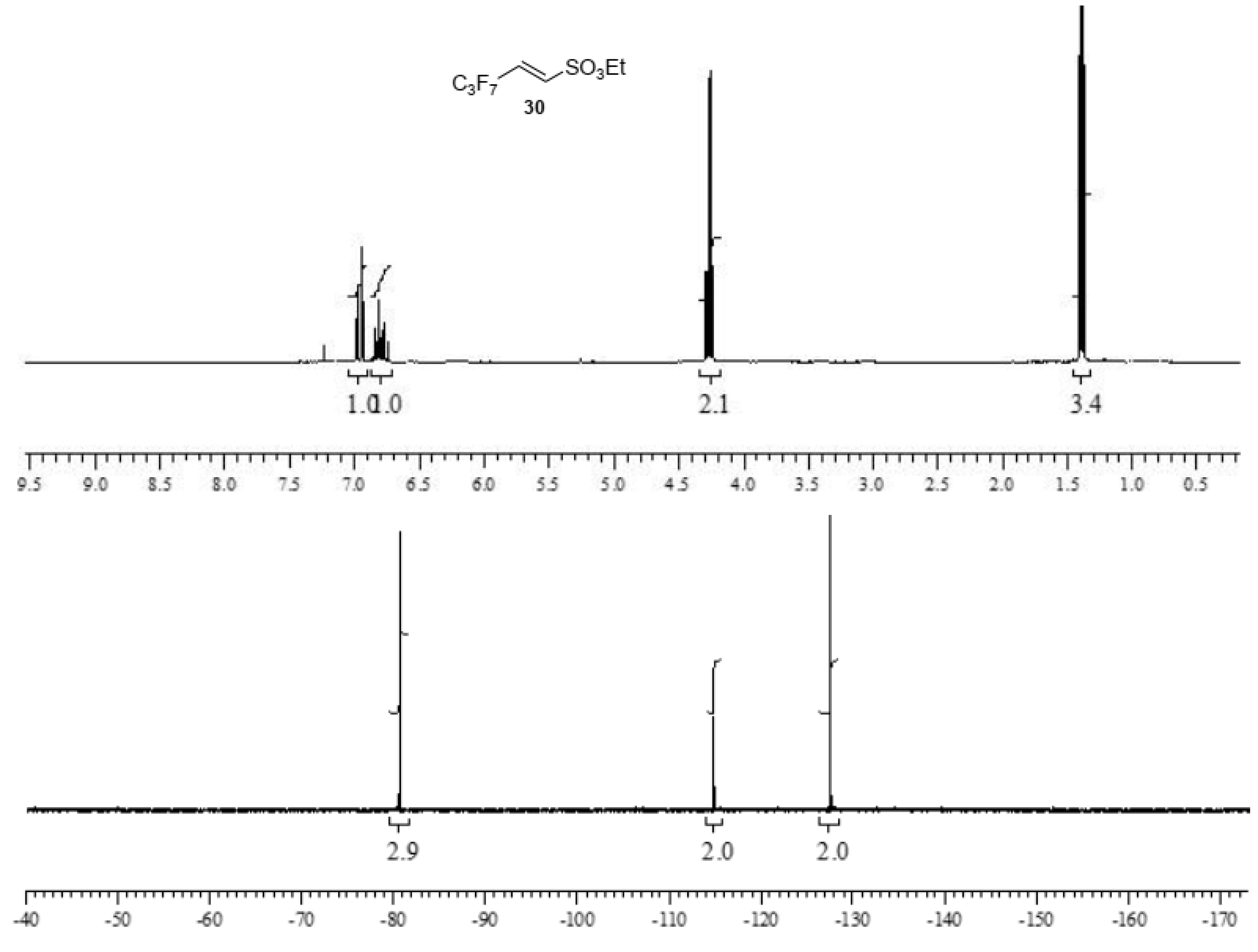
Sulfonic esters 29 and 30 were obtained in moderate to good yields as a white solid (m.p. 65 °C) and an oil (b.p. 135 °C, 10 mmHg), respectively, and were completely characterized as reported in the Supplementary Material (see Section S7). Compounds were identified by 1H- and 19F-NMR spectra (Figure 3 and Figure 4).
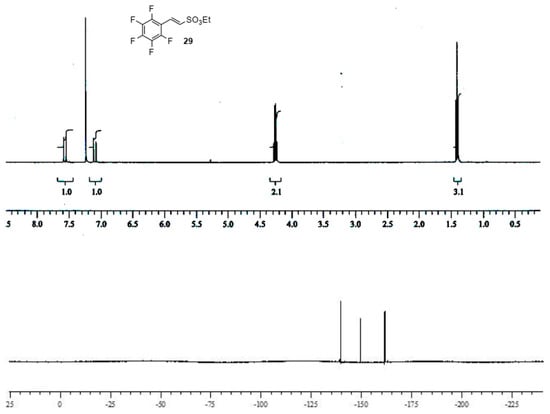
Figure 3.
1H- and 19F-NMR spectra images of (E)-ethyl 2-(perfluorophenyl)ethenesulfonate 29.
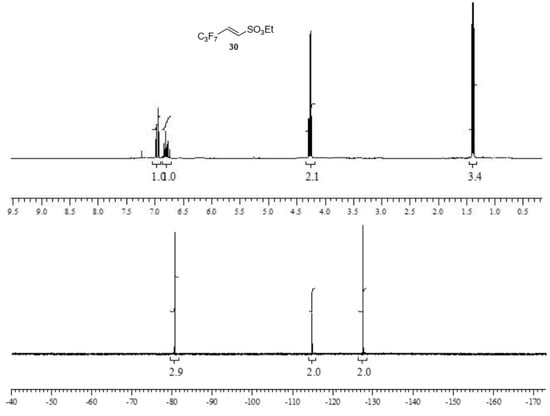
Figure 4.
1H- and 19F-NMR spectra images of (E)-ethyl 3,3,4,4,5,5,5-heptafluoropent-1-ene-1-sulfonate 30.
Below, a summary of perfluorinated monomers synthesized with the three strategies used in this work is listed (Figure 5).
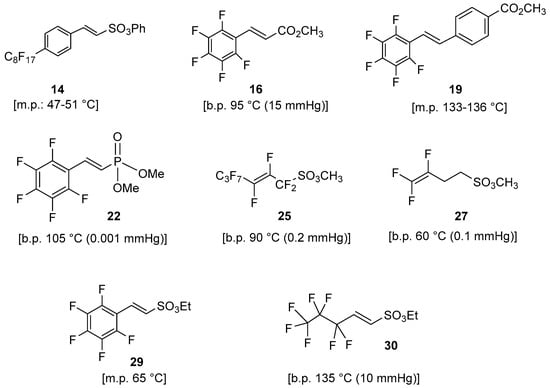
Figure 5.
Summary of synthesized perfluorinated monomers.
All these compounds were subjected to a selection based on their volatility for choosing the best candidate as suitable monomer for the polymerization using low-pressure deposition techniques.
2.2. Thin Film Membrane Deposition by Low-Pressure Plasma
Due to the good thermal stability and high volatility, monomer 27 was chosen as the best candidate for plasma polymerization. Studies are in progress for compounds 25 and 30.
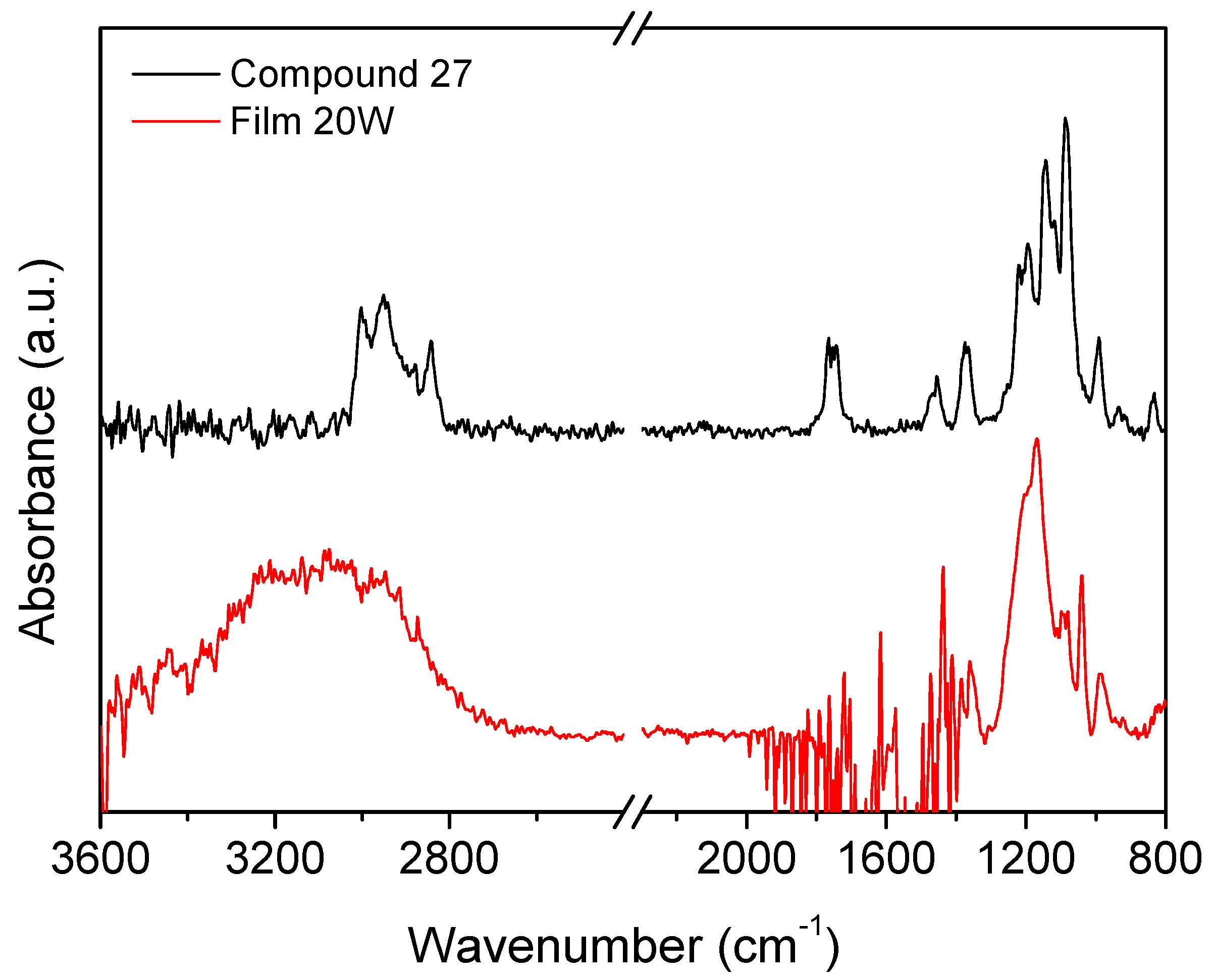
Figure 6 compares the infrared spectra of vapors of compound 27 and the plasma polymerized film obtained at low power (20 W). The main features of the monomer spectrum are the CH3 and CH2 absorption bands between 2800 and 3000 cm−1, the band at 1767 cm−1 assigned to CF2=CF, bending of CH3 and CH2 groups at 1460 cm−1. The band at 1370 cm−1 is ascribed to S=O stretching in the sulfonic ester group. The broad band 950–1300 cm−1 comprises absorptions from CF2 groups at about 1200 cm−1, the sulfonic ester group at 1143 cm−1, and CF at 1090 cm−1. Most of these absorptions can be observed also in the spectrum of the film deposited at the lowest plasma power (20 W), namely those of the sulfonic ester functionality and the CF2 groups. The retention of such functionalities in the deposited film points to a limited fragmentation of the monomer molecule in the plasma at low power. Instead, the high-wavenumber region is dominated by a broad band from 2800 to 3600 cm−1 assigned to OH groups, which was likely formed within the plasma reactor due to some water vapor residues. The CH3 and CH2 stretching band likely overlaps with the OH one, contributing to further broadening of the latter. Finally, the absorption from the double bond CF2=CF in the monomer disappears in the deposited film, indicating that the polymerization reaction has occurred by the rupture of this bond.
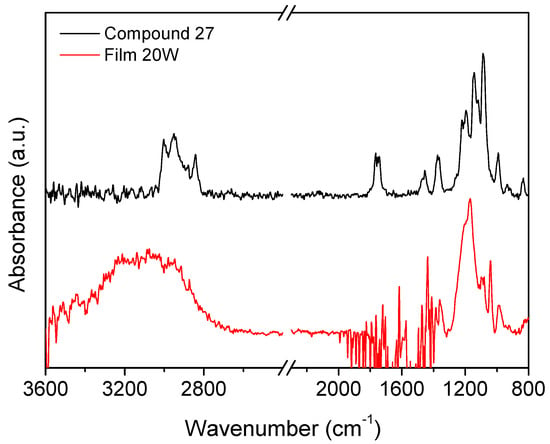
Figure 6.
Comparison between infrared spectra of compound 27 vapors and the plasma polymerized film obtained at 20 W.
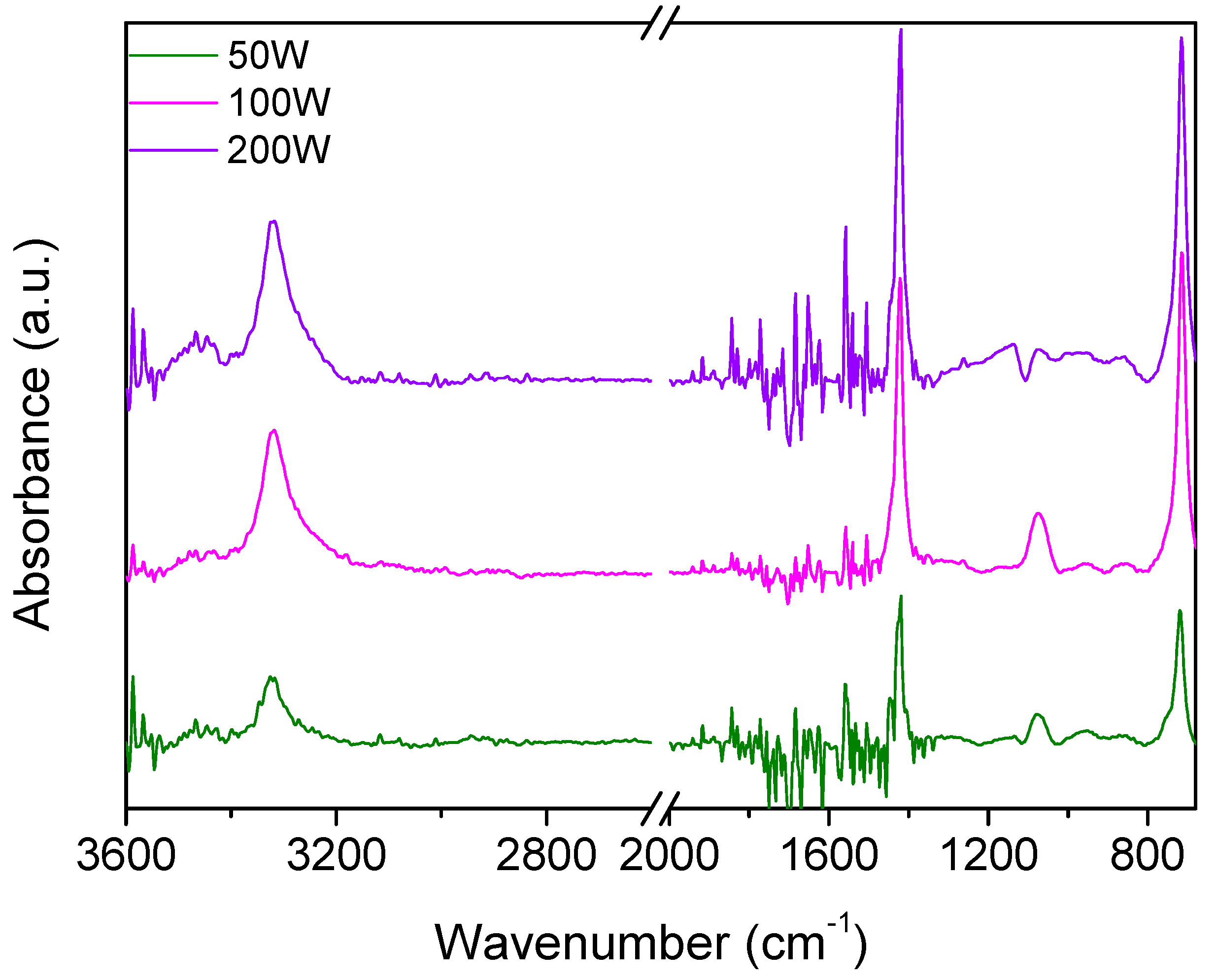
Figure 7 shows the infrared spectra of films deposited at increased plasma powers (50–200 W). In this case, major differences can be observed with respect to the spectra of Figure 4. More specifically, the main absorption band between 900 and 1300 cm−1 ca. is strongly reduced and less defined, with the only band clearly distinguishable being that at 1080 cm−1 assigned to CF groups. The disappearance of absorptions from the π bond, CF2, and sulfonic ester groups characterizing the monomer molecule is in agreement with the more energetic plasma environment generated at higher power values. Furthermore, the spectra are characterized by the absorption from OH groups from 3000 to 3500 cm−1 and a sharp signal at 1418 cm−1, which may be indicative of sulfonyl fluorides or sulfates.
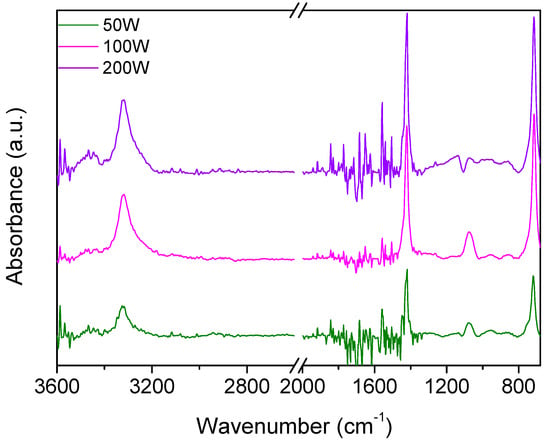
Figure 7.
IR spectra of films deposited from compound 27 at variable plasma powers (50–200 W).
Once more, the formation of these functionalities is reasonable in the energetic plasma environment obtained at higher powers (50–200 W), where new reactive species are formed from the more extensive fragmentation of the precursor molecule. The latter assignment is further confirmed by the increased intensity of the band at higher plasma powers. Finally, the signal at 714 cm−1 seems to be correlated to the one at 1418 cm−1 and can be assigned to S-F. The increase in the plasma power leads to a more extensive fragmentation of the precursor molecule, and this is reflected in the increase in the hydroxyl group absorption and the increase in the S-F absorptions.
To summarize the information coming from the IR characterization, in accordance with the results reported in the literature, the plasma polymerization of compound 27 led to thin film membranes with high monomer retention only at the lowest plasma powers (20 W). Only in this case, in fact, it is possible to drive the polymerization of the monomer mainly by rupture of the double bond and preserve the sulfonic ester functionality in the deposited film. Increasing the power leads to a more extensive monomer fragmentation, defluorination of the produced radicals, and extensive structural rearrangements in the deposited films.
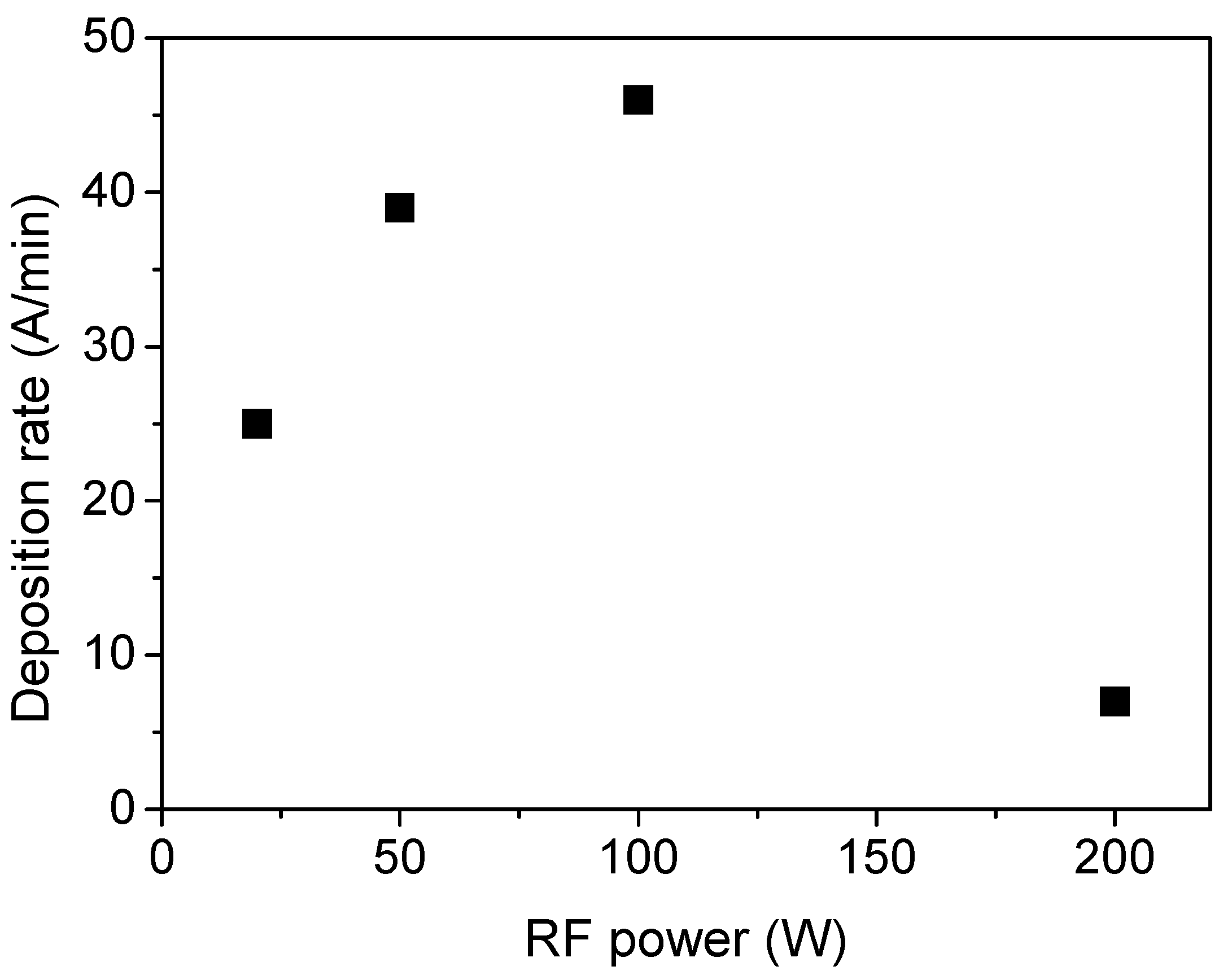
Figure 8 reports the film deposition rate, which is determined as the ratio between film thickness and the deposition time (30 min), as a function of the plasma power. The deposition rate increases up to 100 W and then decreases for higher values. This trend is typical in PECVD processes and can be explained considering that below 100 W, increasing the power of the monomer molecule is fragmented to a greater extent in the plasma, thus generating a higher density of radicals, which are precursors of the growing film. This condition is named the activated growth regime. However, above the threshold power, recombination reactions, etching, and sputtering phenomena of the growing film also occur, which results in a reduced film deposition rate (deactivated growth regime) [42].
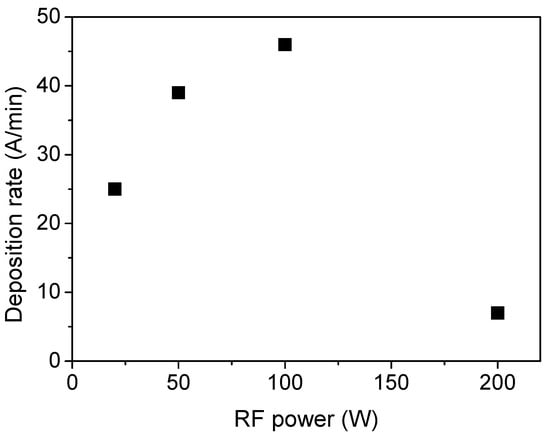
Figure 8.
Film deposition rate as a function of the power delivered to the plasma.
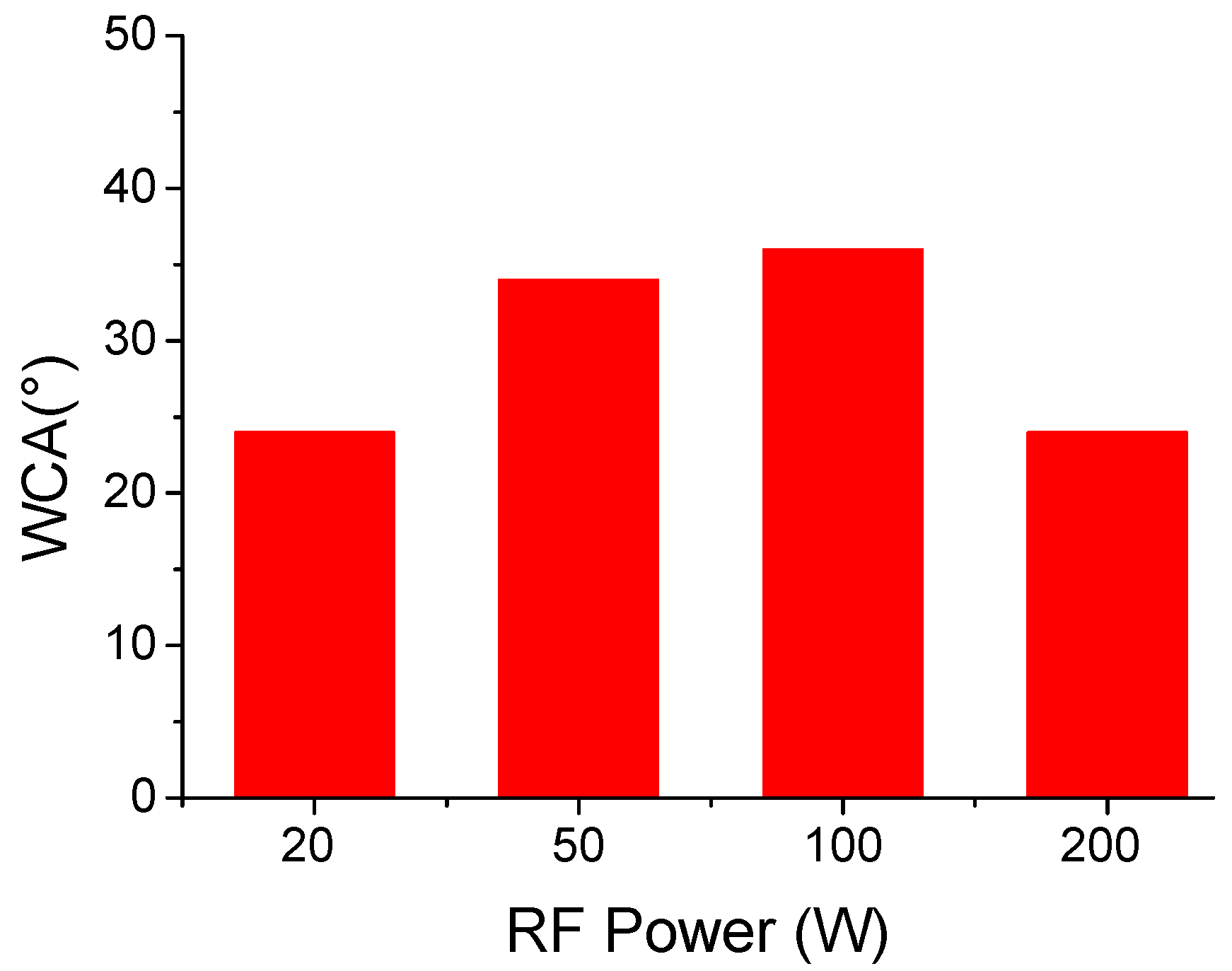
Finally, the wettability of the deposited films was tested through static water contact angle measurements (Figure 9). IR analysis showed that deposited films contain both hydrophilic and hydrophobic moieties, to a different extent, depending on the plasma power used. All deposited films are highly hydrophilic with water contact angles in the range of 25–40°. The film deposited at 20 W with the highest monomer retention is made by hydrophobic moieties such as CF2 groups, as well as hydrophilic ones, such as OH functionalities. The hydrophilicity can be likely due to a surface segregation of the polar functionalities. At higher plasma powers, IR spectra point out to an extensive defluorination of the polymer matrix, which can be, beside the presence of the polar functionalities, the reason for the film’s hydrophilicity.
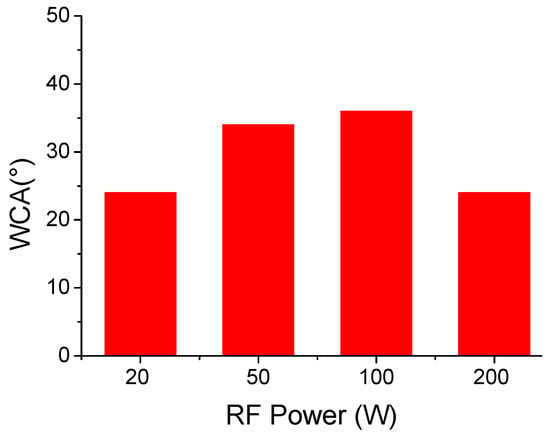
Figure 9.
Static water contact angle of deposited films vs. plasma power.
3. Materials and Methods
3.1. General Remarks
The starting reagents and solvents are commercially available (from Aldrich and ABCR) (see the Supplementary Materials). Solvents (N,N-dimethylformamide (DMA), N,N-dimethylacetamide (DMF), and N-methylpyrrolidone (NMP)) were dried and then distilled before use (see for details Section S1 in the Supplementary Materials).
Pd catalysts of Table 1 were generated in situ from Pd(OAc)2 and triphenylphosphine, while Pd(0) nanoparticles catalysts were prepared according to a known procedure by reduction with LiBH4 (see: ChemSusChem 2012, 5, 109–116).
Reactions were monitored by GLC and GC-MS techniques by using an Agilent 5890 A gas-chromatograph and an Agilent 6850/MSD 5975C instruments with a capillary column HP-5MS (Agilent, l. 30 m, i.d. 0.25 mm, s.p.t. 0.25 μ).
NMR spectra were recorded on a Varian Inova 400 MHz spectrometer. Chemical shift values are given in ppm relative to internal Me4Si.
Identification of the reaction products was accomplished by their preliminary isolation by column chromatography on silica gel (SiO2 50–200 μm, from Baker) or by distillation. Next, the products were identified by comparison of their MS and NMR spectra with those reported in the literature.
For unknown compounds, high-resolution mass spectra were recorded by using a Shimadzu LCMS-IT-TOF instrument with the following settings: mass range 50–1000 m/z, ionization system electrospray ion source in negative ion mode, nebulizer gas nitrogen at 3 bar, dry gas nitrogen at 1.5 L/min and 250 °C, collision gas argon.
3.2. Deposition and Characterization of Thin Film Membranes by PECVD
Film depositions were carried out in a cylindrical parallel plate stainless steel reactor evacuated by a turbomolecular/rotary system. Experiments were carried out feeding the plasma with vapors of compound 27 at variable RF power (20–200 W) and a pressure of 500 mTorr. The vapor flow rate was set with a needle valve to 0.25 sccm. The film deposition time was fixed to 30 min, and double-polished silicon was used as a substrate for deposition.
Film chemical composition was investigated by Fourier Transform Infrared (FTIR) spectroscopy (BRUKER, Equinox 55). Spectra were recorded from 400 to 4000 cm−1 in absorbance mode at 4 cm−1 resolution.
Film water contact angle (WCA) measurements were performed with a manual goniometer (Ramé-Hart, 100) and the reported WCA values were averaged over 5 measurements on each sample with standard deviations of ±1° (for further details see Section S2 of the Supplementary Materials).
3.3. General Procedures for the Synthesis of Perfluorinated Monomers
3.3.1. Procedure for the Heck Coupling in Table 1: Synthesis of Perfluorosulfonic Ester 14
In a typical procedure, a 10 mL round-bottomed flask equipped with a magnetic bar was charged with sulfonate ester or salt (1–4, 0.5 mmol), perfluorohaloarene (5–7, 0.5 mmol), K2CO3 (or TBAOH, 1 mmol), Pd acetate (or Pd-colloids, 3 mol%), and PPh3 (6 mol%) in 5 mL of DMF (DMA, NMP, or water) and heated at 120 °C for 4 h. At the end of the reaction (GC-MS), the mixture was washed with HCl 5% and extracted with dichloromethane. After drying and evaporation of the solvent in vacuo, the products were purified by silica gel column chromatography (eluent petroleum ether/dichloromethane).
All attempts to isolate the expected products of Table 1 from the reaction mixture (sulfonate 8,9,12,13,14) had unsatisfactory results with the sole exception of phenyl sulfonate 14, which was obtained in a 65% of yield (see for details Section S3 in the Supplementary Materials).
(E)-phenyl 2-(4-perfluorooctyl)phenylethene sulfonate 14. Pale yellow solid. m.p.: 47–51 °C 1H-NMR (400 MHz, CDCl3), δ: 6.96 (d, 1 H, J = 16.8 Hz, alpha vinyl proton); 7.20–7.42 (m, 5 H, aryl protons SO3Ph), 7.55 (d, 1 H, J = 16.8 Hz, beta vinyl proton); 7.55 (d, 2 H, J = 8.1 Hz, aryl protons); 7.66 (d, 2 H, J = 8.1 Hz, aryl protons). HRMS (ESI-TOF) m/z [M + H]+: calcd for C22H12F17O3S+ 679.0230, found 679.0238.
3.3.2. Procedure for the Synthesis of Fluorinated Carboxylic Esters 16 and 19 (Scheme 4, Equations (1) and (2))
In a 100 mL round-bottomed flask, equipped with a condenser and a magnetic bar, 1,2,3,4,5-pentafluoro-6-iodobenzene 5 (or methyl 4-iodobenzoate 18, 5 mmol), methyl acrylate 15 (or 1,2,3,4,5-pentafluoro-6-vinylbenzene 17, 5 mmol), K2CO3 (10 mmol), Pd acetate (3 mol%), and PPh3 (6 mol%) were refluxed in 50 mL of DMF. At the end of the reaction (GC-MS, 2 ÷ 4 h), the mixture was washed with HCl 5% and extracted with dichloromethane. After drying and evaporation of the solvent in vacuo, the products were purified by silica gel column chromatography (eluent petroleum ether/dichloromethane).
(E)-methyl 3-(perfluorophenyl)acrylate16. Dark red oil, b.p. 95 °C (15 mmHg) (yield 70 %). 1H-NMR (400 MHz, CDCl3), δ: 3.79 (s, 3H, methoxy); 6.70 (d, 1 H, J = 16.5 Hz, alpha vinyl proton); 7.58 (d, 1 H, J = 16.5 Hz, beta vinyl proton); 13C-NMR (400 MHz, CDCl3), δ: 52.2 (MeO), 110.0 (t, 2JC,F = 13 Hz), 126.0 (t, 3JC,F = 9 Hz), 128.5, 137.95 (d, 1JC,F = 253 Hz); 141.81 (d, 1JC,F = 258 Hz); 145.75 (d, 1JC,F = 254 Hz); 166.52 (carbonyl). 19F-NMR (400 MHz, CDCl3), δ: −148.05 (dd, 2F, 3JF,F = 21 and 4JF,F = 6 Hz), −159.78 (t, 1F, 3JF,F = 21 Hz), −170.29 (td like, 2F, 3JF,F = 21 and 4JF,F = 6 Hz); MS (EI): 252 (M+, 41), 221 (100), 193 (64), 143 (38), 123 (18), 117 (9), 93 (5), 59 (2). HRMS (ESI-TOF) m/z [M + H]+: calcd for C10H6F5O2+ 253.0282, found 253.0271.
(E)-methyl 4-(perfluorostyryl)benzoate19: White solid. 1H-NMR (400 MHz, CDCl3), δ: 3.90 (s, 3 H, methoxy); 7.02 (d, 1 H, J = 16.9 Hz, vinyl proton); 7.40 (d, 1 H, J = 16.9 Hz, vinyl proton); 7.53 (d, 2 H, J = 8.2 Hz, aryl protons); 8.00 (d, 2 H, J = 8.2 Hz, aryl protons); MS (EI): 328 (M+, 72), 297 (100), 269 (18), 250 (18), 219 (51), 199 (5), 192 (4), 148 (9), 109 (14), 77 (3), 51 (4). HRMS (ESI-TOF) m/z [M + H]+: calcd for C16H10F5O2+ 329.0595, found 329.0590.
3.3.3. Procedure for the Oxidative Heck Coupling: Synthesis of Perfluorinated Phosphonyl Ester 22 (Scheme 4, Equation (3))
This ester was prepared according to the literature [31]. In a 25 mL round-bottomed flask Pd(OAc)2 (10 mol%) and Ag2CO3 (2.0 equiv) under N2 were added, which was followed by DMF (2.4 mL) and DMSO (5%, 120 μL) with stirring. Pentafluorobenzene 21 (0.6 mmol, 1.0 equiv) and dimethyl vinylphosphonate 20 (2.0–3.0 equiv) were added subsequently. The mixture was heated at 120 °C (oil bath). After stirring for 12 h, the reaction mixture was cooled to room temperature and diluted with ethyl acetate, washed with 1 N HCl and brine, dried over Na2SO4, filtered, and concentrated. The residue was purified with silica gel chromatography (petroleum ether:ethyl acetate 3:2 as eluent) to provide the pure product (E)-dimethyl 2-(perfluorophenyl)vinylphosphonate 22 as colorless oil (b.p. 105 °C, 0.001 mmHg). 1H NMR (400 MHz, CDCl3) δ 3.74 (s, 3H), 3.76 (s, 3H), 6.56 (t, J = 17.4 Hz, 1H), 7.40 (dd, J = 24.2 Hz, 17.4 Hz, 1H). 13C NMR δ 52.84, 52.89, 110.38 (m), 123.01 (dt, J = 188.5 and 8.4 Hz), 132.78 (d, J = 6.1 Hz), 137.95 (dm, 1JC,F = 254.8), 141.95 (dm, 1JC,F = 257.0 Hz), 145.77 (dm, 1JC,F = 255.6 Hz). 19F NMR δ -140.0 (dd, 3JF,F = 22.0 Hz, 4JF,F = 7.3 Hz, 2F), −150.3 (t, 3JF,F = 19.5 Hz, 1F), −160.88 (td, 3JF,F = 19.5 and 4JF,F = 7.3 Hz, 2F). MS (EI) 302 (M+, 24), 283 (28), 207 (100), 170 (79), 143 (39), 110 (78), 93 (61). HRMS (ESI-TOF) m/z [M + H]+: calcd for C10H9F5O3P+ 303.0204, found 303.0211.
3.3.4. Procedures for the Nucleophilic Substitution: Synthesis of Perfluorinated Sulfonic Esters 25 and 27
Esters 25 and 27 were prepared starting from perfluorohex-1-ene or 4-bromo-1,1,2-trifluorobut-1-ene, respectively, by treatment with sodium sulfite followed by the esterification of sulfonate salts intermediate (23 and 26) with trimethyl orthoformate (see for details Scheme 5, Figure 1 and Section S4 in the Supplementary Materials). Sulfonic esters 25 and 27 were characterized as reported below:
Methyl perfluorohex-2-ene-1-sulfonate25 was isolated as a red oil (b.p. 90 °C, 0.2 mmHg). 19F-NMR (CDCl3) δ ppm (relative to CFCl3): −81.20 (t, 3F, 3JF,F = 8.20 Hz, CF3); −107.22 (dd, 2F, 3JF,F = 24.4 and 4JF,F = 12.2 Hz, CF2-SO3), −119.60 (m, 2F, CF2-CF2-CF=), −128.50 (m, 2F, CF2-CF2-CF=), −152.50 (dt, 1F, 3JF,F = 143.4 and 3JF,F = 24.4 Hz, =CF-CF2-SO3), −153.70 (m, 1F, CF=CF-CF2-SO3). HRMS (ESI-TOF) m/z [M + H]+: calcd for C7H4F11O3S+ 376.9705, found 376.9712.
Methyl 3,4,4-trifluorobut-3-ene-1-sulfonate27 as a dark red oil in 89% of yield. 1H-NMR (CDCl3) δ ppm: 2.67–2.85 (m, 2 H, =CHF-CH2), 3.24 (t, 2 H, J= 7.3 Hz, -CH2-SO3CH3), 3.83 (s, 3 H, SO3CH3). 13C-NMR (CDCl3) δ ppm: 21.05 (d, 2JC,F = 22.9 Hz, =CHF-CH2), 41.13 (s, -CH2-SO3CH3), 55.96 (s, SO3CH3). 125.53 (ddd, 1JC,F = 235.0 Hz, 2JC,F = 53.4 and 18.3 Hz, F2C = CHF-), 153.21 (ddd, 1JC,F = 288.4 and 276.2 Hz, 2JC,F = 45.0 Hz, F2C = CHF-). 19F-NMR (CDCl3) δ ppm: −190.45 (ddt, 1 F, 3JF,F = 116.0 and 33.6 Hz, 3JF,H = 21.4 = CHF-CH2), −135.73 (dd, 1 F, 2JF,F = 116.0, 3JF,H = 82.4, CF2 = CHF-), −116.65 (dd, 1 F, 2JF,F = 82.4, 3JF,F = 33.6, CF2 = CHF-). MS (EI): 204 (0.1), 108 (100), 89 (20), 59 (23), 39 (40). HRMS (ESI-TOF) m/z [M + H]+: calcd for C5H8F3O3S+ 205.0141, found 205.0149.
3.3.5. Procedures for Wittig–Horner Reaction: Synthesis of Perfluorinated Sulfonic Esters 29 and 30
These esters were prepared by adapting a literature procedure [35] that exploits the Wittig–Horner reaction (see for details Section S7 of the Supplementary Materials).
(E)-ethyl 2-(perfluorophenyl)ethenesulfonate29. A solution of ethyl diethylphosphoryl methanesulfonate 28 (1.0 equiv) in dry THF (1 mL × 0.25 mmol of 28) was treated at −78 °C under nitrogen atmosphere with 2.3 M n-BuLi in hexane (approximately 1.05 equiv.). Stirring was continued for 20 min. Then, freshly pentafuorobenzaldehyde (1.1 equiv.) was added. After an additional 45 min. at −78 °C, the solution was allowed to warm to room temperature and stirring was continued for 60 h. The bulk of the solvents was evaporated, and the residue was treated with water (5 mL × 0.25 mmol) and extracted with dichloromethane (3 × 5 mL × 0.25 mmol). The organic layers were dried over MgSO4 and evaporated to afford crude unsaturated sulfonyl ester, which was purified by column chromatography (petroleum ether:ethyl acetate 10:1 as eluent) affording pure (E)-ethyl 2-(perfluorophenyl)ethene sulfonate 29 as white solid (m.p. 65 °C) in a 70% of yield. 1H NMR (CDCl3): 1.40 (t, J = 6.9 Hz, 3 H, CH3); 4.25 (q, J = 6.9, 2 H, CH2); 7.09 (d, J = 15.9, 1 H, vinyl CH); 7.56 (d, J = 15.9, 1 H, vinyl CH). 19F NMR (CDCl3): −139.7 (dt, 3JF,F = 24.4 and 4JF,F = 6.1 Hz, 2 F), −149.6 (t, 3JF,F = 21.3 Hz, 1 F), −161.8 (m, 2 F). HRMS (ESI-TOF) m/z [M + H]+: calcd for C10H8F5O3S+ 303.0109, found 303.0116.
(E)-ethyl 3,3,4,4,5,5,5-heptafluoropent-1-ene-1-sulfonate30. This perfluorinated sulfonic ester was prepared by an analogous procedure from 28 and 2,2,3,3,4,4,4-heptafluorobutanal (CH2Cl2:petroleum ether 7:3 as eluent) in a 60% of yield. The ester was initially obtained as a mixture of E/Z isomer 85/15 in ratio. After distillation (b.p. 135 °C, 10 mmHg), the major E isomer was isolated as red oil. 1H NMR (CDCl3): 1.39 (t, J = 7.1 Hz, CH3), 4.26 (q, J = 7.1, 2 H, CH2), 6.78 (dt, J = 15.4 and 11.2 Hz, vinyl CH), 6.96 (dt, J = 15.4 1.5 Hz, 1 H, vinyl CH). 13C δ 14.95, 69.01, 108.40 (tm, 1JC,F = 264.7 Hz, CF3CF2-), 112.80 (tt, 1JC,F = 255.6 and 2JC,F = 32.0 Hz, CF2C=), 117.52 (qt, 1JC,F = 287.6 and 2JC,F = 33.5 Hz, CF3), 130.4 (t, 2JC,F = 25.2 Hz), 135.8 (t, 3JC,F = 8.4 Hz). 19F NMR (CDCl3): -80.7 (t, 3JF,F = 9.2 Hz, 3 F); -114.9 (quintet, 3JF,F = 9.2 Hz, 2 F); −127.6 (m, 2 F); HRMS (ESI-TOF) m/z [M + H]+: calcd for C7H8F7O3S+ 305.0077, found 305.0071.
4. Conclusions
In conclusion, three main synthetic strategies were explored for synthesizing perfluorinated unsaturated sulfonic, phosphonic, and carboxylic compounds to be used as monomers for future applications in producing proton exchange membranes. Among the synthetic pathways presented, the organometallic approach gave the worst results, with low yields, hard isolation of products, and difficult scale up. In contrast, nucleophilic substitution with sulfite anion, as well as Horner–Wittig condensation strategies, gave moderate to good yields depending on the structure of the target molecules. Studies are still underway to improve these results.
Compound 27 was selected as the best candidate for thin film membrane preparation by PECVD, based on its volatility. Plasma polymerization experiments were carried out at different plasma powers (20–200 W). Comparison of the film chemical composition by IR spectroscopy allowed us to point out that the film prepared in the mildest plasma conditions (20 W) shows the maximum monomer retention in its structure. In this condition, plasma polymerization likely occurs mainly by rupture of the π bond in the monomer molecule. Increasing the power leads to a more extensive monomer fragmentation, defluorination of the radicals and extensive structural rearrangements in the deposited films. Future studies will address the deposition of the thin film by the copolymerization of compound 27 with a different long-chain fluorocarbon and the measurement of the proton exchange capabilities of our thin film membranes and their optimization.
Supplementary Materials
The following are available online.
Author Contributions
Conceptualization, A.M. (Antonio Monopoli), A.M. (Antonella Milella), F.P. and A.N., methodology, M.C. and A.M. (Antonio Monopoli) and P.C.; data curation, P.C., M.C., A.N. including ESI-ToF analysis and A.M. (Antonella Milella) (for plasma results); writing—original draft preparation, A.M. (Antonio Monopoli), A.N. and A.M. (Antonella Milella) (for plasma results); writing—review and editing, A.M. (Antonio Monopoli), supervision, A.M. (Antonio Monopoli), F.F. and A.N. All authors have read and agreed to the published version of the manuscript.
Funding
This research was partially funded by the Italian Ministry of Education (MIUR), grant number PONa3_00369 SISTEMA and EC project SMALLINONE, grant number 515846.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Smitha, B.; Sridhar, S.; Khan, A.A. Solid polymer electrolyte membranes for fuel cell applications—A review. J. Memb. Sci. 2005, 259, 10–26. [Google Scholar] [CrossRef]
- Hogarth, W.H.J.; Diniz Da Costa, J.C.; Lu, G.Q. Solid acid membranes for high temperature (>140 °C) proton exchange membrane fuel cells. J. Power Sources 2005, 142, 223–237. [Google Scholar] [CrossRef]
- Vishnyakov, V.M. Proton exchange membrane fuel cells. Vacuum 2006, 80, 1053–1065. [Google Scholar] [CrossRef]
- Mauritz, K.A.; Moore, R.B. State of understanding of Nafion. Chem. Rev. 2004, 104, 4535–4585. [Google Scholar] [CrossRef]
- Hickner, M.A.; Ghassemi, H.; Kim, Y.S.; Einsla, B.R.; McGrath, J.E. Alternative polymer systems for proton exchange membranes (PEMs). Chem. Rev. 2004, 104, 4587–4611. [Google Scholar] [CrossRef] [PubMed]
- Ous, T.; Arcoumanis, C. Degradation aspects of water formation and transport in Proton Exchange Membrane Fuel Cell: A review. J. Power Sources 2013, 240, 558–582. [Google Scholar] [CrossRef]
- Whiston, M.M.; Azevedo, I.L.; Litster, S.; Whitefoot, K.S.; Samaras, C.; Whitacre, J.F. Expert assessments of the cost and expected future performance of proton exchange membrane fuel cells for vehicles. Proc. Natl. Acad. Sci. USA 2019, 116, 4899–4904. [Google Scholar] [CrossRef] [Green Version]
- Du, L.; Prabhakaran, V.; Xie, X.; Park, S.; Wang, Y.; Shao, Y. Low-PGM and PGM-Free Catalysts for Proton Exchange Membrane Fuel Cells: Stability Challenges and Material Solutions. Adv. Mater. 2021, 33, 1908232. [Google Scholar] [CrossRef] [PubMed]
- Kongkanand, A.; Mathias, M.F. The Priority and Challenge of High-Power Performance of Low-Platinum Proton-Exchange Membrane Fuel Cells. J. Phys. Chem. Lett. 2016, 7, 1127–1137. [Google Scholar] [CrossRef]
- Haider, R.; Wen, Y.; Ma, Z.F.; Wilkinson, D.P.; Zhang, L.; Yuan, X.; Song, S.; Zhang, J. High temperature proton exchange membrane fuel cells: Progress in advanced materials and key technologies. Chem. Soc. Rev. 2021, 50, 1138–1187. [Google Scholar] [CrossRef]
- Xiao, F.; Wang, Y.C.; Wu, Z.P.; Chen, G.; Yang, F.; Zhu, S.; Siddharth, K.; Kong, Z.; Lu, A.; Li, J.C.; et al. Recent Advances in Electrocatalysts for Proton Exchange Membrane Fuel Cells and Alkaline Membrane Fuel Cells. Adv. Mater. 2021, 33, 2006292. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.X.; Swihart, M.T.; Wu, G. Achievements, challenges and perspectives on cathode catalysts in proton exchange membrane fuel cells for transportation. Nat. Catal. 2019, 2, 578–589. [Google Scholar] [CrossRef]
- Dilonardo, E.; Milella, A.; Palumbo, F.; Thery, J.; Martin, S.; Barucca, G.; Mengucci, P.; D’Agostino, R.; Fracassi, F. Plasma deposited Pt-containing hydrocarbon thin films as electrocatalysts for PEM fuel cell. J. Mater. Chem. 2010, 20, 10224–10227. [Google Scholar] [CrossRef]
- Kraytsberg, A.; Ein-Eli, Y. Review of advanced materials for proton exchange membrane fuel cells. Energy Fuels 2014, 28, 7303–7330. [Google Scholar] [CrossRef]
- Pan, M.; Pan, C.; Li, C.; Zhao, J. A review of membranes in proton exchange membrane fuel cells: Transport phenomena, performance and durability. Renew. Sustain. Energy Rev. 2021, 141, 110771. [Google Scholar] [CrossRef]
- Rusanov, A.L.; Kostoglodov, P.V.; Abadie, M.J.M.; Voytekunas, V.Y.; Likhachev, D.Y. Proton-conducting polymers and membranes carrying phosphonic acid groups. Adv. Polym. Sci. 2008, 216, 125–155. [Google Scholar] [CrossRef]
- Kotov, S.V.; Pedersen, S.D.; Qiu, W.; Qiu, Z.M.; Burton, D.J. Preparation of perfluorocarbon polymers containing phosphonic acid groups. J. Fluor. Chem. 1997, 82, 13–19. [Google Scholar] [CrossRef]
- Yamabe, M.; Akiyama, K.; Akatsuka, Y.; Kato, M. Novel phosphonated perfluorocarbon polymers. Eur. Polym. J. 2000, 36, 1035–1041. [Google Scholar] [CrossRef]
- Souzy, R.; Ameduri, B.; Boutevin, B.; Virieux, D. Synthesis of new aromatic perfluorovinyl ether monomers containing phosphonic acid functionality. J. Fluor. Chem. 2004, 125, 1317–1324. [Google Scholar] [CrossRef]
- Wehbi, M.; Mehdi, A.; Negrell, C.; David, G.; Alaaeddine, A.; Améduri, B. Phosphorus-Containing Fluoropolymers: State of the Art and Applications. ACS Appl. Mater. Interfaces 2020, 12, 38–59. [Google Scholar] [CrossRef]
- Primachenko, O.N.; Odinokov, A.S.; Marinenko, E.A.; Kulvelis, Y.V.; Barabanov, V.G.; Kononova, S.V. Influence of sulfonyl fluoride monomers on the mechanism of emulsion copolymerization with the preparation of proton-conducting membrane precursors. J. Fluor. Chem. 2021, 244, 109736. [Google Scholar] [CrossRef]
- Brault, P. Review of Low Pressure Plasma Processing of Proton Exchange Membrane Fuel Cell Electrocatalysts. Plasma Process. Polym. 2016, 13, 10–18. [Google Scholar] [CrossRef] [Green Version]
- Bhosale, A.C.; Ghosh, P.C.; Assaud, L. Preparation methods of membrane electrode assemblies for proton exchange membrane fuel cells and unitized regenerative fuel cells: A review. Renew. Sustain. Energy Rev. 2020, 133, 110286. [Google Scholar] [CrossRef]
- Milella, A.; Palumbo, F.; Dilonardo, E.; Barucca, G.; Cosma, P.; Fracassi, F. Single step plasma deposition of platinum-fluorocarbon nanocomposite films as electrocatalysts of interest for micro fuel cells technology. Plasma Process. Polym. 2014, 11, 1068–1075. [Google Scholar] [CrossRef]
- Bosso, P.; Milella, A.; Armenise, V.; Fanelli, F.; Fracassi, F. Hybrid perfluorocarbon/carboxylic acid thin films via plasma deposition of hexafluoropropene and acrylic acid mixtures. Vacuum 2021, 184, 109933. [Google Scholar] [CrossRef]
- Roualdes, S.; Rouessac, V.; Durand, J. Plasma Membranes. Compr. Membr. Sci. Eng. 2010, 1, 159–197. [Google Scholar] [CrossRef]
- Jiang, Z.; Jiang, Z.J. Plasma techniques for the fabrication of polymer electrolyte membranes for fuel cells. J. Memb. Sci. 2014, 456, 85–106. [Google Scholar] [CrossRef]
- Song, M.K.; Kim, Y.T.; Fenton, J.M.; Russell Kunz, H.; Rhee, H.W. Chemically-modified Nafion®/poly(vinylidene fluoride) blend ionomers for proton exchange membrane fuel cells. J. Power Sources 2003, 117, 14–21. [Google Scholar] [CrossRef]
- Ahrens, T.; Kohlmann, J.; Ahrens, M.; Braun, T. Functionalization of fluorinated molecules by transition-metal-mediated C-F bond activation to access fluorinated building blocks. Chem. Rev. 2015, 115, 931–972. [Google Scholar] [CrossRef]
- Mastrorilli, P.; Monopoli, A.; Dell’Anna, M.M.; Latronico, M.; Cotugno, P.; Nacci, A. Ionic Liquids in Palladium-Catalyzed Cross-Coupling Reactions. In Ionic Liquids (ILs) in Organometallic Catalysis; Springer: Berlin/Heidelberg, Germany, 2013; pp. 237–285. [Google Scholar]
- Cotugno, P.; Monopoli, A.; Ciminale, F.; Cioffi, N.; Nacci, A. Pd nanoparticle catalysed one-pot sequential Heck and Suzuki couplings of bromo-chloroarenes in ionic liquids and water. Org. Biomol. Chem. 2012, 10, 808–813. [Google Scholar] [CrossRef] [PubMed]
- Monopoli, A.; Afzal, A.; Di Franco, C.; Ditaranto, N.; Cioffi, N.; Nacci, A.; Cotugno, P.; Torsi, L. Design of novel indium oxide supported gold nanocatalysts and their application in homocoupling of arylboronic acids. J. Mol. Catal. A Chem. 2014, 386, 101–107. [Google Scholar] [CrossRef]
- Calò, V.; Nacci, A.; Monopoli, A.; Cotugno, P. Heck reactions with palladium nanoparticles in ionic liquids: Coupling of aryl chlorides with deactivated olefins. Angew. Chem.-Int. Ed. 2009, 48, 6101–6103. [Google Scholar] [CrossRef]
- Prakash, G.K.S.; Jog, P.V.; Krishnan, H.S.; Olah, G.A. A domino approach (hydrolysis/dehydrohalogenation/heck coupling) for the synthesis of styrene sulfonate salts. J. Am. Chem. Soc. 2011, 133, 2140–2143. [Google Scholar] [CrossRef]
- Battace, A.; Zair, T.; Doucet, H.; Santelli, M. Heck vinylations using vinyl sulfide, vinyl sulfoxide, vinyl sulfone, or vinyl sulfonate derivatives and aryl bromides catalyzed by a palladium complex derived from a tetraphosphine. Synthesis 2006, 2006, 3495–3505. [Google Scholar] [CrossRef]
- Nardello, V.; Aubry, J.M.; Johnston, P.; Bulduk, I.; De Vries, A.H.M.; Alsters, P.L. Facile preparation of the water-soluble singlet oxygen traps anthracene-9,10-divinylsulfonate (AVS) and anthracene-9,10-diethylsulfonate (AES) via a Heck reaction with vinylsulfonate. Synlett 2005, 2005, 2667–2669. [Google Scholar] [CrossRef]
- Monopoli, A.; Calò, V.; Ciminale, F.; Cotugno, P.; Angelici, C.; Cioffi, N.; Nacci, A. Glucose as a clean and renewable reductant in the pd-nanoparticle-catalyzed reductive homocoupling of bromo- and chloroarenes in water. J. Org. Chem. 2010, 75, 3908–3911. [Google Scholar] [CrossRef]
- Zhang, X.; Fan, S.; He, C.Y.; Wan, X.; Min, Q.Q.; Yang, J.; Jiang, Z.X. Pd(OAc)2 catalyzed olefination of highly electron-deficient perfluoroarenes. J. Am. Chem. Soc. 2010, 132, 4506–4507. [Google Scholar] [CrossRef] [PubMed]
- Groβ, U.; Engler, G. Reaktionen von fluoralkenen II. Über reaktionen von F-hept-1-en mit s-nucleophilen. J. Fluor. Chem. 1985, 29, 425–430. [Google Scholar] [CrossRef]
- Padmapriya, A.A.; Just, G.; Lewis, N.G. A New Method for the Esterification of Sulphonic Acids. Synth. Commun. 1985, 15, 1057–1062. [Google Scholar] [CrossRef]
- Carretero, J.C.; Demillequand, M.; Ghosez, L. Synthesis of α, β-unsaturated sulphonates via the Wittig-Horner reaction. Tetrahedron 1987, 43, 5125–5134. [Google Scholar] [CrossRef]
- Milella, A. Plasma Processing of Polymers. In Encyclopedia of Polymer Science and Technology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2008; pp. 1–41. [Google Scholar] [CrossRef]

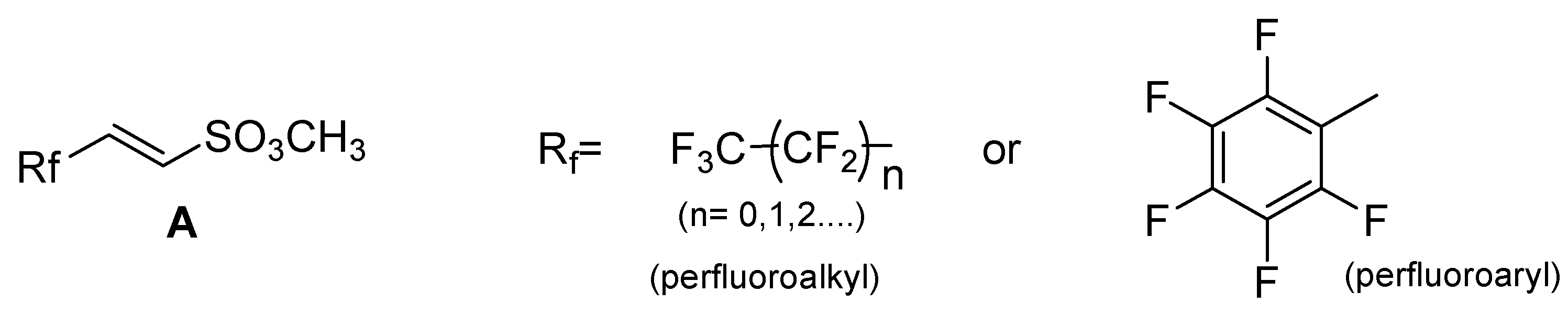
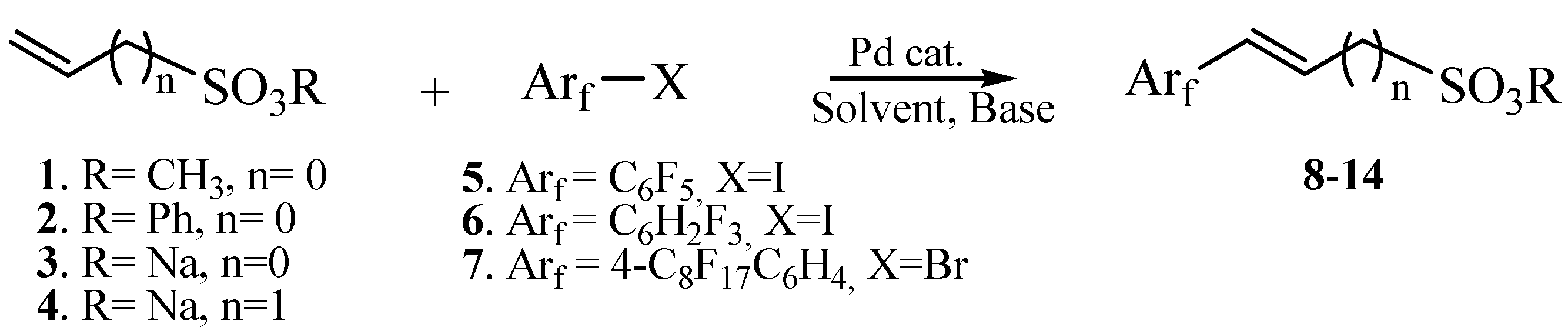
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).