Abstract
Neurological and neurodegenerative diseases are debilitating conditions, and frequently lack an effective treatment. Monoacylglycerol lipase (MAGL) is a key enzyme involved in the metabolism of 2-AG (2-arachidonoylglycerol), a neuroprotective endocannabinoid intimately linked to the generation of pro- and anti-inflammatory molecules. Consequently, synthesizing selective MAGL inhibitors has become a focus point in drug design and development. The purpose of this review was to summarize the diverse synthetic scaffolds of MAGL inhibitors concerning their potency, mechanisms of action and potential therapeutic applications, focusing on the results of studies published in the past five years. The main irreversible inhibitors identified were derivatives of hexafluoroisopropyl alcohol carbamates, glycol carbamates, azetidone triazole ureas and benzisothiazolinone, whereas the most promising reversible inhibitors were derivatives of salicylketoxime, piperidine, pyrrolidone and azetidinyl amides. We reviewed the results of in-depth chemical, mechanistic and computational studies on MAGL inhibitors, in addition to the results of in vitro findings concerning selectivity and potency of inhibitors, using the half maximal inhibitory concentration (IC50) as an indicator of their effect on MAGL. Further, for highlighting the potential usefulness of highly selective and effective inhibitors, we examined the preclinical in vivo reports regarding the promising therapeutic applications of MAGL pharmacological inhibition.
1. Introduction
Monoacylglycerol lipase (MAGL; EC 3.1.1.23) is a membrane-bound α/β serine hydrolase, which selectively metabolizes 2-monoacylglycerols [1]. Its main substrate is 2-arachidonoylglycerol (2-AG), an endogenous agonist of the cannabinoid receptors 1 and 2 (CB1R and CB2R) and a precursor of arachidonic acid (AA), and, consequently, of downstream-derived eicosanoids, thus interfering with the eicosanoid signaling [2]. 2-AG can be metabolized by multiple enzymes; however, MAGL is the main enzyme involved in its metabolism, regulating its hydrolysis rate into AA and glycerol [2].
2-AG is one of the most abundant endocannabinoids and plays an essential role in the regulation of many physiological processes. It modulates neuroplasticity and neuroprotection by mediating retrograde suppression of inhibitory or excitatory currents in GABAergic and glutamatergic synapses [3] by activating CB1R receptors [4]. Furthermore, CB1R receptor activation by 2-AG is associated with modulation of several other neurotransmissions: dopaminergic, noradrenergic, serotonergic, with consequences on behavior (e.g., food intake, addiction), memory and pain perception [1,5]. 2-AG appears to also regulate metabolism, cell proliferation, pain sensation, and reproductive and cardiovascular functions [6,7], by activating CB2R receptors and other targets (peroxisome proliferator-activated receptors, transient receptor potential vanilloid-1) [7] (Figure 1).
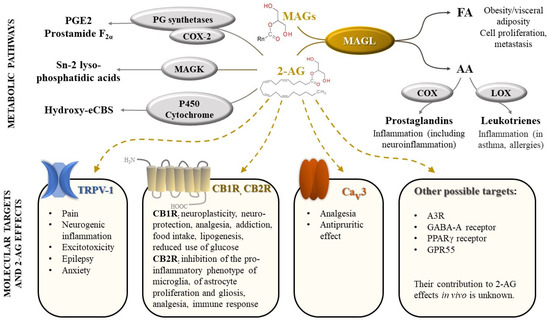
Figure 1.
Monoacylglycerol lipase (MAGL), an enzyme with central role in a complex biological network, regulating cannabinoid signaling and arachidonic acid metabolism. Legend: FA-fatty acids; AA-arachidonic acid; COX—cyclooxygenase; LOX—lipoxygenase; MAGs—monoacylglycerols (n = 8–18); 2-AG—2-arachidonoylglycerol; PG—prostaglandin; PGE2—prostaglandin E2; MAGK—monoacylglycerol kinase; eCBs—endocannabinoids; TRPV-1—transient receptor potential vanilloid type 1; CB1R—cannabinoid receptors 1; CB2R—cannabinoid receptors 2; Cav3 T-type calcium channel; A3R—adenosine receptor 3; GABA-A receptors- γ-Aminobutyric acid type A receptors; PPAR-γ receptors—Peroxisome proliferator-activated γ receptors; GPR55—G protein-coupled receptor 55.
Activation of immune cells (e.g., macrophages) leads to an increase in 2-AG production, which, in turn, decreases pro-inflammatory mediators and re-establishes homeostasis [8]. However, the main metabolite of 2-AG, AA, is well known for its involvement in inflammation, among other physiological processes, acting as a substrate for several enzymes such as cyclooxygenases (COX-1 and COX-2), thus being converted into pro-inflammatory prostaglandins (PGs) and thromboxanes [8,9]. MAGL appears to be the main enzyme involved in AA synthesis in the brain, liver, and lung [10,11].
In addition to 2-AG, MAGL hydrolyzes other monoacylglycerides, affecting the levels of free fatty acids, which later serve as a precursor pool for pro-tumorigenic signaling lipids [1].
Therefore, the inhibition of MAGL appears to be a promising therapeutic target for various pathologies, such as neurodegenerative and neuropsychiatric diseases, chronic pain or cancer [1,12]. In recent years, a plethora of MAGL inhibitors were synthetized, often containing reactive carbamate or urea moieties, resulting in an irreversible inactivation of the enzyme by covalent modification of the active serine residue, an interaction well-described in previously published reviews [1,13].
The main advantage of covalent inhibition is that it offers a sustained pharmacological response [14]. Some recent identified irreversible inhibitors present a good pharmacokinetic profile and are highly effective in reducing inflammation in vivo [15,16]. However, chronic administration of irreversible inhibitors was associated with negative in vivo effects, such as CB1R receptor downregulation and physical dependence. Therefore, the focus of recent studies was the development of reversible inhibitors, lacking these unwanted effects and with an optimal pharmacokinetic profile.
In this review, we aim to revise the latest data regarding MAGL inhibitors, focusing on their most important structural features. We highlight the results of in-depth chemical, mechanistic and computational studies on MAGL inhibitors, and the most promising therapeutic uses reported in recent in vivo studies, focusing on papers published between 2015 and 2021—a period of intense research in this emerging field.
2. Molecular Characterization of MAGL and Mechanism of Catalysis
Biologically organized as a dimer, this 33 kDa enzyme possesses three main structural features: a catalytic triad, a nucleophilic elbow, and an oxyanion hole. The core consists of a β-sheet with seven parallel and one antiparallel strands, surrounded by six α-helices, with the active site formed by three amino acid residues: Ser122, Asp239 and His269, which are located between β-strands and α-helices of the α/β-hydrolase fold [17,18], at the bottom of the long channel under the lid domain of the enzyme [19].
The serine residue (Ser) is placed in the conserved sequence Gly-His-Ser-Met-Gly, representative of α/β-serine hydrolases, also known as “a nucleophilic elbow”, which basically forms a pocket, connecting to the exterior via a connection channel. The core is covered by a highly variable domain—a cap [13]. Inside the cap domain and lining the active site access, a highly lipophilic α-helix allows anchoring of the enzyme to the cell membrane, thus making it possible for the protein to recruit its lipophilic substrates. Several hydrophobic residues cover the connection channel, interacting with the arachidonoyl moiety of 2-AG and ensuring substrate specificity [12,20]. This flexible hydrophobic tunnel becomes more polar towards the catalytic site, allowing the accommodation of the glycerol group. Another narrow tunnel, perpendicular to the connection channel, connects the catalytic site with the outside of the protein and is coated by Pro178, Ser181, Leu184, Tyr194, Arg202 and Ala203, allowing the glycerol moiety, released after substrate hydrolysis, to exit [12].
MAGL selectively hydrolyses 2-monoacylglycerols with different fatty acid chain lengths (from C8:0 to C18:0) and saturation. MAGL preferentially hydrolyses unsaturated compared to saturated substrates (e.g., arachidonoyl glycerol vs. palmitoyl glycerol) [12]. It does not cleave diacylglycerols, triacylglycerols [21], cholesterol esters or prostaglandin glycerol esters [22].
The MAGL catalytic mechanism consists of two different phases: the binding of the substrate and the hydrolysis of the ester bond [12], and involves two main reactions: acylation and diacylation. Catalysis starts by an acylation [23]: once the substrate reaches the active site, Ser122 triggers a nucleophilic attack to the carbonyl of the substrate, forming a complex with it [16]. This attack is facilitated by an acid-base mechanism in which Ser122 is activated by a proton transfer that starts when Asp239 deprotonates His269, which takes another proton from the hydroxyl group of Ser122 [12]. Consequently, the hydroxyl residue of the serine, with subsequently increased nucleophilicity, attacks the carbonyl group of the substrate [23]. The tetrahedral intermediate formed during the transition state is placed in a cavity of the enzyme named the “oxyanion hole” [16]. The oxyanion hole stabilizes the charge distribution and reduces the state energy of the tetrahedral intermediate by forming at least two hydrogen bonds [23]. The release of glycerol as the leaving group from the tetrahedral intermediate leads to an acyl-enzyme complex [12]. Then, the diacylation step takes place with the nucleophilic attack of an activated water molecule to the carbonyl group of the acylated complex, releasing AA and regenerating the enzyme [12,23].
3. Development of Pharmacophore Models for MAGL Inhibitors
To date, the usage of computer-aided drug design (CADD) tools has been widely preferred in the rational drug discovery of MAGL inhibitors, with several research groups employing CADD approaches on several chemical scaffolds to discover and identify novel MAGL inhibitors. Afzal et al. proposed the 3D QSAR pharmacophore model for MAGL inhibitors using a ligand-based pharmacophore generation [24]. Accordingly, the pharmacophore comprised three features: one hydrogen bond acceptor, one positive center (e.g., piperazine) and three aromatic rings. The same group further investigated the structural features needed to inhibit MAGL. Using hybrid ligand and structure-based approaches, they established three key essential features for MAGL inhibition: an amphiphilic benzene-fused heterocyclic ring (to form a π–π stacking interaction with Tyr194 and also interact with the polar and non-polar amino acids) linked with a bulky hydrophobic fragment (to fill up the hydrophobic space and to interact with the hydrophobic amino acids) connected through a linker having a carbonyl functional group, either in cyclic or acyclic amide form, or in a heterocyclic system (to form an H-bond with one or more key amino acid residues, Ala51, Met123 and Ser122 of the oxyanion hole in the active site) [25].
Examination of the MAGL X-ray structure, combined with the analysis of the structural elements of diphenylmethane inhibitors, led to the development of a novel 3D pharmacophore model, focused on the correct distance and reciprocal orientation of the aromatic rings within the inhibitor structure. The key feature is the longer distance between the centroids of the aromatic systems (~7 Å). The spatial arrangement of the aryl moieties, constrained by a dihedral angle of 119.4°, is critical to maximize the interaction with the hydrophobic pockets of the enzyme, and provides the correct accommodation of the molecule within the catalytic site [26].
Jha and co-workers (2021) developed a pharmacophore-based virtual screening (VS) protocol aimed at the identification of new reversible MAGL inhibitors, which was based on the latest X-ray structure of MAGL in a complex with a reversible non-covalent inhibitor [27]. Accordingly, the pharmacophore comprises five mandatory features (four hydrogen-bond acceptors and a hydrophobic feature—one aromatic ring group) and three optional hydrophobic features (a chlorine, a terminal aromatic ring group and a thiazole ring). This pharmacophore model provided high predictability for estimating the activities of a variety of compounds. When employing all eight features, the virtual screening results provided 5707 new hits from amongst 276,150 molecules, thereby proving that the model is highly selective. Moreover, the authors highlighted the importance of the structural water as an indirect anchoring point to MAGL binding site [28].
From the analysis of available data concerning MAGL reversible inhibition, the key pharmacophoric elements that appear to be important for inhibitory activity are: (a) a carbonyl group able to interact in the oxyanion hole of the enzyme, (b) an aromatic portion also comprising a heterocyclic nucleus involved in van der Waals interactions in a closed hydrophobic region of the binding site, and (c) a second aromatic portion, which interacts in the opening [29].
4. MAGL Inhibitors
During the past decade, various irreversible and reversible MAGL inhibitors were discovered. Although the first-generation inhibitors were capable of blocking MAGL in in vitro pharmacological assays and showed beneficial effects in preclinical tests, their lack of selectivity led to various side effects, such as CB1R desensitization, tolerance and physical dependence. Nevertheless, the structure optimization of these inhibitors (e.g., insertion of hexafluoroisopropanol as a leaving group) inspired the further development of selective MAGL inhibitors, with a better safety profile [13,30].
In this review, we focus on presenting the compounds designed during the past five years, and derivatives with proven high selectivity for MAGL, rather than other enzymes involved in endocannabinoid processing, which hold promise for optimal pharmacokinetics and improved efficacy in disorders, such as neurodegenerative diseases, chronic pain and chronic inflammatory diseases. Although we used the half maximal inhibitory concentration (IC50) as an indicator of each compound’s effect on MAGL, one should consider that this parameter is only a relative indicator of the inhibitor efficacy because testing protocols often differ [31].
4.1. Covalent Irreversible Inhibitors
Irreversible well-characterized inhibitors (Figure 2) contain an electrophilic moiety which inactivates the enzyme by bonding covalently, typically to the reactive side chain of an amino acid in the active site [1]. The electrophilic fragment is often called a warhead in medicinal chemistry and its choice can significantly reduce the toxicity and increase the selectivity towards similar targets [32]. The most used warheads in the development of MAGL covalent inhibitors are carbamate, urea, maleimide or oxadiazolone [1].
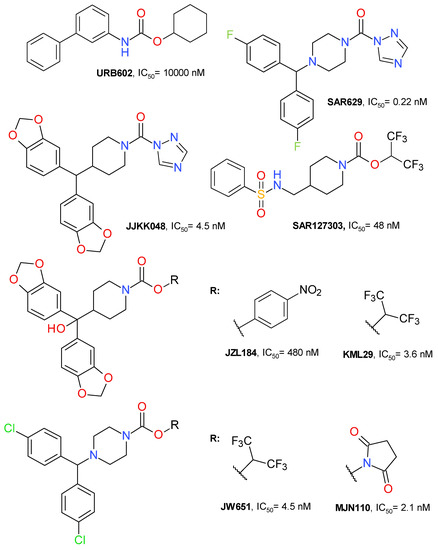
Figure 2.
Representative irreversible MAGL inhibitors.
The first selective MAGL inhibitor identified was JZL184, a 4-nitrophenyl carbamate derivative. KML29 is an analogue of JZL184 with hexafluoroisopropyl alcohol (HFIP) as the leaving group. This change improved the inhibitory potency and reduced the off-target effects. JW651 is also a HFIP carbamate and is structurally similar to KML29. SAR127303 is a highly potent HFIP piperidine carbamate inhibitor of MAGL. MJN110 is the N-hydroxysuccinimidyl (NHS) analogue of JW651 with a superior potency than KML29 [33,34]. The chemical structures of all these irreversible inhibitors are depicted in Figure 2.
The inhibitors must be highly lipophilic in order to interact with the target MAGL binding pocket, which is correlated with poor water solubility. This is a challenge for further development and leads to safety risks due to increased metabolic clearance/off-target activities [1]. Newer derivatives, reviewed in this paper, are designed by carefully selecting the warhead and by establishing an equilibrium between lipophilicity and hydrophilicity for an optimal pharmacokinetic profile.
A clear, simple classification of the MAGL inhibitors is difficult to establish. Most irreversible inhibitors are based on the carbamate scaffold. One criterion for classification may be the nature of the alcohol bounded by the carbamate unity. The research on the mechanism by which carbamates inhibit MAGL highlighted the importance of a low pKa value for the alcohol; HFIP is the most used.
HFIP has a pKa of 9.3 and its structure is similar to that of the glycerol moiety of AG, the natural MAGL substrate. HFIP carbamate derivatives have good aqueous chemical stability over a pH range of 2 to 8. HFIP is released after MAGL’s carbamoylation and its safety profile is supported by the clinical experience with sevoflurane [35]. Administered as a volatile anesthetic, sevoflurane is mainly metabolized to fluoride and HFIP, which is rapidly glucuronidated and excreted as HFIP-glucuronide in urine [36].
In general, the HFIP carbamate offers a high selectivity towards serine hydrolases, but there are some common off-targets that include the serine hydrolases fatty acid amide hydrolase (FAAH), α/β hydrolase domain containing 6 (ABHD6), phospholipase A2 group VII (PLA2G7), and the carboxyesterases (CES) [37]. ABHD6 is connected to the endocannabinoid signaling by its capacity to hydrolyze 1-arachidonoyl-glycerol (1-AG) and 2-AG [38].
Another important scaffold, although not used as often as the carbamate template, is represented by the urea derivatives. SAR629 (Figure 2) is a derivative of urea substituted with piperazine and triazole that inhibits MAGL at nanomolar concentrations by forming a carbamylated adduct through Ser122 residue [12,39]. JJKK048 (Figure 2) is also a triazole urea derivative with a structure that is highly similar to those of carbamate derivatives JLZ184 and KLM29. It potently inhibits MAGL with an IC50 value of 0.36 nM and is selective in rapport with FAAH and ABHD6 [39]. Not all triazole ureas inhibit MAGL in an irreversible manner [40].
4.1.1. Azetidine HFIP Carbamates
Butler et al. (2017) used azetidine as a linker between the HFIP carbamate moiety and the lipophilic scaffold [14]. A secondary heterocyclic ring was added directly to the azetidine ring, in which the substitution in the 3 position was essential to provide a linear configuration. All the used heterocycles—oxadiazole, pyrimidine, piperazine and pyrazole—provided potent MAGL inhibitors, but the latter was chosen for development (Figure 3). The use of the pyrazole ring to link the active electrophilic group with a hydrophilic tail led to an increased inhibitory potency and optimized ligand efficiency and fit quality values [14].
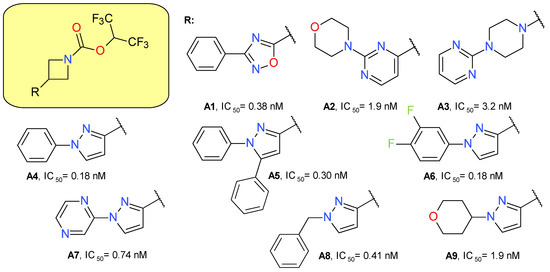
Figure 3.
Irreversible MAGL inhibitors—azetidine carbamate derivatives.
The pyrazole derivative A4 was tested at both 1 and 10 µM, showing significant inhibition against ABHD6, CES 1 and 2, and PLA2G7. The inhibitory effects on FAAH were observed only for the 10 µM concentration.
The administration of compound A4 in mice (10 mg/kg subcutaneously) increased 2-AG brain concentration up to five-fold and the effect duration was an average of 8 h. The major disadvantage of the compound was the rapid clearance, leading the research group to hypothesize that ring cores larger than azetidine will increase the stability of the inhibitor and of its adduct with MAGL. The piperidine analog of A4 proved to have a prolonged pharmacodynamic effect [14].
4.1.2. Piperazine HFIP Carbamates
The piperazine HFIP carbamate scaffold was previously used to develop JW651, a benzhydrylpiperazine derivative that potently inhibits MAGL with an IC50 of 38 nM, with no significant effect on other brain serine hydrolases at concentrations up to 10 μM. Oral administration in C57Bl/6J mice at 5 mg/kg augmented several-fold the brain levels of 2-AG and reduced accordingly the AA levels without affecting the N-arachidonoylethanolamide (AEA) concentration [37].
Cisar et al. (2018) designed and synthesized various biaryl- and monoaryl-methyl analogues of JW651 as potent MAGL inhibitors. The substituted pyrazolylmethyl piperazines derivatives presented a potent inhibitory effect on MAGL and favorable lipophilicity, but a low selectivity towards ABHD6 and PLA2G7. The structure B1 represents the most potent MAGL inhibitor of these series (Figure 4). The replacement of the pyrazolylmethyl fragment with benzyl maintained MAGL inhibitory potency but reduced the selectivity towards ABHD6. The introduction of a morpholine fragment (B2) improved the selectivity profile. The pyrrolidine benzyl derivative B3 demonstrated both high potency and selectivity for MAGL. Compounds B2 and B3 completely inhibited MAGL action in the brain of mice administered a 5 mg/kg dose by oral gavage [35].
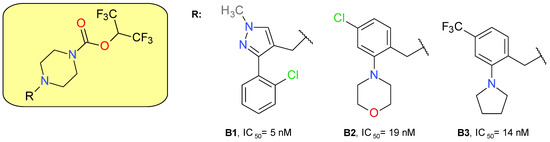
Figure 4.
Irreversible MAGL inhibitors—piperazine carbamate derivatives.
A mass spectrometry-based assay on B3, named ABX-1431, and recently Lu AG06466, confirmed the irreversible inhibition of MAGL by the carbamoylation of the catalytic Ser122 residue [35]. B3 is a lipophilic basic amine molecule, but its activity on the hERG channel is weak (IC20 = 7 μM) [41].
4.1.3. Azabicyclo[3.1.0]Hexane Trifluoromethyl Glycol Carbamates
McAllister et al. (2018) developed a new series of MAGL inhibitors based on the pyrazole azetidine scaffold of compounds such as A4 by replacing the rapidly hydrolysable azetidine with azabicyclo[3.1.0]hexane. In order to optimize the compounds’ solubility and their pharmacokinetic parameters, various types of carbamates were attached to the bicyclic ring (Figure 5). Compound C1, a HFIP carbamate, demonstrated the best inhibitory potency on MAGL and a very good selectivity in rapport with FAAH, but a very high lipophilic character and a low solubility. The analog p-nitrophenol carbamate (C2) produced a potent inhibitory effect on both MAGL and FAAH [42]. The low selectivity of C2 towards FAAH confirms previous results on p-nitrophenol carbamates such as JZL184 [43]. The use of NHS carbamate (C3) decreased the inhibitory potency but provided very good selectivity on MAGL compared to FAAH, CES 1 and 2, and ABHD6.
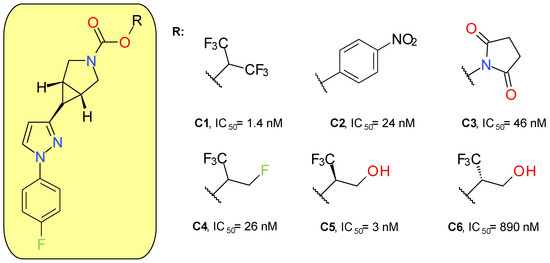
Figure 5.
Irreversible MAGL inhibitors—azabicyclo[3.1.0]hexane trifluoromethyl glycol carbamates and related structures.
Applying small changes to HFIP provided compounds with better pharmacokinetic profiles. The authors underlined that branching in the leaving group pocket is an important structural feature for maintaining selectivity compared to other enzymes involved in endocannabinoid processing. Compounds C5 and C6 were designed to better mimic 2-AG, the natural enzyme substrate. The racemic mixture of C5 and C6 demonstrated very good potency (IC50 = 6 nM) and selectivity. The major difference between the activities of the two enantiomers indicates that, in addition to carbamate reactivity, the binding interactions between the enzyme and the leaving group are also important factors [42].
The lead compound of this series (C5) was named PF-06795071 and was assayed on a panel of related serine hydrolases displaying no significant effects, with the exception of CES 1 (80% inhibition at 10 μM). Following intravenous administration in rats and dogs, C5 displayed a short half-life of approximately 1 to 2 h [42]. A 18F-labeled analogue of C5 was synthesized and administered to Sprague Dawley rats, demonstrating a good brain uptake and distribution [44].
4.1.4. Azetidone Triazole Ureas
A series of new urea MAGL inhibitors were developed using as a template JJKK048 and SAR629. The trans-3,4-diarylsubstituted β-lactam structural motif was used to obtain an optimal configuration for MAGL inhibition (Figure 6). 4-Fluorophenyl and the methylene-3,4-dioxyphenyl moieties provide the key hydrophobic interactions with the enzyme, which is reflected in an increased inhibition potency of human MAGL. Compound D1 presented the best potency on MAGL and close to 900-fold selectivity compared to FAAH. Its enantiomer, the 3S, 4R isomer, displayed eight-fold lower potency on MAGL and less selectivity than FAAH. The exchange of the fluorine atom with a methoxy group (D2, D3) slightly reduced the MAGL potency and significantly reduced the selectivity towards FAAH [26].
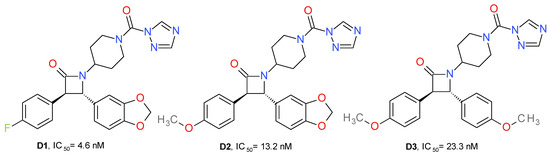
Figure 6.
Irreversible MAGL inhibitors—trans-3,4-diarylsubstituted β-lactam derivatives.
Potency is also related to the pKa of the leaving group’s conjugated acid. Accordingly, the triazole moiety offered the best pharmacokinetic profile (pKa = 10). This can be observed because the imidazole analogue of D1 presented no significant inhibitory effects on MAGL. Similarly, the HFIP and p-nitrophenyl carbamates analogues of D1 had lower potencies [26].
4.1.5. Benzisothiazolinone Derivatives
In the structure of MAGL there are three important cysteine residues—Cys242, Cys201 and Cys208—that regulate the enzyme’s catalytic activity. Cys242 is situated near the catalytic Ser122 and represents a target of several covalent inhibitors of MAGL. The group of maleimide derivatives irreversibly inhibit MAGL by forming a covalent S-alkylated adduct through a Michael addition reaction. N-arachidonoyl maleimide has a medium inhibitory effect on MAGL, with an IC50 value near 1 μM [30]. Disulfiram and several related disulfide derivatives also target the Cys residues of MAGL, but the mechanism consists of the formation of disulfide bonds [45].
Based on the structure of octhilinone and octylbenzisothiazolinone (E1), two potent irreversible inhibitors of MAGL with IC50 values of 88 and 20 nM, respectively [13], Castelli et al. (2020) designed a series of N-substituted benzisothiazolinone derivatives. Compound E2 (Figure 7) potently inhibits the activity of MAGL. This effect was significantly lower on MAGL mutated at various Cys residues. The authors demonstrated that E2 forms a reducible disulfide bond with Cys201, and not Cys242. Structurally related compounds irreversibly inhibited the bacterial sortase A enzyme by the same mechanism, i.e., the formation of a disulfide bond with the catalytic Cys residue [13].
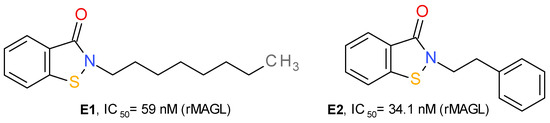
Figure 7.
Irreversible MAGL inhibitors—benzisothiazolinone derivatives.
4.2. Reversible Inhibitors
The first reported reversible MAGL inhibitors were euphol and pristimerin; however, the two terpenoids lack selectivity, and thus were not considered as potential scaffolds for further development of MAGL inhibitors [46].
The first developed reversible, non-competitive MAGL inhibitors, benzo[d][1,3]dioxol-5-ylmethyl 6-([1,1′-biphenyl]-4-yl)-hexanoate and (4-(4-chlorobenzoyl)piperidin-1-yl)(3-hydroxyphenyl)methanone, were effective in vivo (they improved the clinical outcome of multiple sclerosis in a mouse model of autoimmune encephalomyelitis [47] and relieved the neuropathic hypersensitivity induced by oxaliplatin [48], respectively), with no impairment of motor and cognitive functions, thus confirming the hypothesis that reversible MAGL inhibitors possess a better safety profile than irreversible inhibitors [47,48].
4.2.1. Salicylketoxime Derivatives
Bononi et al. a used salicylketoxime scaffold, with a peripheral phenolic ring substituted with a fluorine atom in a position ortho to the hydroxyl group [46]. The potency of inhibition increased in direct proportion with the number of methylene units in the alkyl group of the ketoxime moiety, up to nanomolar inhibition values (Figure 8). The highest inhibitory activity was reported for the compounds with a linear saturated ketoximic alkyl chain, which can be explained by the structural similarity to the endogenous substrate 2-AG. Inserting simple aromatic groups in the ketoxime moiety (e.g., phenyl ring or a benzyl group) decreased the inhibitory effect. The most active compounds, F1 and F2, were shown to possess good antiproliferative potency against cancer cells, particularly in ovarian and breast cancer [46].
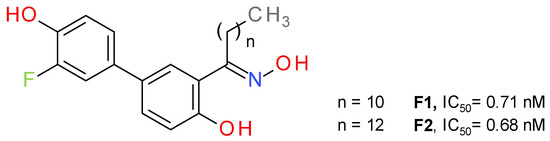
Figure 8.
Reversible MAGL inhibitors—salicylketoxime derivatives.
4.2.2. Piperidine Derivatives
Piperidine derivatives possessing an amidic carbonyl group are promising scaffolds for the development of reversible MAGL inhibitors. This moiety forms two H-bonds with the nitrogen backbone of Ala51 and Met123. The piperidyl is directed toward an open cavity of the protein and interacts with Leu148, Leu213 and Leu241 [49]. Various derivatives using this scaffold were identified and their inhibitory efficacy was assessed.
Granchi et al. (2016) identified 1-benzoylpiperidine derivatives as candidates for developing selective MAGL inhibitors. The first identified inhibitor was a piperidine molecule substituted in position 4 with 4-chlorobenzoyl and with 4-methoxybenyoyl at its N-atom. The replacement of the piperidine central ring with a piperazine abolished the effect on MAGL. The substituent in position 4 was maintained and several modifications at the N-benzoyl scaffold resulted in 4-(4-chlorobenzoyl)piperidin-1-yl)(4-hydroxyphenyl)methanone (G1), with an IC50 of 840 nM (Figure 9). It possessed reversible interaction properties and antiproliferative activity in cancer cells [50].
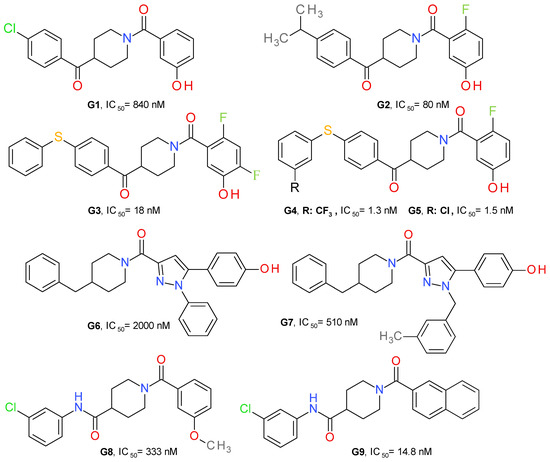
Figure 9.
Reversible MAGL inhibitors—piperidine derivatives.
Further development of more potent inhibitors starting from the above-mentioned lead, was reported. A fluorine atom in para to the phenolic hydroxyl group on the amidic phenyl ring or to the amide carbonyl group appears to be beneficial for MAGL inhibition potency, such as compound G2. The methoxylated analog of G2 displayed a higher IC50 value (2600 nM) compared to G2, thus confirming that the hydroxyl group is essential for the interaction with MAGL. The docking studies demonstrated the role of the hydroxyl group by bonding to Glu53 and His272 [51].
The class of 1-benzoylpiperidine-based MAGL inhibitors was expanded and several structure-activity relationships were deduced. The exchange of the fluorine atom in G2 with other groups, such as triflouromethyl, nitro or amino, proved to be detrimental for the MAGL affinity. The substitution of the chlorine in the structure of G1 with benzyl or phenylsulfide, but not phenylsulfone, improved the compounds’ potency. The addition of two fluorine atoms to the N-benzoyl fragment led to compound G3 (Figure 8), a potent and reversible MAGL inhibitor with low effects on FAAH and ABHD6 [52]. Compounds G4 and G5 were obtained after testing various substituents on the phenylsulfide fragment of G3 [16].
Tabrizi et al. identified the 1,5-diphenylpyrazole-3-carboxamide scaffold as an option for further development. Several rounds of structural investigations were carried out by maintaining a fixed central pyrazole core and varying the substituent present on the nitrogen atom (phenyl or methyl group) and the fragment linked to the carbonyl group in position 3. Derivatives with a p-hydroxyl group on the benzene attached in position 5 possessed higher inhibitory activity compared with other substitution patterns. The hydroxyl permits the formation of an H-bond with the oxygen of Pro178. The best result was achieved by linking the carbonyl with the nitrogen of 4-benzylpiperidine, as can be seen in the structure of compound G6 (Figure 9). The elimination of the p-hydroxyl or its replacement with a chlorine atom reduces the potency, demonstrating its role in the inhibition of MAGL. The next optimization stage focused on changing the 1-pyrazole substitution with different aromatic moieties, such as the benzyl, phenylethyl or phenylpropyl. Compound G7 emerged as the most active of this series of derivatives. The docking studies indicated that m-methylbenzyl connected to pyrazole is oriented to the surface of the binding site and forms lipophilic interactions with Ala151 and Leu241. The compound can be considered to be a structural analogue of G1 [48].
The compound G7 is relatively selective towards other endocannabinoids, degrading enzymes producing a 7% inhibition of FAAH at 10 μM and presenting no significant binding affinities the cannabinoid receptors.
Zhi et al. (2020) designed amide derivatives of 1-benzoylpiperidine as selective and reversible MAGL inhibitors. Compound G8 (Figure 9) emerged as a first lead structure. The docking studies indicated the importance of the carbonyl in position 1 of the piperidine ring by forming two H-bonds with the nitrogen backbone of Ala51 and Met123. The chlorine atom showed hydrophobic interaction with Val191 and Tyr194, and its presence is correlated with an increased inhibitory potency. When the chorine was replaced with a hydroxyl group in the structure of G8, the inhibitory activity was reduced significantly.
The optimization process led to a further increase in the inhibitory effect, particularly when a larger hydrophobic group was attached to the carbonyl (naphtalen vs. benzene), allowing a better occupancy of the larger hydrophobic cavity and the establishment of various lipophilic interactions. The m-chloroaniline fragment of G9 bonds to the hydrophobic subpocket enclosed by side chains of Val191, Tyr194, Val270 and Lys273, whereas the naphthalene moiety forms lipophilic interactions with Leu148, Leu213 and Leu241 [49]. G9 was selective towards MAGL and presented no significant binding affinities with the cannabinoid receptors. It demonstrated a very low toxicity in the mouse fibroblasts (L929) assay.
4.2.3. Pyrrolidone Derivatives
A high-throughput screening identified 1-(4-phenoxyphenyl)-2-pyrrolidone as a weak inhibitor of MAGL. Adding a pyrimidinyl piperazine fragment to the 2-pyrrolidone ring led to compound H1 having a significantly increased inhibitory effect. The pyrimidinyl piperazine scaffold was previously used in other types of MAGL inhibitors, such as the compound A3 (Figure 3). Using a structure-based drug design approach, a novel series of reversible MAGL inhibitors was developed, starting from the lead piperazinyl-pyrrolidin-2-one core [27].
Changing the pyrimidine ring with benzene or benzoyl proved to be unfavorable. The 2-thiazole-carbonyl fragment provided an approximately 3.5-fold increase in potency. This fragment was selected to occupy the amphiphilic pocket of the enzyme and used in further investigations of this series. The next step was focused on the nature of the N-pyrrolidine substituent. The conformational change of 4-phenoxyphenyl to its 3-phenoxyphenyl isomer improved the inhibitory effect on MAGL. Among the derivatives with substituents at position 3 of the central benzene ring, the chloro-derivatives showed the best profile. Despite the high potency of compound H2 (Figure 10), it showed a low metabolic stability. To improve the metabolic stability, a series of successive structural modulations led to the optimal biaryl moiety, the 3-fluoro-5-(2-methylpyridin-3-yl)phenyl fragment. The authors returned to the initial pyrimidinyl piperazine scaffold and obtained the compound H3 [27].
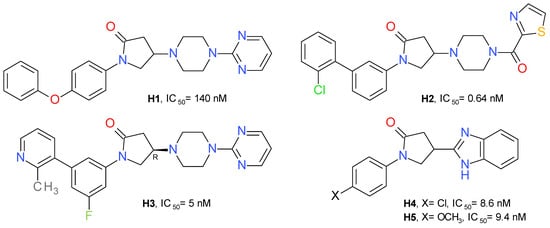
Figure 10.
Reversible MAGL inhibitors—pyrrolidone derivatives.
Oral administration of H3 (10 mg/kg) lead to a 25% decrease in AA concentration and a 340% increase in 2-AG in the brain of mice, compared to the control [27].
A series of variously substituted N-phenyl-pyrrolidin-2-one linked with benzothiazol or benzimidazole was synthesized and assayed on human MAGL. The benzimidazole derivatives produced better inhibition on MAGL. Compound H4 determined a potent inhibition on MAGL with an IC50 value of 8.6 nM, and its methoxy analogue, compound H5 (Figure 10), presented a similar inhibitory profile, but a better selectivity towards FAAH [53]. Structurally similar compounds were prepared linking benzoxazole in the position 4 of the pyrrolidone scaffold. The best inhibitory effects were observed for the compounds substituted at the nitrogen atom with 4-nitrobezene and 4-benzenesulfonamide [54].
4.2.4. Azetidinyl Amides
Zhu et al. (2020) developed a series of 3-piperazinyl-azetidine diamides with potent inhibitory effects on MAGL. The derivatives containing a 2-thiazole-carbonyl fragment connected to piperazine were, in general, more active than the compounds having a benzoyl fragment. The lipophilicity of the azetidinyl amide substituent influenced the MAGL inhibitory potential. The best compound was chosen based on the IC50 value and the percentage 2-AG brain accumulation measured in homogenized rat brain incubated with 1 μM of each test substance. Compound I1 (Figure 11) has little effect on FAAH (IC50 = 4 μM) and no significant interaction with receptors CB1R or CB2R.
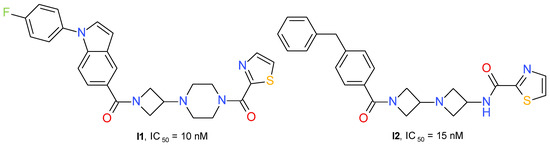
Figure 11.
Reversible MAGL inhibitors—azetidinyl amide derivatives.
A structurally similar series was developed using the diazetidinyl diamide scaffold. The compounds can be considered to be 3-aminoazetidine equivalents of the piperazine derivatives. The thiazole derivatives group presented higher potency in the MAGL enzyme assay than the corresponding analogs containing a phenyl fragment. The thiazole-amide of I2 (Figure 11) forms a hydrogen bond to the side chain guanidine group of Arg57 and a π-π stacking interaction with Tyr194. The existence of an azetidine-amide carbonyl allows access into the oxyanion hole of MAGL and forms a hydrogen bond with the backbone amide NH of Met123, adjacent to the catalytic Ser122 [55].
4.2.5. Various Structures
Dato et al. (2020) developed various compounds (Figure 12) targeting MAGL and presented ω-quinazolinonylalkyl aryl ureas as a new class of inhibitors of MAGL, exhibiting IC50 values in the range of 20–41 µM. The compound J1, 1-(3,5-bis(trifluoromethyl)phenyl)-3-(4-(4-oxo-3,4-dihydroquinazolin-2-yl)butyl)urea, was the most active inhibitor. This class of compounds was shown to interact with MAGL in a reversible manner, exhibiting a mixed-type inhibition with a predominant competitive behavior [56]. 2-sulfonacetamide (J4) and squareamide (J3) derivatives possessed lower IC50.
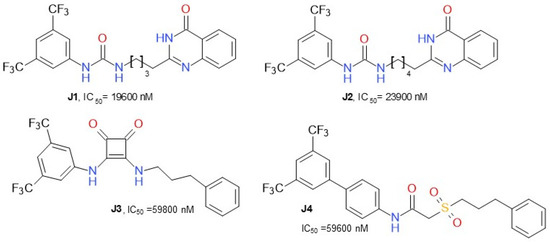
Figure 12.
Reversible MAGL inhibitors—various structures.
5. Potential Therapeutic Applications of MAGL Inhibition
As presented above, MAGL cleaves 2-AG and other monoacylglycerols [12,16], thus influencing the levels of several other lipid molecules with pro-inflammatory and pro-neoplastic functions. In addition, 2-AG activates several other receptors, leading to intricate, interconnected signaling pathways [12]. Consequently, MAGL is emerging as a multifaceted therapeutic target for a steadily increasing list of maladies, some of which are currently lacking targeted or highly efficient curative treatments [12,16].
MAGL inhibition is associated with neuroprotective effects [12,16], partly by reducing central pro-inflammatory mediators’ levels (discussed below), and partly by enhancing 2-AG activation of CB1R/ CB2R receptors and of down-stream signaling pathways [57].
Because it catalyzes the formation of AA from 2-AG, MAGL can be included in the category of pro-inflammatory enzymes. As a major cerebral source of AA, it is linked to the synthesis of prostaglandin E2 (PGE2) in both basal and pro-inflammatory settings. Mice lacking MAGL exhibited highly elevated brain 2-AG levels with concomitant decreases in arachidonic acid levels, and its oxidative metabolites PGE2 and PGD2, indicating MAGL is a key player in neuroinflammatory responses [10,58]. Chronic inflammation is a central process involved in a high number of metabolic disorders, neurodegenerative and neurological, malignant diseases, and autoimmune diseases [59].
CB1R activation modulates neurotransmitter release, neurogenesis, and synaptic plasticity, and affects neuronal functions through interaction with glial cells. 2-AG can activate astrocytic CB1R receptors, leading to neurotransmitter release, thus modulating neuronal functions beyond the originating synapse [60]. In addition, CB1R receptor activation was shown to activate the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt)/mammalian target of the rapamycin (mTOR) complex 1 pathway, which, in turn, induces brain-derived neurotrophic factor (BDNF) expression [57]. Moreover, the activation of CB1R receptors (by 2-AG) can induce autophagy, facilitating the elimination of protein aggregates [61].
CB2R activation decreases microglial activation and neuroinflammatory response [62]. Furthermore, 2-AG was shown to modulate other substrates such as peroxisome proliferator-activated receptors (PPARs, NR1C3) [63,64] or the G-protein receptor 55 (GPR55) [65], receptors found to reduce glial reactivity, and consequently local inflammatory events.
Genetic animal models showed MAGL deletion inhibits the progression of neurodegenerative diseases such as Huntington disease [66], Parkinson disease [10] and Alzheimer disease [67].
The elements of the cannabinoid system are expressed almost ubiquitously throughout nociceptive pathways, indicating the system modulates nociceptive signaling [68]. Genetic deletion of MAGL significantly increases central and peripheral 2-AG, leading to the downregulation and the desensitization of central and peripherally expressed CB1R receptors. CB1R desensitization was observed in brain areas controlling emotional and stress-related states, pain sensation, memory and learning, whereas brain areas associated with motor function were not affected by it. Consequently, genetic mouse models of MGL deficiency do not show analgesic properties [69,70]. By comparison, partial pharmacological MAGL inactivation resulted in significant analgesic effect in various models of pain, as further discussed below.
Modulation of endocannabinoids’ levels may offer an alternative for the treatment of psychiatric disorders, as a consequence of CB1R-mediated reduction of neuroinflammation and of the dopaminergic system modulation [71]. Neuronal and glial-derived endocannabinoid-signals are associated with cognitive dysfunction and with the pathogenesis of epilepsy [58], making MAGL a pharmacological target for epilepsy treatment.
MAGL is involved in energy balance by mobilizing cellular lipid stores in adipose and other tissues [72] and by regulating 2-AG concentration [17]. CB1R activation by 2-AG determines energy accumulation [17]. Centrally, it potentiates hypothalamic orexigenic pathways, and peripherally it enhances fat uptake in adipose tissue, increases de novo lipogenesis in the liver and decreases energy expenditure in muscle [17].
In diet-induced obesity models, MAGL ablation prevented the development of glucose intolerance and insulin resistance [73]. No changes in the body weight of MAGL−/− mice after 12 weeks of very high-fat feeding were observed, suggesting the desensitizing effects of cannabinoid receptors [73]. The results were further confirmed by other authors, who also underlined the liver content of certain species of saturated and unsaturated MAGs were highly enriched in MAGL−/− mice [72,74]. In addition to producing a leaner phenotype, global MAGL deletion led to an improved serum metabolic profile [72].
Switching to a lipogenic phenotype is part of the multitude of metabolic changes accompanying a cell’s malignant transformation [75]. The increased expression of MAGL in aggressive malignant cells was linked to its ability to modulate a network of pro-oncogenic lipids [16,75,76,77]. MAGL overexpression by non-aggressive cancer cells, which can be achieved following a high-fat diet, alters their phenotype, increasing malignancy. These effects can be reversed in vitro, by administering a MAGL inhibitor [48,50,78]. MAGL inhibition was shown to decrease proliferation and increase apoptosis, via the upregulation of Bax and downregulation of Bcl-2 and Cyclin D1 expression [47,49].
MAGL was found to be upregulated in androgen-independent prostate cancer cells [79], malignant melanoma cells [76] and hepatic carcinoma cells [80,81], with pro-tumorigenic effects, by modulating both endocannabinoid and fatty acid metabolism/signaling. Its inhibition led to a decrease in cancer aggressiveness, which was reversed by a CB1R receptor antagonist or fatty acids administration [79].
5.1. Irreversible Pharmacological Inhibition of MAGL
Irreversible MAGL pharmacological inhibition in rodents was shown to have a great utility in addressing various disorders, supporting the hypothesis that MAGL represents an attractive therapeutic target.
5.1.1. Diseases of the Central Nervous System
KML29 (Figure 2) delayed the onset and progression of amyotrophic lateral sclerosis by reducing inflammation and increasing BDNF expression [82]. MAGL inhibition (JZL184-Figure 2) in primary mouse striatal astrocytes resulted in a decreased mutant huntingtin-induced synthesis of TNF-α via CB1R receptor activation [66]. Further, blocking MAGL expression led to cells resistant to mutant huntingtin-induced neuronal loss [66].
MAGL inhibition resulted in the suppression of β-amyloid synthesis and accumulation, and decreased β-site amyloid precursor protein cleaving enzyme 1 (BACE1), in a mouse model of Alzheimer’s disease. Due to reducing neuroinflammation and neurodegeneration, the hippocampal synaptic structure and function was maintained, and long-term synaptic plasticity, spatial learning and memory were improved [83,84,85].
Thus, MAGL inhibition promotes neuron survival in chronic neurodegenerative disorders, a property that preclinical studies show can also be extended to acute degeneration conditions (e.g., stroke and brain trauma). In a middle cerebral artery occlusion model of stroke, the administration of the MAGL inhibitor JZL184 (Figure 2), alone or in combination with the tissue plasminogen activator, determined the reduction of brain edema and infarct volume, reducing neuronal loss, in a CB1R-dependent manner [86]. The same inhibitor prevented neuropathologic changes and promoted neurologic recovery in a closed head injury model [62].
The blockade of MAGL resulted in anxiolytic effects in mice [87,88] and rats [89]. Regarding stress-induced anxiety, MAGL inhibition resulted in improved excitatory-inhibitory balance in the ventromedial prefrontal cortex, promoting traumatic stress resilience in rats [90]. Stress-related mental illness was associated with neuroinflammation, and stress-induced behavior was linked to PGE2 synthesis. In a social defeat stress model, PGE2 synthesis in the subcortical region, but not the cortical region, was linked to the activities of MAGL and COX-1/2 in a TLDR2/2-dependent manner [91].
Both anxiolytic and antidepressant effects observed are partly secondary to increased corticosterone secretion, whereas the enhanced locomotor activity is an intrinsic effect of augmented 2-AG signaling [92]. MAGL inhibitors were reported to exert a significant antidepressant effect, as reported in chronic corticosterone-exposed mice via GABAergic synaptic disinhibition [92].
MAGL inhibitors reduce symptoms associated with chronic stress such as hyperalgesia [88] and reduce intestinal permeability, the latter via modulation of claudins 1, 2 and 5, and occludin expression [93].
MAGL inhibition was useful in reducing seizures in both status epilepticus [94] and focal seizures [95] models. It prevented associated neuronal cell loss and cognitive impairment, in addition to reducing seizure-induced IL-1β and COX-2 expression [94].
Inhibition of MAGL is a viable target in treating nicotine addiction. A significant correlation between enzyme expression and withdrawal intensity was found in a murine model. Furthermore, administration of the JZL184 inhibitor (Figure 2) dose-dependently reduced withdrawal signs in a CB1R-dependent manner [96].
KML29 (Figure 2), administered systemically, produces six-fold elevations in brain 2-AG without markedly elevating AEA levels, and possesses anti-allodynic effects in a chronic constriction injury model [97].
Significant antinociception has been demonstrated in rodent models of peripheral inflammatory pain [55,95,98,99], visceral and gastrointestinal pain [95,100], neuropathic pain [97,101,102] and chemotherapy-induced neuropathy [103,104]. The selective inhibitor B3 (Figure 4) proved useful in suppressing pain-associated behavior in a rat formalin pain model [35]. KML29 (Figure 2) resulted in significantly lower inflammation and pain in a monoiodoacetate model of osteoarthritis, with reduced leukocyte infiltration, joint pain, withdrawal threshold and development of secondary allodynia [98]. In a similar osteoarthritis model, treatment with the inhibitor MJN110 (Figure 2) reversed weight-bearing asymmetry, lowered paw withdrawal thresholds (acute treatment) and resulted in anti-nociceptive tolerance (repeated treatment) [105]. MJN110 also blocked the expression of membrane-associated PGE synthase-1 in the ipsilateral dorsal horn of the spinal cord of the monoiodoacetate-treated rats [105]. Various inhibitors reduced chronic constriction, injury-induced and paclitaxel-induced mechanical allodynia, and thermal hyperalgesia, as a consequence of CB1R/CB2R activation [89,90,91]. In models of acute pain, effects of MAGL inhibition appear to be largely mediated by CB1R, whereas in inflammatory and neuropathic pain models, the analgesic effect is based on the activation of both CB1R and CB2R.
5.1.2. Inflammatory Diseases
Pharmacological inhibition of MAGL leads to reduction of neuroinflammatory responses in a manner associated with 2-AG increase and with AA, IL1β, IL6 or TNFα decrease in various animal models, such as LPS-induced neuroinflammation [106], MPTP-induced Parkinson, and genetic models of Alzheimer and SLA [62,82,106]
Nonetheless, the brain region-specific effects of MAGL should be considered when targeting its pharmacologic inhibition, because pro-inflammatory outcomes were reported. In mice with genetic deletion of the enzyme or treated with a specific inhibitor (JZL184), microglial reactivity was observed in the cerebellum, but not in the hippocampus. This resulted in increased mRNA for pro-inflammatory molecules, such as the enzyme COX-2, and impairment of motor coordination [107].
Inhibition of MAGL generation of AA exerted an CB1R receptors-independent antipyretic effect in mice, in an IL-1β- or LPS-induced fever model [108]. However, MAGL deletion is not required for the maintenance of the basal CBT and temperature homeostasis, but peripherally attenuates induced fever response [108].
A milder but still significant increase in 2-AG levels and in other monoacylglycerols was observed in the thymus, spleen and liver of MAGL KO mice [58], supporting the importance of MAGL in the regulation of the peripheral inflammatory response.
MAGL inhibitors exerted beneficial effects in animal models of chronic inflammatory illnesses, such as complete Freund reactive induced arthritis [98] and collagen-induced arthritis [99].
In mice fed a Western diet, MAGL knockout led to decreased hepatic inflammation [109]. MAGL deletion had protective effects on hepatocytes in different models of acute liver injury, an effect mediated via 2-AG CB2R-enhanced signaling and modulation of eicosanoid pathways, leading to a decrease in neutrophil-mediated liver damage [109]. Similar outcomes were observed in a preclinical model of hepatic injury associated with inflammation [110]. Furthermore, enhancement of CB2R activation appears to prompt fibrosis regression due to autophagy-mediated anti-inflammatory mechanisms in macrophages [109].
Regarding skeletal muscle post-injury healing, the MAGL inhibitor JZL184 (Figure 2) yielded an anti-inflammatory effect, decreasing neutrophil and macrophage infiltration and pro-inflammatory cytokine expression. However, this resulted in a delayed myofiber regeneration and promotion of fibrosis. These effects were reported to be CB1R receptors- and CB2R receptors-dependent [111].
The use of the MAGL inhibitor URB602 (Figure 2) was tested in a mice model of lung ischemia-reperfusion injury, which is a postoperative complication that can occur post-lung transplantation/cardiopulmonary bypass. Pre-treatment resulted in several protective effects: decreased wet to dry lung weight ratio score and improved oxygenation index, in addition to increased levels of 2-AG and decreased levels of PGI2, TXB2, LTB4, IL-6 and TNF-α [112]. In an LPS-induced acute lung injury murine model, MAGL inhibition with JZL184 decreased leukocyte migration, vascular permeability, and cytokine/chemokine and adhesion molecules levels in the bronchoalveolar lavage fluid [113].
5.1.3. Other Possible Applications
Obesity and Metabolic Diseases
Preclinical studies with MJN110 (Figure 2) showed that MAGL inhibitors’ administration is associated with reduced food intake in rats [114]. Taking into account the above-mentioned effects, and also that systemic oxidative stress, which is reported to possess a negative impact on lipids, proteins and endothelial function [115], is decreased by 2-AG [116], inhibiting MAGL may prove a useful strategy for the treatment of obesity and its comorbidities.
Neoplastic Maladies
Preclinical studies have confirmed the results of the in vitro studies, with JZL184 (Figure 2) showing antiproliferative efficacy in breast and prostate cancers, and in osteosarcoma [117], consequent to CB1R/CB2R activation and a decrease in inflammatory markers. Furthermore, in a xenograft and azoxymethane-induced colon cancer model, MAGL inhibition led to downregulation of VEGF and FGF-2, thus justifying the antiangiogenic and antimetastatic effect determined by MAGL inhibition.
All preclinical studies presenting the therapeutic utility of the irreversible MAGL inhibition are summarized in Table 1.

Table 1.
Preclinical studies assessing MAGL irreversible inhibitors’ utility, conducted between 2015 and 2021.
One aspect to be taken into account relates to the inhibitors’ selectivity towards MAGL rather than other endocannabinoid processing enzymes such as FAAH or ABHD6. MAGL inhibitor JZL184 (Figure 2) has been shown to have low specificity and thus blocking activities of other hydrolases such as fatty acid amide hydrolase and carboxylesterases [58], producing an increase in the central levels of 2-AG and OEA [87]. In consequence, repetitive, high doses of this inhibitor, e.g., 40 mg/kg [103,104], due to the persistent cerebral endocannabinoids, increase impaired endocannabinoid-dependent synaptic plasticity, leading to physical dependence and desensitization of brain CB1R receptors. The partial blockade of MAGL using low doses (4 mg/kg) of JZL184 for ~7 days did not lead to CB1R receptor desensitization or behavioral tolerance [103]. It was reported to decrease locomotor activity but did not induce catalepsy and hypothermia [101].
Compounds with higher MAGL selectivity generally lack cannabinoid adverse effects. Thus, KML29, an inhibitor with higher selectivity than JZL184, did show analgesic tolerance at high doses but did not induce locomotor activity decrease, tail immersion, or body temperature differences vs. control [97], nor catalepsy or hypothermia [101].
The tolerance observed in preclinical trials may explain the results of the clinical trials assessing the efficacy of B3 (Figure 4; currently LuAg06466, known formerly as ABX-1431), a highly potent and selective MAGL inhibitor, suitable for once-per-day oral administration, in Tourette syndrome [41]. When tested in a randomized, double-blind, placebo-controlled crossover, exploratory phase 1b study, a single dose was shown to improve tics and premonitory urges in adults with this disease [126]. However, administration of this compound for a longer period of time (12 weeks) showed no improvement in tic severity, premonitory urges, quality of life, and common psychiatric comorbidities when compared with a placebo [126].
It is hypothesized that using reversible inhibitors will improve the safety profile, preventing the manifestation of the adverse reactions mentioned above, including that of tolerance [1].
5.2. Reversible Pharmacological Inhibition of MAGL
Although various reversible inhibitors were identified, information from preclinical testing is scarce and further testing is needed, as seen in Table 2. The reversible inhibitors show pharmacodynamic effects similar to those of irreversible inhibitors in neuropathic and inflammatory pain [47,53,55] and for ameliorating characteristics of reserpine-induced depression [49]. In a previous study using a multiple sclerosis murine model, a reversible and selective MAGL inhibitor proved useful in stalling disease progression, with no CB1R receptors-related undesired effects [47]. We must underline that, for G7, repeated treatment was also tested: this reversible MAGL inhibitor showed complete lack of tolerance development, with long-lasting efficacy of the compound after repeated treatments.
Table 2.
Preclinical studies assessing MAGL reversible inhibitors utility, conducted between 2015 and 2021.
Figure 13 summarizes the potential therapeutic applications of MAGL inhibitors and portrays the potential mechanisms of action, based on the results of preclinical studies presented above.
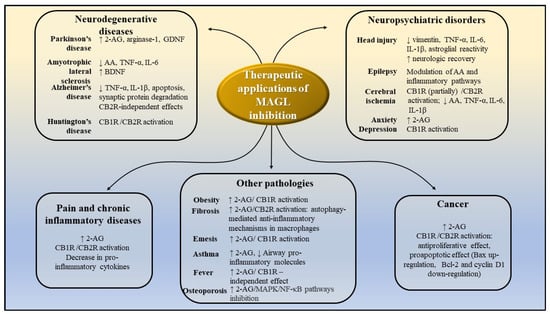
Figure 13.
Potential therapeutic application of MAGL inhibitors, as demonstrated by preclinical studies. AA-arachidonic acid; 2-AG-2-arachidonoylglycerol; CB1R—cannabinoid receptors 1; CB2R—cannabinoid receptors 2; IL-1β-interleukin-1β; IL-6—interleukin 6; TNF-α -tumor necrosis factor α; MAPK- mitogen-activated protein kinase; NF-kB—nuclear factor kappa-light-chain-enhancer of activated B cells; GDNF—glial cell line-derived neurotrophic factor; BDNF—brain-derived neurotrophic factor.
6. Conclusions
Several preclinical studies showed beneficial effects of MAGL blockades in various disorders, such as neurodegenerative and refractory neuropsychiatric disorders, chronic pain, and cancer. Thus, synthesizing selective MAGL inhibitors has become a focus point in drug design and development.
In the present review, we summarized the diverse synthetic scaffolds of MAGL inhibitors reported between 2015 and 2021, in connection with their potency. We presented their structural features critical for high selectivity and increased inhibitory potency, with the purpose of offering new insight for the development of novel inhibitors.
Irreversible inhibitors—developed from scaffolds such as hexafluoroisopropyl alcohol carbamates, glycol carbamates, azetidone triazole ureas and benzisothiazolinone—possess sustained pharmacological response and increased potency, whereas reversible inhibitors—derivatives of salicylketoxime, piperidine, pyrrolidone and azetidinyl amides—are expected to ensure a superior safety profile.
By modulating both endocannabinoid signaling and arachidonic acid metabolism, MAGL represents an attractive target in medicinal chemistry, and its inhibition may prove key to treating various pathologies currently lacking a satisfactory treatment. However, one should note that, due to the complexity of the biological networks in which MAGL functions, the extent of the consequences and effects generated by its pharmacological inhibition cannot be fully understood.
Author Contributions
Conceptualization, A.Z.; data curation, A.Z., A.U., D.P.M. and G.M.N.; writing—original draft preparation, A.Z., D.R., D.P.M. and A.U.; writing—review and editing, A.Z. and G.M.N.; funding acquisition, A.Z. All authors have read and agreed to the published version of the manuscript.
Funding
The authors acknowledge the financial support offered by Romanian National Authority for Scientific Research CNDI-UEFISCDI through grant PN-III-P1-1.1-PD-2019-0574, no. 179/2020. The costs to publish in open access were supported by the aforementioned grant.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Not applicable.
References
- Deng, H.; Li, W. Monoacylglycerol lipase inhibitors: Modulators for lipid metabolism in cancer malignancy, neurological and metabolic disorders. Acta Pharm. Sin. B 2020, 10, 582–602. [Google Scholar] [CrossRef] [PubMed]
- Dinh, T.P.; Carpenter, D.; Leslie, F.M.; Freund, T.F.; Katona, I.; Sensi, S.L.; Kathuria, S.; Piomelli, D. Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc. Natl. Acad. Sci. USA 2002, 99, 10819–10824. [Google Scholar] [CrossRef] [PubMed]
- Tanimura, A.; Yamazaki, M.; Hashimotodani, Y.; Uchigashima, M.; Kawata, S.; Abe, M.; Kita, Y.; Hashimoto, K.; Shimizu, T.; Watanabe, M.; et al. The endocannabinoid 2-arachidonoylglycerol produced by diacylglycerol lipase α mediates retrograde suppression of synaptic transmission. Neuron 2010, 65, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Castillo, P.E.; Younts, T.J.; Chávez, A.E.; Hashimotodani, Y. Endocannabinoid signaling and synaptic function. Neuron 2012, 76, 70–81. [Google Scholar] [CrossRef]
- Busquets-Garcia, A.; Bains, J.; Marsicano, G. CB1 receptor signaling in the brain: Extracting specificity from ubiquity. Neuropsychopharmacology 2018, 43, 4–20. [Google Scholar] [CrossRef]
- Maccarrone, M.; Bab, I.; Bíró, T.; Cabral, G.A.; Dey, S.K.; Di Marzo, V.; Konje, J.C.; Kunos, G.; Mechoulam, R.; Pacher, P.; et al. Endocannabinoid signaling at the periphery: 50 years after THC. Trends Pharmacol. Sci. 2015, 36, 277–296. [Google Scholar] [CrossRef]
- Di Marzo, V.; Bifulco, M.; De Petrocellis, L. The endocannabinoid system and its therapeutic exploitation. Nat. Rev. Drug Discov. 2004, 3, 771–784. [Google Scholar] [CrossRef]
- Alhouayek, M.; Masquelier, J.; Muccioli, G.G. Controlling 2-arachidonoylglycerol metabolism as an anti-inflammatory strategy. Drug Discov. Today 2014, 19, 295–304. [Google Scholar] [CrossRef]
- Rouzer, C.A.; Marnett, L.J. Endocannabinoid oxygenation by cyclooxygenases, lipoxygenases, and cytochromes P450: Cross-talk between the eicosanoid and endocannabinoid signaling pathways. Chem. Rev. 2011, 111, 5899–5921. [Google Scholar] [CrossRef]
- Nomura, D.K.; Morrison, B.; Blankman, J.L.; Long, J.Z.; Kinsey, S.G.; Marcondes, M.C.G.; Ward, A.M.; Hahn, Y.K.; Lichtman, A.H.; Conti, B.; et al. Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science 2011, 334, 809–813. [Google Scholar] [CrossRef] [PubMed]
- Nomura, D.K.; Hudak, C.S.; Ward, A.M.; Burston, J.J.; Issa, R.S.; Fisher, K.J.; Abood, M.E.; Wiley, J.L.; Lichtman, A.H.; Casida, J.E. Monoacylglycerol lipase regulates 2-arachidonoylglycerol action and arachidonic acid levels. Bioorganic Med. Chem. Lett. 2008, 18, 5875–5878. [Google Scholar] [CrossRef]
- Gil-Ordóñez, A.; Martin-Fontecha, M.; Ortega-Gutiérrez, S.; López-Rodríguez, M.L. Monoacylglycerol lipase (MAGL) as a promising therapeutic target. Biochem. Pharmacol. 2018, 157, 18–32. [Google Scholar] [CrossRef]
- Scalvini, L.; Piomelli, D.; Mor, M. Monoglyceride lipase: Structure and inhibitors. Chem. Phys. Lipids 2016, 197, 13–24. [Google Scholar] [CrossRef]
- Butler, C.R.; Beck, E.M.; Harris, A.; Huang, Z.; McAllister, L.A.; Ende, C.W.A.; Fennell, K.; Foley, T.L.; Fonseca, K.; Hawrylik, S.J.; et al. Azetidine and piperidine carbamates as efficient, covalent inhibitors of monoacylglycerol lipase. J. Med. Chem. 2017, 60, 9860–9873. [Google Scholar] [CrossRef] [PubMed]
- Granchi, C.; Caligiuri, I.; Minutolo, F.; Rizzolio, F.; Tuccinardi, T. A patent review of monoacylglycerol lipase (MAGL) inhibitors (2013–2017). Expert Opin. Ther. Patents 2017, 27, 1341–1351. [Google Scholar] [CrossRef] [PubMed]
- Bononi, G.; Poli, G.; Rizzolio, F.; Tuccinardi, T.; Macchia, M.; Minutolo, F.; Granchi, C. An updated patent review of monoacylglycerol lipase (MAGL) inhibitors (2018-present). Expert Opin. Ther. Patents 2021, 31, 153–168. [Google Scholar] [CrossRef] [PubMed]
- Grabner, G.; Zimmermann, R.; Schicho, R.; Taschler, U. Monoglyceride lipase as a drug target: At the crossroads of arachidonic acid metabolism and endocannabinoid signaling. Pharmacol. Ther. 2017, 175, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Gutierrez, S.; Viso, A.; Cisneros, J.A. The medicinal chemistry of agents targeting monoacylglycerol lipase. Curr. Top. Med. Chem. 2008, 8, 231–246. [Google Scholar] [CrossRef]
- Tyukhtenko, S.; Ma, X.; Rajarshi, G.; Karageorgos, I.; Anderson, K.W.; Hudgens, J.W.; Guo, J.J.; Nasr, M.L.; Zvonok, N.; Vemuri, K.; et al. Conformational gating, dynamics and allostery in human monoacylglycerol lipase. Sci. Rep. 2020, 10, 1–16. [Google Scholar] [CrossRef]
- Basavarajappa, B.S. Critical enzymes involved in endocannabinoid metabolism. Protein Pept. Lett. 2007, 14, 237–246. [Google Scholar] [CrossRef] [PubMed]
- King, A.R.; Lodola, A.; Carmi, C.; Fu, J.; Mor, M.; Piomelli, D. A critical cysteine residue in monoacylglycerol lipase is targeted by a new class of isothiazolinone-based enzyme inhibitors. Br. J. Pharmacol. 2009, 157, 974–983. [Google Scholar] [CrossRef]
- Wouters, J.; Lambert, D. A review on the monoacylglycerol lipase: At the interface between fat and endocannabinoid signalling. Curr. Med. Chem. 2010, 17, 2588–2607. [Google Scholar] [CrossRef]
- Casas-Godoy, L.; Duquesne, S.; Bordes, F.; Sandoval, G.; Marty, A. Lipases: An overview. Methods Mol. Biol. 2012, 861, 3–30. [Google Scholar] [CrossRef] [PubMed]
- Bawa, S.; Afzal, O.; Kumar, S.; Kumar, R.; Jaggi, M. 3D-QSAR study of benzotriazol-1-yl carboxamide scaffold as monoacylglycerol lipase inhibitors. J. Pharm. Bioallied Sci. 2014, 6, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Afzal, O.; Kumar, S.; Kumar, R.; Firoz, A.; Jaggi, M.; Bawa, S. Docking based virtual screening and molecular dynamics study to identify potential monoacylglycerol lipase inhibitors. Bioorganic Med. Chem. Lett. 2014, 24, 3986–3996. [Google Scholar] [CrossRef] [PubMed]
- Brindisi, M.; Maramai, S.; Gemma, S.; Brogi, S.; Grillo, A.; Mannelli, L.D.C.; Gabellieri, E.; Lamponi, S.; Saponara, S.; Gorelli, B.; et al. Development and pharmacological characterization of selective blockers of 2-arachidonoyl glycerol degradation with efficacy in rodent models of multiple sclerosis and pain. J. Med. Chem. 2016, 59, 2612–2632. [Google Scholar] [CrossRef]
- Aida, J.; Fushimi, M.; Kusumoto, T.; Sugiyama, H.; Arimura, N.; Ikeda, S.; Sasaki, M.; Sogabe, S.; Aoyama, K.; Koike, T. Design, synthesis, and evaluation of piperazinyl pyrrolidin-2-ones as a novel series of reversible monoacylglycerol lipase inhibitors. J. Med. Chem. 2018, 61, 9205–9217. [Google Scholar] [CrossRef] [PubMed]
- Jha, V.; Biagi, M.; Spinelli, V.; Di Stefano, M.; Macchia, M.; Minutolo, F.; Granchi, C.; Poli, G.; Tuccinardi, T. Discovery of monoacylglycerol lipase (MAGL) inhibitors based on a pharmacophore-guided virtual screening study. Molecules 2020, 26, 78. [Google Scholar] [CrossRef] [PubMed]
- Xiong, F.; Ding, X.; Zhang, H.; Luo, X.; Chen, K.; Jiang, H.; Luo, C.; Xu, H. Discovery of novel reversible monoacylglycerol lipase inhibitors via docking-based virtual screening. Bioorganic Med. Chem. Lett. 2021, 41, 127986. [Google Scholar] [CrossRef]
- Tuo, W.; Leleu-Chavain, N.; Spencer, J.; Sansook, S.; Millet, R.; Chavatte, P. Therapeutic potential of fatty acid amide hydrolase, monoacylglycerol lipase, and N-acylethanolamine acid amidase inhibitors. J. Med. Chem. 2016, 60, 4–46. [Google Scholar] [CrossRef]
- Nitulescu, G.; Margina, D.; Zanfirescu, A.; Olaru, O.; Nitulescu, G. Targeting bacterial sortases in search of anti-virulence therapies with low risk of resistance development. Pharmaceuticals 2021, 14, 415. [Google Scholar] [CrossRef]
- Martin, J.; MacKenzie, C.J.; Fletcher, D.; Gilbert, I.H. Characterising covalent warhead reactivity. Bioorganic Med. Chem. 2019, 27, 2066–2074. [Google Scholar] [CrossRef]
- Niphakis, M.J.; Cognetta, A.B., III; Chang, J.W.; Buczynski, M.W.; Parsons, L.H.; Byrne, F.; Burston, J.J.; Chapman, V.; Cravatt, B.F. Evaluation of NHS carbamates as a potent and selective class of endocannabinoid hydrolase inhibitors. ACS Chem. Neurosci. 2013, 4, 1322–1332. [Google Scholar] [CrossRef]
- Chang, J.W.; Niphakis, M.J.; Lum, K.M.; Cognetta, A.B., III; Wang, C.; Matthews, M.L.; Niessen, S.; Buczynski, M.W.; Parsons, L.H.; Cravatt, B.F. Highly selective inhibitors of monoacylglycerol lipase bearing a reactive group that is bioisosteric with endocannabinoid substrates. Chem. Biol. 2012, 19, 579–588. [Google Scholar] [CrossRef]
- Cisar, J.S.; Weber, O.D.; Clapper, J.R.; Blankman, J.L.; Henry, C.L.; Simon, G.M.; Alexander, J.P.; Jones, T.K.; Ezekowitz, R.A.B.; O’Neill, G.P.; et al. Identification of ABX-1431, a selective inhibitor of monoacylglycerol lipase and clinical candidate for treatment of neurological disorders. J. Med. Chem. 2018, 61, 9062–9084. [Google Scholar] [CrossRef]
- Kharasch, E.D. Biotransformation of sevoflurane. Anesth. Analg. 1995, 81, 27S–38S. [Google Scholar] [CrossRef]
- Chang, J.W.; Cognetta, A.B., III; Niphakis, M.J.; Cravatt, B.F. Proteome-wide reactivity profiling identifies diverse carbamate chemotypes tuned for serine hydrolase inhibition. ACS Chem. Biol. 2013, 8, 1590–1599. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Kaplan, J.; Stella, N. ABHD6: Its place in endocannabinoid signaling and beyond. Trends Pharmacol. Sci. 2019, 40, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Aaltonen, N.; Savinainen, J.; Ribas, C.R.; Rönkkö, J.; Kuusisto, A.; Korhonen, J.; Navia-Paldanius, D.; Häyrinen, J.; Takabe, P.; Käsnänen, H.; et al. Piperazine and piperidine triazole ureas as ultrapotent and highly selective inhibitors of monoacylglycerol lipase. Chem. Biol. 2013, 20, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.Z.; Ahenkorah, S.; Vaara, M.; Staszewski, M.; Adams, Y.; Laitinen, T.; Navia-Paldanius, D.; Parkkari, T.; Savinainen, J.; Walczyński, K.; et al. Loratadine analogues as MAGL inhibitors. Bioorganic Med. Chem. Lett. 2015, 25, 1436–1442. [Google Scholar] [CrossRef]
- Jiang, M.; Van der Stelt, M. Activity-based protein profiling delivers selective drug candidate ABX-1431, a monoacylglycerol lipase inhibitor, to control lipid metabolism in neurological disorders. J. Med. Chem. 2018, 61, 9059–9061. [Google Scholar] [CrossRef] [PubMed]
- McAllister, L.A.; Butler, C.R.; Mente, S.; O’Neil, S.V.; Fonseca, K.R.; Piro, J.R.; Cianfrogna, J.A.; Foley, T.L.; Gilbert, A.M.; Harris, A.R.; et al. Discovery of trifluoromethyl glycol carbamates as potent and selective covalent monoacylglycerol lipase (MAGL) inhibitors for treatment of neuroinflammation. J. Med. Chem. 2018, 61, 3008–3026. [Google Scholar] [CrossRef] [PubMed]
- Long, J.Z.; Jin, X.; Adibekian, A.; Li, W.; Cravatt, B.F. Characterization of tunable piperidine and piperazine carbamates as inhibitors of endocannabinoid hydrolases. J. Med. Chem. 2010, 53, 1830–1842. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, Z.; Mori, W.; Fu, H.; Schafroth, M.A.; Hatori, A.; Shao, T.; Zhang, G.; Van, R.S.; Zhang, Y.; Hu, K.; et al. Design, synthesis, and evaluation of 18F-labeled monoacylglycerol lipase inhibitors as novel positron emission tomography probes. J. Med. Chem. 2019, 62, 8866–8872. [Google Scholar] [CrossRef]
- Omran, Z. New disulfiram derivatives as MAGL-selective inhibitors. Molecules 2021, 26, 3296. [Google Scholar] [CrossRef]
- Bononi, G.; Granchi, C.; Lapillo, M.; Giannotti, M.; Nieri, D.; Fortunato, S.; El Boustani, M.; Caligiuri, I.; Poli, G.; Carlson, K.E.; et al. Discovery of long-chain salicylketoxime derivatives as monoacylglycerol lipase (MAGL) inhibitors. Eur. J. Med. Chem. 2018, 157, 817–836. [Google Scholar] [CrossRef]
- Hernandez-Torres, G.; Cipriano, M.; Heden, E.; Bjorklund, E.; Canales, A.; Zian, D.; Feliu, A.; Mecha, M.; Guaza, C.; Fowler, C.J.; et al. A reversible and selective inhibitor of monoacylglycerol lipase ameliorates multiple sclerosis. Angew. Chem. Int. Ed. Engl. 2014, 53, 13765–13770. [Google Scholar] [CrossRef]
- Tabrizi, M.A.; Baraldi, P.G.; Baraldi, S.; Ruggiero, E.; De Stefano, L.; Rizzolio, F.; Mannelli, L.D.C.; Ghelardini, C.; Chicca, A.; Lapillo, M.; et al. Discovery of 1,5-diphenylpyrazole-3-carboxamide derivatives as potent, reversible, and selective monoacylglycerol lipase (MAGL) inhibitors. J. Med. Chem. 2018, 61, 1340–1354. [Google Scholar] [CrossRef]
- Zhi, Z.; Zhang, W.; Yao, J.; Shang, Y.; Hao, Q.; Liu, Z.; Ren, Y.; Li, J.; Zhang, G.; Wang, J. Discovery of aryl formyl piperidine derivatives as potent, reversible, and selective monoacylglycerol lipase inhibitors. J. Med. Chem. 2020, 63, 5783–5796. [Google Scholar] [CrossRef] [PubMed]
- Granchi, C.; Rizzolio, F.; Palazzolo, S.; Carmignani, S.; Macchia, M.; Saccomanni, G.; Manera, C.; Martinelli, A.; Minutolo, F.; Tuccinardi, T. Structural optimization of 4-chlorobenzoylpiperidine derivatives for the development of potent, reversible, and selective monoacylglycerol lipase (MAGL) inhibitors. J. Med. Chem. 2016, 59, 10299–10314. [Google Scholar] [CrossRef] [PubMed]
- Granchi, C.; Lapillo, M.; Glasmacher, S.; Bononi, G.; Licari, C.; Poli, G.; el Boustani, M.; Caligiuri, I.; Rizzolio, F.; Gertsch, J.; et al. Optimization of a benzoylpiperidine class identifies a highly potent and selective reversible monoacylglycerol lipase (MAGL) inhibitor. J. Med. Chem. 2019, 62, 1932–1958. [Google Scholar] [CrossRef]
- Granchi, C.; Bononi, G.; Ferrisi, R.; Gori, E.; Mantini, G.; Glasmacher, S.; Poli, G.; Palazzolo, S.; Caligiuri, I.; Rizzolio, F.; et al. Design, synthesis and biological evaluation of second-generation benzoylpiperidine derivatives as reversible monoacylglycerol lipase (MAGL) inhibitors. Eur. J. Med. Chem. 2021, 209, 112857. [Google Scholar] [CrossRef] [PubMed]
- Altamimi, A.S.A.; Bawa, S.; Athar, F.; Hassan, Q.; Riadi, Y.; Afzal, O. Pyrrolidin-2-one linked benzofused heterocycles as novel small molecule monoacylglycerol lipase inhibitors and antinociceptive agents. Chem. Biol. Drug Des. 2020, 96, 1418–1432. [Google Scholar] [CrossRef]
- Afzal, O.; Altamimi, A.; Shahroz, M.; Sharma, H.; Riadi, Y.; Hassan, Q. Analgesic and anticancer activity of benzoxazole clubbed 2-pyrrolidinones as novel inhibitors of monoacylglycerol lipase. Molecules 2021, 26, 2389. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Connolly, P.J.; Zhang, Y.-M.; McDonnell, M.E.; Bian, H.; Lin, S.-C.; Liu, L.; Zhang, S.-P.; Chevalier, K.M.; Brandt, M.R.; et al. The discovery of azetidine-piperazine di-amides as potent, selective and reversible monoacylglycerol lipase (MAGL) inhibitors. Bioorganic Med. Chem. Lett. 2020, 30, 127243. [Google Scholar] [CrossRef]
- Dato, F.M.; Neudorfl, J.M.; Gutschow, M.; Goldfuss, B.; Pietsch, M. Omega-quinazolinonylalkyl aryl ureas as reversible inhibitors of monoacylglycerol lipase. Bioorg. Chem. 2020, 94, 103352. [Google Scholar] [CrossRef] [PubMed]
- Blazquez, C.; Chiarlone, A.; Bellocchio, L.; Resel, E.; Pruunsild, P.; García-Rincón, D.; Sendtner, M.; Timmusk, T.; Lutz, B.; Galve-Roperh, I.; et al. The CB1 cannabinoid receptor signals striatal neuroprotection via a PI3K/Akt/mTORC1/BDNF pathway. Cell Death Differ. 2015, 22, 1618–1629. [Google Scholar] [CrossRef]
- Long, J.Z.; Nomura, D.K.; Cravatt, B.F. Characterization of monoacylglycerol lipase inhibition reveals differences in central and peripheral endocannabinoid metabolism. Chem. Biol. 2009, 16, 744–753. [Google Scholar] [CrossRef]
- Margină, D.; Ungurianu, A.; Purdel, C.; Tsoukalas, D.; Sarandi, E.; Thanasoula, M.; Tekos, F.; Mesnage, R.; Kouretas, D.; Tsatsakis, A. Chronic inflammation in the context of everyday life: Dietary changes as mitigating factors. Int. J. Environ. Res. Public Health 2020, 17, 4135. [Google Scholar] [CrossRef] [PubMed]
- Navarrete, M.; Díez, A.; Araque, A. Astrocytes in endocannabinoid signalling. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130599. [Google Scholar] [CrossRef]
- Costa, L.; Amaral, C.; Teixeira, N.; Correia-Da-Silva, G.; Fonseca, B.M. Cannabinoid-induced autophagy: Protective or death role? Prostagland. Other Lipid Mediat. 2016, 122, 54–63. [Google Scholar] [CrossRef]
- Choi, S.-H.; Arai, A.L.; Mou, Y.; Kang, B.; Yen, C.C.-C.; Hallenbeck, J.; Silva, A.C. Neuroprotective effects of MAGL (monoacylglycerol lipase) inhibitors in experimental ischemic stroke. Stroke 2018, 49, 718–726. [Google Scholar] [CrossRef] [PubMed]
- Iannotti, F.; Vitale, R. The endocannabinoid system and PPARs: Focus on their signalling crosstalk, action and transcriptional regulation. Cells 2021, 10, 586. [Google Scholar] [CrossRef] [PubMed]
- Pistis, M.; O’Sullivan, S.E. The role of nuclear hormone receptors in cannabinoid function. Cannabinoid Pharmacol. 2017, 80, 291–328. [Google Scholar] [CrossRef]
- Yang, H.; Zhou, J.; Lehmann, C. GPR55—A putative “type 3” cannabinoid receptor in inflammation. J. Basic Clin. Physiol. Pharmacol. 2016, 27, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Calvo, A.; Bajo-Grañeras, R.; Maroto, I.B.; Zian, D.; Grabner, G.; García-Taboada, E.; Resel, E.; Zechner, R.; Zimmermann, R.; Ortega-Gutiérrez, S.; et al. Astroglial monoacylglycerol lipase controls mutant huntingtin-induced damage of striatal neurons. Neuropharmacology 2019, 150, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Piro, J.R.; Benjamin, D.I.; Duerr, J.M.; Pi, Y.; Gonzales, C.; Wood, K.M.; Schwartz, J.W.; Nomura, D.K.; Samad, T.A. A dysregulated endocannabinoid-eicosanoid network supports pathogenesis in a mouse model of Alzheimer’s disease. Cell Rep. 2012, 1, 617–623. [Google Scholar] [CrossRef]
- Woodhams, S.G.; Chapman, V.; Finn, D.P.; Hohmann, A.G.; Neugebauer, V. The cannabinoid system and pain. Neuropharmacology 2017, 124, 105–120. [Google Scholar] [CrossRef]
- Imperatore, R.; Morello, G.; Luongo, L.; Taschler, U.; Romano, R.; De Gregorio, D.; Belardo, C.; Maione, S.; Di Marzo, V.; Cristino, L. Genetic deletion of monoacylglycerol lipase leads to impaired cannabinoid receptor CB1 R signaling and anxiety-like behavior. J. Neurochem. 2015, 135, 799–813. [Google Scholar] [CrossRef]
- Navia-Paldanius, D.; Aaltonen, N.; Lehtonen, M.; Savinainen, J.R.; Taschler, U.; Radner, F.P.; Zimmermann, R.; Laitinen, J.T. Increased tonic cannabinoid CB1R activity and brain region-specific desensitization of CB1R Gi/o signaling axis in mice with global genetic knockout of monoacylglycerol lipase. Eur. J. Pharm. Sci. 2015, 77, 180–188. [Google Scholar] [CrossRef]
- Ogawa, S.; Kunugi, H. Inhibitors of fatty acid amide hydrolase and monoacylglycerol lipase: New targets for future antidepressants. Curr. Neuropharmacol. 2015, 13, 760–775. [Google Scholar] [CrossRef] [PubMed]
- Douglass, J.D.; Zhou, Y.X.; Wu, A.; Zadrogra, J.A.; Gajda, A.M.; Lackey, A.I.; Lang, W.; Chevalier, K.M.; Sutton, S.W.; Zhang, S.-P.; et al. Global deletion of MGL in mice delays lipid absorption and alters energy homeostasis and diet-induced obesity. J. Lipid Res. 2015, 56, 1153–1171. [Google Scholar] [CrossRef] [PubMed]
- Taschler, U.; Radner, F.; Heier, C.; Schreiber, R.; Schweiger, M.; Schoiswohl, G.; Preiss-Landl, K.; Jaeger, D.; Reiter, B.; Koefeler, H.C.; et al. Monoglyceride lipase deficiency in mice impairs lipolysis and attenuates diet-induced insulin resistance. J. Biol. Chem. 2011, 286, 17467–17477. [Google Scholar] [CrossRef] [PubMed]
- Tardelli, M.; Bruschi, F.V.; Claudel, T.; Fuchs, C.; Auer, N.; Kunczer, V.; Stojakovic, T.; Scharnagl, H.; Habib, A.; Grabner, G.; et al. Lack of monoacylglycerol lipase prevents hepatic steatosis by favoring lipid storage in adipose tissue and intestinal malabsorption. J. Lipid Res. 2019, 60, 1284–1292. [Google Scholar] [CrossRef]
- Nomura, D.K.; Long, J.Z.; Niessen, S.; Hoover, H.S.; Ng, S.-W.; Cravatt, B.F. Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 2010, 140, 49–61. [Google Scholar] [CrossRef]
- Baba, Y.; Funakoshi, T.; Emoto, K.; Masugi, Y.; Ekmekcioglu, S.; Amagai, M.; Mori, M.; Tanese, K. Expression of monoacylglycerol lipase as a marker of tumour invasion and progression in malignant melanoma. J. Eur. Acad. Dermatol. Venereol. 2017, 31, 2038–2045. [Google Scholar] [CrossRef]
- Li, X.; Gao, S.; Li, W.; Liu, Z.; Shi, Z.; Qiu, C.; Jiang, J. Effect of monoacylglycerol lipase on the tumor growth in endometrial cancer. J. Obstet. Gynaecol. Res. 2019, 45, 2043–2054. [Google Scholar] [CrossRef]
- Ma, M.; Bai, J.; Ling, Y.; Chang, W.; Xie, G.; Li, R.; Wang, G.; Tao, K. Monoacylglycerol lipase inhibitor JZL184 regulates apoptosis and migration of colorectal cancer cells. Mol. Med. Rep. 2016, 13, 2850–2856. [Google Scholar] [CrossRef]
- Xiang, W.; Shi, R.; Kang, X.; Zhang, X.; Chen, P.; Zhang, L.; Hou, A.; Wang, R.; Zhao, Y.; Zhao, K.; et al. Monoacylglycerol lipase regulates cannabinoid receptor 2-dependent macrophage activation and cancer progression. Nat. Commun. 2018, 9, 2574. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Z.; Lian, Z.; Liao, R.; Chen, Y.; Qin, Y.; Wang, J.; Jiang, Q.; Wang, X.; Gong, J. Monoacylglycerol lipase: A novel potential therapeutic target and prognostic indicator for hepatocellular carcinoma. Sci. Rep. 2016, 6, 35784. [Google Scholar] [CrossRef]
- Zhu, W.; Zhao, Y.; Zhou, J.; Wang, X.; Pan, Q.; Zhang, N.; Wang, L.; Wang, M.; Zhan, D.; Liu, Z.; et al. Monoacylglycerol lipase promotes progression of hepatocellular carcinoma via NF-kappaB-mediated epithelial-mesenchymal transition. J. Hematol. Oncol. 2016, 9, 127. [Google Scholar] [CrossRef] [PubMed]
- Pasquarelli, N.; Engelskirchen, M.; Hanselmann, J.; Endres, S.; Porazik, C.; Bayer, H.; Buck, E.; Karsak, M.; Weydt, P.; Ferger, B.; et al. Evaluation of monoacylglycerol lipase as a therapeutic target in a transgenic mouse model of ALS. Neuropharmacology 2017, 124, 157–169. [Google Scholar] [CrossRef]
- Pihlaja, R.; Takkinen, J.; Eskola, O.; Vasara, J.; López-Picón, F.R.; Haaparanta-Solin, M.; Rinne, J.O. Monoacylglycerol lipase inhibitor JZL184 reduces neuroinflammatory response in APdE9 mice and in adult mouse glial cells. J. Neuroinflammation 2015, 12, 81. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, C. Alleviation of neuropathology by inhibition of monoacylglycerol lipase in APP transgenic mice lacking CB2 receptors. Mol. Neurobiol. 2017, 55, 4802–4810. [Google Scholar] [CrossRef]
- Hashem, J.; Hu, M.; Zhang, J.; Gao, F.; Chen, C. Inhibition of 2-arachidonoylglycerol metabolism alleviates neuropathology and improves cognitive function in a tau mouse model of Alzheimer’s disease. Mol. Neurobiol. 2021, 58, 4122–4133. [Google Scholar] [CrossRef]
- Rahmani, M.-R.; Shamsizadeh, A.; Moghadam-Ahmadi, A.; Bazmandegan, G.; Allahtavakoli, M. JZL184, as a monoacylglycerol lipase inhibitor, down-regulates inflammation in a cannabinoid pathway dependent manner. Biomed. Pharmacother. 2018, 103, 1720–1726. [Google Scholar] [CrossRef]
- Bedse, G.; Bluett, R.J.; Patrick, T.A.; Romness, N.K.; Gaulden, A.D.; Kingsley, P.J.; Plath, N.; Marnett, L.J.; Patel, S. Therapeutic endocannabinoid augmentation for mood and anxiety disorders: Comparative profiling of FAAH, MAGL and dual inhibitors. Transl. Psychiatry 2018, 8, 92. [Google Scholar] [CrossRef]
- Lomazzo, E.; Bindila, L.; Remmers, F.; Lerner, R.; Schwitter, C.; Hoheisel, U.; Lutz, B. Therapeutic potential of inhibitors of endocannabinoid degradation for the treatment of stress-related hyperalgesia in an animal model of chronic pain. Neuropsychopharmacology 2014, 40, 488–501. [Google Scholar] [CrossRef] [PubMed]
- Alteba, S.; Zer-Aviv, T.M.; Tenenhaus, A.; Ben David, G.; Adelman, J.; Hillard, C.J.; Doron, R.; Akirav, I. Antidepressant-like effects of URB597 and JZL184 in male and female rats exposed to early life stress. Eur. Neuropsychopharmacol. 2020, 39, 70–86. [Google Scholar] [CrossRef] [PubMed]
- Worley, N.B.; Varela, J.A.; Gaillardetz, G.P.; Hill, M.N.; Christianson, J.P. Monoacylglycerol lipase alpha inhibition alters prefrontal cortex excitability and blunts the consequences of traumatic stress in rat. Neuropharmacology 2020, 166, 107964. [Google Scholar] [CrossRef]
- Nie, X.; Kitaoka, S.; Shinohara, M.; Kakizuka, A.; Narumiya, S.; Furuyashiki, T. Roles of toll-like receptor 2/4, monoacylglycerol lipase, and cyclooxygenase in social defeat stress-induced prostaglandin E2 synthesis in the brain and their behavioral relevance. Sci. Rep. 2019, 9, 17548. [Google Scholar] [CrossRef]
- Wang, Y.; Gu, N.; Duan, T.; Kesner, P.; Blaskovits, F.; Liu, J.; Lu, Y.; Tong, L.; Gao, F.; Harris, C.; et al. Monoacylglycerol lipase inhibitors produce pro- or antidepressant responses via. hippocampal CA1 GABAergic synapses. Mol. Psychiatry 2017, 22, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, X.; Yang, C.; Zhao, S. Effect of monoacylglycerol lipase inhibition on intestinal permeability in chronic stress model. Biochem. Biophys. Res. Commun. 2020, 525, 962–967. [Google Scholar] [CrossRef] [PubMed]
- Terrone, G.; Pauletti, A.; Salamone, A.; Rizzi, M.; Villa, B.R.; Porcu, L.; Sheehan, M.J.; Guilmette, E.; Butler, C.R.; Piro, J.R.; et al. Inhibition of monoacylglycerol lipase terminates diazepam-resistant status epilepticus in mice and its effects are potentiated by a ketogenic diet. Epilepsia 2017, 59, 79–91. [Google Scholar] [CrossRef]
- Griebel, G.; Pichat, P.; Beeské, S.; Leroy, T.; Redon, N.; Jacquet, A.; Françon, D.; Bert, L.; Even, L.; Lopez-Grancha, M.; et al. Selective blockade of the hydrolysis of the endocannabinoid 2-arachidonoylglycerol impairs learning and memory performance while producing antinociceptive activity in rodents. Sci. Rep. 2015, 5, 7642. [Google Scholar] [CrossRef]
- Muldoon, P.P.; Chen, J.; Harenza, J.L.; Abdullah, R.A.; Sim-Selley, L.J.; Cravatt, B.F.; Miles, M.F.; Chen, X.; Lichtman, A.H.; Damaj, M.I. Inhibition of monoacylglycerol lipase reduces nicotine withdrawal. Br. J. Pharmacol. 2015, 172, 869–882. [Google Scholar] [CrossRef]
- Crowe, M.S.; Wilson, C.D.; Leishman, E.; Prather, P.L.; Bradshaw, H.B.; Banks, M.L.; Kinsey, S.G. The monoacylglycerol lipase inhibitor KML29 with gabapentin synergistically produces analgesia in mice. Br. J. Pharmacol. 2017, 174, 4523–4539. [Google Scholar] [CrossRef]
- Philpott, H.T.; McDougall, J.J. Combatting joint pain and inflammation by dual inhibition of monoacylglycerol lipase and cyclooxygenase-2 in a rat model of osteoarthritis. Arthritis Res. 2020, 22, 9. [Google Scholar] [CrossRef] [PubMed]
- Nass, S.R.; Steele, F.F.; Ware, T.B.; Libby, A.H.; Hsu, K.-L.; Kinsey, S.G. Monoacylglycerol lipase inhibition using JZL184 attenuates paw inflammation and functional deficits in a mouse model of inflammatory arthritis. Cannabis Cannabinoid Res. 2021, 6, 233–241. [Google Scholar] [CrossRef]
- Sakin, Y.S.; Dogrul, A.; Ilkaya, F.; Seyrek, M.; Ulas, U.H.; Gülşen, M.; Bagci, S. The effect of FAAH, MAGL, and dual FAAH/MAGL inhibition on inflammatory and colorectal distension-induced visceral pain models in rodents. Neurogastroenterol. Motil. 2015, 27, 936–944. [Google Scholar] [CrossRef] [PubMed]
- Ignatowska-Jankowska, B.; Wilkerson, J.L.; Mustafa, M.; Abdullah, R.; Niphakis, M.; Wiley, J.L.; Cravatt, B.F.; Lichtman, A.H. Selective monoacylglycerol lipase inhibitors: Antinociceptive versus cannabimimetic effects in mice. J. Pharmacol. Exp. Ther. 2015, 353, 424–432. [Google Scholar] [CrossRef]
- Kamimura, R.; Hossain, M.Z.; Unno, S.; Ando, H.; Masuda, Y.; Takahashi, K.; Otake, M.; Saito, I.; Kitagawa, J. Inhibition of 2-arachydonoylgycerol degradation attenuates orofacial neuropathic pain in trigeminal nerve-injured mice. J. Oral Sci. 2018, 60, 37–44. [Google Scholar] [CrossRef]
- Curry, Z.; Wilkerson, J.L.; Bagdas, D.; Kyte, S.L.; Patel, N.; Donvito, G.; Mustafa, M.A.; Poklis, J.L.; Niphakis, M.J.; Hsu, K.-L.; et al. Monoacylglycerol lipase inhibitors reverse paclitaxel-induced nociceptive behavior and proinflammatory markers in a mouse model of chemotherapy-induced neuropathy. J. Pharmacol. Exp. Ther. 2018, 366, 169–183. [Google Scholar] [CrossRef]
- Thomas, A.; Okine, B.N.; Finn, D.P.; Masocha, W. Peripheral deficiency and antiallodynic effects of 2-arachidonoyl glycerol in a mouse model of paclitaxel-induced neuropathic pain. Biomed. Pharmacother. 2020, 129, 110456. [Google Scholar] [CrossRef]
- Burston, J.J.; Mapp, P.I.; Sarmad, S.; Barrett, D.; Niphakis, M.J.; Cravatt, B.F.; Walsh, D.; Chapman, V. Robust anti-nociceptive effects of monoacylglycerol lipase inhibition in a model of osteoarthritis pain. Br. J. Pharmacol. 2016, 173, 3134–3144. [Google Scholar] [CrossRef] [PubMed]
- Piro, J.R.; Suidan, G.L.; Quan, J.; Pi, Y.; O’Neill, S.M.; Ilardi, M.; Pozdnyakov, N.; Lanz, T.A.; Xi, H.; Bell, R.D.; et al. Inhibition of 2-AG hydrolysis differentially regulates blood brain barrier permeability after injury. J. Neuroinflamm. 2018, 15, 142. [Google Scholar] [CrossRef]
- Martínez-Torres, S.; Cutando, L.; Pastor, A.; Kato, A.; Sakimura, K.; De la Torre, R.; Valjent, E.; Maldonado, R.; Kano, M.; Ozaita, A. Monoacylglycerol lipase blockade impairs fine motor coordination and triggers cerebellar neuroinflammation through cyclooxygenase-2. Brain Behav. Immun. 2019, 81, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Alavez, M.; Nguyen, W.; Mori, S.; Moroncini, G.; Viader, A.; Nomura, D.K.; Cravatt, B.F.; Conti, B. Monoacylglycerol lipase regulates fever response. PLoS ONE 2015, 10, e0134437. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Mulvihill, M.M.; Mukhopadhyay, P.; Xu, H.; Erdélyi, K.; Hao, E.; Holovac, E.; Haskó, G.; Cravatt, B.F.; Nomura, D.K.; et al. Monoacylglycerol lipase controls endocannabinoid and eicosanoid signaling and hepatic injury in mice. Gastroenterology 2013, 144, 808–817.e15. [Google Scholar] [CrossRef] [PubMed]
- Habib, A.; Chokr, D.; Wan, J.; Hegde, P.; Mabire, M.; Siebert, M.; Ribeiro-Parenti, L.; Le Gall, M.; Lettéron, P.; Pilard, N.; et al. Inhibition of monoacylglycerol lipase, an anti-inflammatory and antifibrogenic strategy in the liver. Gut 2018, 68, 522–532. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.-K.; Zhang, M.; Tian, Z.-L.; Wang, M.; Zhao, R.; Wang, L.-L.; Li, S.-S.; Liu, M.; Li, J.-Y.; Zhang, M.-Z.; et al. The monoacylglycerol lipase inhibitor JZL184 decreases inflammatory response in skeletal muscle contusion in rats. Eur. J. Pharmacol. 2015, 761, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Yao, H.; Cheng, Y.; Gong, D.; Liao, X.; Wang, R. Effects of monoacylglycerol lipase inhibitor URB602 on lung ischemia-reperfusion injury in mice. Biochem. Biophys. Res. Commun. 2018, 506, 578–584. [Google Scholar] [CrossRef]
- Abohalaka, R.; Bozkurt, T.E.; Nemutlu, E.; Onder, S.C.; Sahin-Erdemli, I. The effects of fatty acid amide hydrolase and monoacylglycerol lipase inhibitor treatments on lipopolysaccharide-induced airway inflammation in mice. Pulm. Pharmacol. Ther. 2020, 62, 101920. [Google Scholar] [CrossRef] [PubMed]
- Sticht, M.A.; Lau, D.J.; Keenan, C.M.; Cavin, J.-B.; Morena, M.; Vemuri, V.K.; Makriyannis, A.; Cravatt, B.F.; Sharkey, K.A.; Hill, M.N. Endocannabinoid regulation of homeostatic feeding and stress-induced alterations in food intake in male rats. Br. J. Pharmacol. 2019, 176, 1524–1540. [Google Scholar] [CrossRef] [PubMed]
- Gradinaru, D.; Margina, D.; Borsa, C.; Ionescu, C.; Ilie, M.; Costache, M.; Dinischiotu, A.; Prada, G.-I. Adiponectin: Possible link between metabolic stress and oxidative stress in the elderly. Aging Clin. Exp. Res. 2016, 29, 621–629. [Google Scholar] [CrossRef]
- Elmazoglu, Z.; Rangel-Lopez, E.; Medina-Campos, O.N.; Pedraza-Chaverri, J.; Tunez, I.; Aschner, M.; Santamaria, A.; Karasu, C. Cannabinoid-profiled agents improve cell survival via. reduction of oxidative stress and inflammation, and Nrf2 activation in a toxic model combining hyperglycemia+Abeta1-42 peptide in rat hippocampal neurons. Neurochem. Int. 2020, 140, 104817. [Google Scholar] [CrossRef]
- Marino, S.; de Ridder, D.; Bishop, R.T.; Renema, N.; Ponzetti, M.; Sophocleous, A.; Capulli, M.; Aljeffery, A.; Carrasco, G.; Gens, M.D.; et al. Paradoxical effects of JZL184, an inhibitor of monoacylglycerol lipase, on bone remodelling in healthy and cancer-bearing mice. EBioMedicine 2019, 44, 452–466. [Google Scholar] [CrossRef]
- Pasquarelli, N.; Porazik, C.; Bayer, H.; Buck, E.; Schildknecht, S.; Weydt, P.; Witting, A.; Ferger, B. Contrasting effects of selective MAGL and FAAH inhibition on dopamine depletion and GDNF expression in a chronic MPTP mouse model of Parkinson’s disease. Neurochem. Int. 2017, 110, 14–24. [Google Scholar] [CrossRef]
- Covey, D.P.; Dantrassy, H.M.; Yohn, S.E.; Castro, A.; Conn, P.J.; Mateo, Y.; Cheer, J.F. Inhibition of endocannabinoid degradation rectifies motivational and dopaminergic deficits in the Q175 mouse model of Huntington’s disease. Neuropsychopharmacology 2018, 43, 2056–2063. [Google Scholar] [CrossRef]
- Bernal-Chico, A.; Canedo, M.; Manterola, A.; Victoria Sanchez-Gomez, M.; Perez-Samartin, A.; Rodriguez-Puertas, R.; Matute, C.; Mato, S. Blockade of monoacylglycerol lipase inhibits oligodendrocyte excitotoxicity and prevents demyelination In Vivo. Glia 2015, 63, 163–176. [Google Scholar] [CrossRef]
- Zhang, J.; Teng, Z.; Song, Y.; Hu, M.; Chen, C. Inhibition of monoacylglycerol lipase prevents chronic traumatic encephalopathy-like neuropathology in a mouse model of repetitive mild closed head injury. J. Cereb. Blood Flow. Metab. 2015, 35, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Pagano, E.; Borrelli, F.; Orlando, P.; Romano, B.; Monti, M.; Morbidelli, L.; Aviello, G.; Imperatore, R.; Capasso, R.; Piscitelli, F.; et al. Pharmacological inhibition of MAGL attenuates experimental colon carcinogenesis. Pharmacol. Res. 2017, 119, 227–236. [Google Scholar] [CrossRef]
- Parker, L.A.; Niphakis, M.J.; Downey, R.; Limebeer, C.L.; Rock, E.M.; Sticht, M.A.; Morris, H.; Abdullah, R.A.; Lichtman, A.H.; Cravatt, B.F. Effect of selective inhibition of monoacylglycerol lipase (MAGL) on acute nausea, anticipatory nausea, and vomiting in rats and Suncus murinus. Psychopharmacology 2015, 232, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Parker, L.A.; Limebeer, C.L.; Rock, E.M.; Sticht, M.A.; Ward, J.; Turvey, G.; Benchama, O.; Rajarshi, G.; Wood, J.T.; Alapafuja, S.O.; et al. A comparison of novel, selective fatty acid amide hydrolase (FAAH), monoacyglycerol lipase (MAGL) or dual FAAH/MAGL inhibitors to suppress acute and anticipatory nausea in rat models. Psychopharmacology 2016, 233, 2265–2275. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhou, C.; Qi, D.; Gao, Y.; Zhu, M.; Tao, T.; Sun, X.; Xiao, J. Inhibiting monoacylglycerol lipase suppresses RANKL-induced osteoclastogenesis and alleviates ovariectomy-induced bone loss. Front. Cell Dev. Biol. 2021, 9, 640867. [Google Scholar] [CrossRef]
- Muller-Vahl, K.R.; Fremer, C.; Beals, C.; Ivkovic, J.; Loft, H.; Schindler, C. Monoacylglycerol lipase inhibition in tourette syndrome: A 12-week, randomized, controlled study. Mov. Disord. 2021, 36. [Google Scholar] [CrossRef]













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).