1. Introduction
Calcium pyrophosphate deposition (CPPD) is a metabolic disease primarily affecting the elderly population, and it is characterized by the deposition of calcium pyrophosphate (CaPP) crystals in synovial fluid. The presence of crystals may not result in symptoms, but be present as an incidental finding of chondrocalcinosis (CC) during X-ray imaging, or present as painless lumps. However, they can also contribute to acute CaPP crystal arthritis or chronic arthropathy with structural changes in osteoarthritis [
1,
2]. Historically, because of the similar symptoms, CC was confused with gout. It was only distinguished by McCarty et al. in 1962 as a separate disorder, and a new name was proposed: pseudogout [
3]. CPPD is common, and the occurrence is correlated with age [
4]. In the UK, prevalence was approximately 10–15% in a community sample of 65–75 year-olds; and above 40% of the over-80 population suffer from CPPD associated symptoms [
5,
6,
7]. It had a prevalence of 0.42% in an Italian population survey, making it the fourth most common musculoskeletal condition [
8].
There are 12 known crystallographic forms of CaPP crystals, but only monoclinic and triclinic forms are found in vivo. In vitro crystallization studies have shown that initial deposits of CaPP are amorphous or orthorhombic, and that a triclinic form is the final form of interconversion among different crystal forms [
9].
Treatment for chondrocalcinosis is done in response to symptoms. An acute form is treated with rest, in situ application of ice, joint aspiration, colchicine and intra-articular corticosteroid injections [
10], immunosuppressive agents [
11,
12], and surgical joint replacement in severe cases of arthropathy [
11,
13]. Attempts to shed CaPP crystals with disodium EDTA and Mg
2+—potent in vitro solubilizers—were a therapeutic failure [
14]. Some dissolution of CaPP crystal in vitro has been achieved with the use of polyphosphates and by the combined activity of alkaline phosphatase and inorganic pyrophosphatase [
15,
16]; however, the enzymes turned out to be unsuccessful in vivo and are not currently used in the treatment of CPPD [
17]. Therefore, at present, there are no treatments to safely eliminate CaPP crystals once deposited [
10].
Nonenzymatic hydrolysis of pyrophosphate occurs at one of the lowest rates found in nature. This is caused by the presence of many canonical structures of that molecule that shield the central phosphorus atom from the eventual nucleophilic attack. In biological systems, a pyrophosphate is hydrolyzed by inorganic pyrophosphatase and other enzymes performing phosphoryl transfer reactions. To polarize the phosphate moiety, making the central phosphorus atom more electrophilic and more vulnerable to nucleophilic attack, these enzymes require the presence of a divalent cation, such as Mg
2+ or Mn
2+ [
18,
19].
Here, we are describing an attempt to find a peptide able to dissolve/hydrolyze CaPP from a random phage-display-delivered library. The method included two selection steps based on the affinities and catalytic properties of bacteriophages and subsequent testing of chemically synthesized peptides based on their phage-displayed counterparts. Activity assays were mostly based on ion chromatography of left-over crystals. We also studied the impacts of Mg2+ and Ca2+ on the reaction.
3. Materials and Methods
3.1. Methodology
Two ready-to-use libraries, 1 × 1013 PFU/mL (plaque-forming units per mL) of M13 bacteriophages with peptides displayed on the pIII protein, were chosen for the study: New England Biolabs’ PhD-C7C and PhD-12 with the architecture of N’-ACX7C-GGGS-pIII-C’ with disulfide bonds between cysteines, and N’-X12-GGGS-pIII-C’. X stands for any random proteinogenic amino acid. Since the goal of the study was to recover the catalytically active peptides, screening of the phage library was divided into two stages: affinity selection and catalytic selection.
In affinity selection, the displayed peptide is expected to bind to the immobilized alendronic acid (4-amino-1-hydroxy-1-phosphonobutyl)phosphonic acid), a pyrophosphate analog (
Figure 7), via the divalent cation Mg
2+ (needed for the binding and catalysis). We chose this molecule over crystal, as the potential dissolution/hydrolysis could have happened during the binding process, resulting in a lack of affinity. Bisphosphonates such as alendronic acid do not undergo such hydrolysis. Subsequent washing with a buffer leaves only phages with a strong interaction between peptide and alendronate. The detachment was done through the introduction of a chelating agent—EDTA. Obtained this way, bacteriophages were amplified from blue colonies, leveled to the same titer, purified, and dried. The amounts of protein in samples were assessed by the Bradford method according to a BSA standard. Dissolved bacteriophages were used for catalytic selection. Then, 700 μg of bacteriophagal protein was introduced to the reaction mixture with Mg
2+ and CaPP crystals in Tris buffer, pH 7.5. The reaction was carried out for 42 days at 50 °C to prevent any growth of possible microorganisms delivered through contamination, and relative phosphate/pyrophosphate levels in the supernatant were monitored during that period. As blind probes, we used a sample with no phages at all (-f), a sample with no phages and no Mg
2+ (-f-M), and a sample from the host alone without the M13 phage subjected to the same method of bacteriophages isolation as all the other samples, in case any of the bacterial enzymes having hydrolytic properties. At the end of the incubation, Mg
2+/Ca
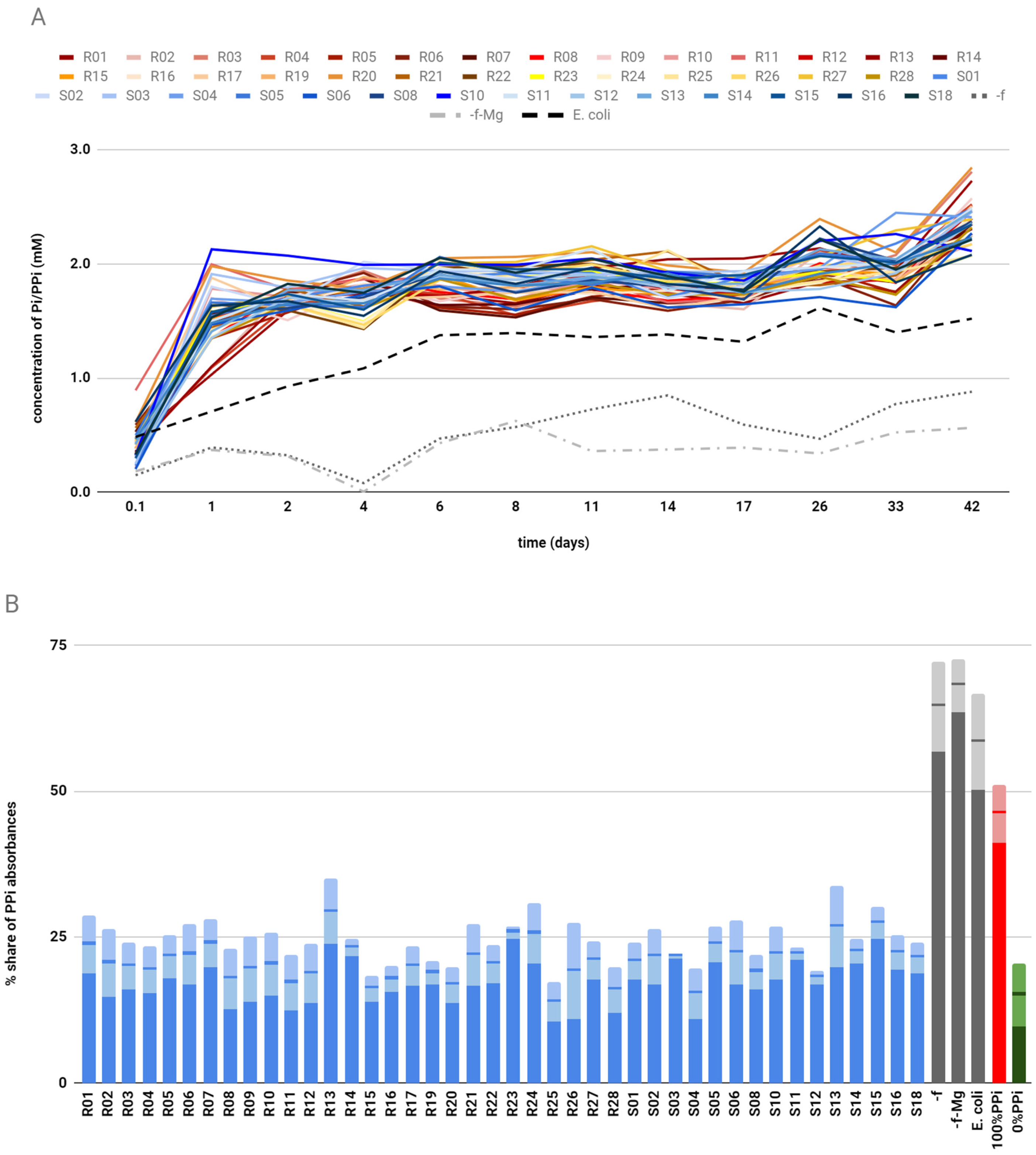
2+ levels in the supernatant and volume of the reaction mixture leftover were also measured. The remaining precipitate was lyophilized and the ratio of PPi to Pi amounts was measured using ion-exchange chromatography. Bacteriophages exhibiting the lowest PPi percentages were chosen for DNA isolation and sequencing. The selection process, including affinity and catalytic assays for bacteriophages, is graphically summarized in
Figure 8.
Five peptides, chosen on the basis of their catalytic performance, were chemically synthesized as standalone peptides. Theu had a four amino acid-long linker, GGGS (or a one amino acid-long linker in the case of PhD-12-G3), and a 10 amino acid-long fragment of native bacteriophagal protein—AETVESSLAK—since the potential active site could have formed between native capsid protein and the displayed peptide.
Synthesized peptides were tested for CaPP hydrolysis properties along with peptide cyclo(RPDDHR) (C3) expressing some pyrophosphatase/synthetase properties [
24]: serine–histidine (SH) dipeptide known from its hydrolytic abilities [
25,
26] and SGAGKT (W1) peptide designed to resemble a proteinaceous Walker A motif (p-loop) to bind inorganic phosphate ions [
27]. Additionally, a sample with a 30 times higher concentration of Mg
2+ (-p30xM) was prepared, as high concentrations of this ion are also known to speed up the hydrolysis of pyrophosphate [
28]. The experiment was carried out for 8 days; Pi/PPi and Mg
2+/Ca
2+ levels were monitored during this time. The precipitate left over after the reaction was subsequently lyophilized and analyzed by ion-exchange chromatography. The final results are percentage shares of absorbance of fractions with pyrophosphate with regard to the sums of PPi and Pi absorbances.
3.2. CaPP Crystallization
All chemicals for this study have been supplied from Merck Life Science, Poznań, Poland, unless stated otherwise. Triclinic calcium pyrophosphate crystallization was carried out in 40 mM Tris buffer (pH 7.5, 40 mM CaCl
2, 50 mM Na
2P
2O
7·10H
2O) over 4 months at 37 °C. It has been shown that initially some monoclinic and amorphous forms are formed during the crystallization (1 and 7% respectively) [
29], so time seems to be a crucial factor, as it leads to the interconversion of all the other forms to biologically occurring triclinic form [
9]. Before usage, crystals were filtered, washed with deionized water, and dried over vacuum.
3.3. Affinity Assay
Alendronic acid was immobilized on NovaSyn® TG carboxy resin via peptide bond formation between the carboxyl residue of the resin and amine moiety of alendronic acid. First, active esters of carboxylic resin were generated in MES buffer, 50 mM, pH 6. Five equivalents of EDC were used. Subsequently, the resin was washed 3 times with the buffer, and 1.5 eq of alendronic acid in the MES buffer was added. The reaction was carried out overnight. Based on the mass difference of the dried resin before and after the reaction, the yield of the product was 92%.
Two read-to-use random libraries of M13 bacteriophages, PhD-C7C and PhD-12, 1 × 1013 PFU/mL were purchased from New England Biolabs (Halifax, Canada); and E. coli ER2738 was used as a host strain. Both bacteriophages and host bacteria strains were handled according to the producers’ manuals.
Affinity selection: 100 mg of alendronate-functionalized resin was placed into a plastic centrifuge tube filter and washed several times with the washing buffer (50 mM Tris, pH 7.5, with 50 mM concentration of Mg2+). To the alendronate resin dispersed in 500 μL washing buffer, 10 μL of bacteriophage library (approximately 1 × 1011 PFU) was added. The mixture was gently mixed and left for incubation for 20 min. Subsequently, the solution was removed by centrifugation (10 min, 2000 RPM). The unbound phages were removed by washing the resin 10 times with 500 μL of washing buffer. In the end, the detachment of phages was achieved by washing the resin twice with 600 μL of eluting buffer (50 mM Tris, pH 7.5, with 0.25 M EDTA). Since a high concentration of EDTA can destroy bacteriophages, they were precipitated. To do so, ⅙ volume of 20% PEG/2.5 M NaCl was added to the eluent, and bacteriophages were allowed to precipitate at 4 °C for 20 min. Tubes were spun at 14,000 rpm for 10 min and the supernatant was discarded. The invisible pellet was suspended in 1000 μL of TBS. Plating was done on agar/LB/IPTG/Xgal/Tet.
Bacteriophage titration of the eluent showed that the number of PFU varied from 5 to 15 from the initial 1011 of subjected to the affinity selection bacteriophages. The whole procedure was repeated 3 times for each library to collect the total of 47 viable bacteriophages (19 from the PhD-C7C named S01 to S19 and 28 from the PhD-12 library named R01 to R28). Three-day-old blue plaques from the bacterial lawn were stabbed with a sterile pipette tip and added to a diluted 1:100 overnight culture of a host strain in LB/Tet for amplification. After 4.5 h of incubation at 37 °C, the culture was spun 3 times for 10 min at 12,000× g, 4 °C, and each time the upper 80% of supernatant was transferred to a fresh tube. One sixth of the volume of 20% PEG/2.5 M NaCl was added to the remaining supernatant, and bacteriophages were allowed to precipitate at 4 °C overnight. On the next day, the tubes were spun at 14,000 rpm for 10 min and the supernatant was discarded. The invisible pellet was suspended in 100 μL of TBS. From this stock, bacteriophages were further used for catalytic selection and ssDNA isolation.
All the chemicals for the Bradford assay were bought from ThermoFisher (Schwerte, Germany) and the assay was done for the 10 μL of amplified and purified bacteriophages. The concentration assessed varied between 150 and 350 μg per sample according to the BSA standards.
3.4. Catalytic Assay for Bacteriophages
For the catalytic selection of bacteriophages, 700 μg of bacteriophagal protein was used. The reaction mixture consisted of 20 μL of 200 mM Tris buffer, pH 7.5; 5 μL of 100 mM of MgCl
2; and 5 μL of CaPP crystals suspended in water. The concentration of PPi in the suspension was found by Chen’s phosphorus microdetermination method [
30] to be approximately 25 mM. The final volume of the reaction mixture was 100 μL. The experiment was carried out for 42 days at 50 °C. During this time, levels of Pi/PPi were monitored. At the end of the experiment, levels of Mg
2+/Ca
2+ were measured [
31] in the centrifuged pellet. The volume of the reaction mixture at the end of the experiment was taken into consideration while estimating concentrations of Pi/PPi and Mg
2+/Ca
2+. Probes that dried out due to human error or a factory defect of the Eppendorf tube were disqualified from further steps. Pellet and remnants of the reaction cocktail were used for the ion-exchange chromatography using the Bisaz et al. modified method [
30] in order to determine relative quantities of PPi and Pi after the reaction.
3.5. ssDNA Isolation and Cloning
To isolate ssDNA, bacteriophages were lysed with 4 M NaI in TE buffer pH 8.0 for 1 h. Subsequently, ssDNA was precipitated in 70% EtOH. Before sequencing the ssDNA isolated from bacteriophages, it was amplified using PCR Mix Plus Green (A&A Biotechnology, Gdansk, Poland); forward primer 5′-TTAGTGGTACCTTTCTATTCTC-3′; reverse primer 5′-GTATGGGATTTTGCTAAACAAC-3′. Twenty-five cycles of denaturation (95 °C, 30 s), annealing (52 °C, 30 s), and elongation (72 °C, 60 s) were performed, along with 3 min of initial denaturation and 3 min of final elongation. Purification of the PCR product was performed using affinity chromatography with a commercially available GeneMatrix DNA clean-up kit (EURx, Gdańsk, Poland) in accordance with the manual. Sequencing was performed by Oligo.pl with the same primers as for amplification.
3.6. Peptide Synthesis
All amino acid derivatives and coupling reagents were purchased from Iris Biotech GmbH (Marktredwitz, Germany) unless stated otherwise. Peptide synthesis was performed in solid phase using the standard Fmoc strategy. Various peptides were synthesized on different types of resins depending on their sequences, the presence of disulfide bonds, and challenges in synthesis. Peptides from PhD-12 and PhD-12-3G libraries were synthesized on Wang resin. Peptides from the PhD-C7C library were synthesized on the Fmoc–Ala–Wang resin, as the direct coupling of Fmoc–Cys(Mmt)–OH to the Wang resin is not advised due to racemization—it resulted in an extension of the sequence with Ala at the C-terminus (
Table 1). Extended versions of peptides with fragments of pIII protein delivered from PhD-12 and PhD-12-3G libraries and peptide W1 were synthesized on FmocLys(Boc)–Wang resin. Extended versions of peptides with cysteine bridges (library PhD-C7C) were synthesized as amides on Rink–amide resin because their synthesis in the carboxylic form on Wang resin was unsuccessful. (
Table 1). Loading of the first amino acid in the case of not preloaded Wang resin was achieved in DMF with catalytic quantities of DMAP. Symmetrical anhydride of the amino acid derivative was used. The anhydride of the protected amino acid was generated in dry DCM with 0.5 equivalents (eq.) of DIC and 1 eq. of amino acid derivative. In the case of the Rink–amide resin, the first amino acid was loaded with 3 eq. PyBOP, 3 eq. 6Cl-HOBt, and 1%DIPEA. The reaction was carried out twice for 2 h. Fmoc deprotection was done in 20% piperidine in DMF. Elongation of a peptide chain was achieved with 3 eq. of amino acid derivative, 3 eq. Oxyma Pure, and 3 eq. DIC in DMF. A Kaiser test was performed before every coupling and Fmoc deprotection. Peptide C3 synthesis was described elsewhere [
24]. SerHis peptide was obtained from Bachem (Bubendorf, Switzerland). Disulfide bond formation was achieved in the solid phase. First, the Mmt group was removed by treating the resin with 2% TFA in DCM. Multiple washes were performed till the color was completely washed out. Cysteine oxidation was done by treatment with 2 eq. of NCS (Merck Life Science, Poznań, Poland) in DMF for 15 min at room temperature. Full deprotection was achieved by incubation in a solution of TFA/TIS/Water 95/2.5/2.5
v/v for 2 h. Crude deprotected peptides were purified using preparative liquid chromatography on the c12 Jupiter Proteo column. The peak with a mass corresponding to the peptide was isolated and lyophilized. Purity was confirmed by HPLC-MS. Mass spectra were acquired on the Shimadzu LCMS-IT-TOF mass spectrometer with electrospray ionization (ESI) equipped with Jupiter Proteo column I.D. 2.0 × 250 mm, phase A: miliQ water/0.1% FA, phase B: gradient grade acetonitrile/0.1% FA, detection at 210 nm, fixed on scan mode. Elution was with linear increase from 0% to 70% of phase B in 35 min, flow = 0.2 mL/min.
3.7. Catalytic Assay for Peptides
For confirmation of catalytic properties of peptides, 1.8 μL of 80 mM stock peptide was used. The reaction mixture consisted of 20 μL of 200 mM Tris buffer, pH 7.5; 5 μL of 100 mM of MgCl2; and 5 μL of CaPP crystals suspended in water, 10% (w/w). The concentration of PPi in the suspension was assessed by phosphorus microdetermination to be approximately 1000 mM +/− 50 mM. The final volume of the reaction mixture was 100 μL. During the experiment, both Pi/PPi and Mg2+/Ca2+ levels in the supernatant over the centrifuged pellet were monitored. After 8 days of the experiment, the pellet and remnants of the reaction cocktail were lyophilized and used for the ion-exchange chromatography in order to determine relative quantities of PPi and Pi after the reaction.
Results of phosphate and pyrophosphate percentages in the pellet left over after the reaction were first specified as percentage shares of absorbance of pyrophosphate in elution fractions with regard to a sum of absorbances for both fractions. However, as a small part of pyrophosphate was also washed out with a fraction in the case of some phosphate, the percentage shares of absorbance for elution fractions of pyrophosphate do not coincide with the percentages of pyrophosphate in a sample. Thus, the introduction of a standard curve was necessary to assess the percentage of hydrolysis. The standard curve was created by preparing a 10% (w/w) suspension of calcium pyrophosphate and calcium phosphate. Subsequently, mixtures of adequate volumes of well-mixed suspensions were made to give samples corresponding to 100%, 80%, 60%, 40%, 20%, and 0% of pyrophosphate to phosphate mixes. These standard solutions were subjected to ion chromatography to assess the percentage shares of absorbance. Standard runs were subjected to ion-exchange chromatography for each round of result collection.
The determination of PPi content in a pellet left over from the reaction was achieved by using an anion-exchange resin as described by Bisaz et al., with modifications [
32]. Firstly, the Dowex 1 × 8 resin was washed with 4 M HCl and subsequently with water until the pH of the eluent reached over 4. Equal amounts of resin were divided into small 2 mL columns. The swollen resin was 20 mm in height. The reaction mixtures were created after the samples were freeze-dried, dissolved in 100 μL 10% trichloroacetic acid (TCA), and applied to the column. After 15 min of incubation at room temperature, the column was washed with 1.9 mL of water. Phosphoric acid was collected while washing the resin with 0.05% HCl, and pyrophosphoric acid was collected while washing the resin with 0.5% HCl. Eight fractions of 1 mL each were collected: two after applying the sample and washing the resin with water, four after washing the resin with 0.05% HCl, and two after washing it with 0.5% HCl. The amount of Pi/PPi was determined by microdetermination of phosphorus using 50 μL of a sample.
The amounts of phosphates (both Pi and PPi) in the soluble fraction were monitored using modified Chen’s phosphorus microdetermination method [
30]. For monitoring of phosphates levels during the reaction, 1 μL from the top of the centrifuged reaction mixture (or water for blind probe) was placed directly on the 96-well plate and filled up to 50 μL. Subsequently, 150 μL of a mixture of equal volumes of 6 N sulfuric acid, 0.8 M ammonium molybdate, and 10% ascorbic acid was added. After 1 h of incubation at 50 °C for the development of the color, the absorbance at 820 nm was measured against a blind probe with water. For the determination of phosphate levels in fractions after ion-exchange chromatography, 50 μL of the fraction was used. The correlation of absorbance to phosphate concentration was determined through calibration with known amounts of sodium phosphate and sodium pyrophosphate.
The determination of divalent cations was based on the reaction between o-cresolphthalein complexone (o-CPC) and Mg
2+/Ca
2+ in alkaline media described in [
31]. First, 5 μL from the top of the centrifuged reaction mixture (or water for blind probe) was placed directly on the 96-well plate, which was topped up to 200 μL with an o-cresolphthalein complexone 0.018% (
w/v) in 0.125 M ammonia/ammonium hydroxide buffer, pH 10.5. After 5 min of color development, the absorbance against a blind probe was measured at 570 nm. For the catalytic properties of the bacteriophages, the final values are medians from 4 separate measurements and 3 reads of absorbance each. The concentration was estimated on the basis of known concentrations of MgCl
2. For the monitoring of Mg
2+/Ca
2+ levels during the confirmation of the catalytic properties of peptides, the results are medians of 3 reads of absorbance from one measurement each. The concentration was estimated on the basis of known concentrations of MgCl
2.
All the spectrophotometric assays were performed with a NanoStar BMG spectrophotometer on a 96-well plate with at least two blind samples. A single read was a median from 16 measuring points. Before every sample was taken, reaction cocktails were spun for 4 min at 4 °C, and the required volume was taken from the top of the mixture to prevent accidental pellet intake. Before further incubation, the pellet was gently dispersed via an ultrasonic bath.
3.8. Molecular Dynamics
Molecular dynamics studies for R25 and R25-pIII were performed with Yasara 20.8.23, with the Amber IPQ15 force field [
33], wall boundary conditions, default simulation cell size, and water molecules density 0.997 g/mL. The structure of pIII protein was taken from Protein Data Bank, accession number LG3P. During molecular dynamics, the first 10 amino acids from pIII protein were allowed to undertake the simulation along with side chains of the amino acids in the proximity of the R25 peptide. The rest of the pIII was fixed in space. We chose this fragment as it seemed like it did not form strong hydrogen bonds with the rest of the pIII protein, nor did its residues create any hydrophobic core. This is potentially could unfold and form an active site along with the displayed peptide. Additionally, fragment AETVESSLAK was the fragment we added to extended versions of peptides. Local energy minima of R25 and R25-pIII structures were calculated using the steepest descent energy minimization function.
3.9. R25 Affinity Assay
The affinity of R25 peptide was tested on an alendronate-functionalized resin and CaPP crystals in the presence of Mg2+ and Ca2+ ions. For this purpose, 100 mg of alendronate-functionalized resin or CaPP crystals were placed into a plastic centrifuge tube filter and washed several times with the washing buffer (20 mM Tris pH 7.5). For samples with the presence of Mg2+ or Ca2+ ions, 50 mM of the corresponding divalent cations was present in the buffer solution; then, 10 μL of R25 stock (80 mM) was added to 200 μL of proper buffer, and the mixture was incubated for 20 min. During incubation, probes were gently mixed. Afterward, the solution was removed by centrifugation (10 min, 2000 RPM). Washing was repeated two more times. Subsequently, the solid phase was washed 3 times with the eluting buffer (50 mM Tris pH 7.5, with 0.25 M EDTA). Two fractions, 600 μL each (from washing buffer and eluting buffer), were obtained from every sample. Fractions were subsequently frozen, lyophilized, and dissolved in 100 μL of miliQ water. Finally, 10 μL from each sample was analyzed via HPLC: Jupiter Proteo column; phase A: miliQ water/0.1% TFA; phase B: gradient grade acetonitrile/0.1% TFA; detection at 214 nm. Elution had a linear increase from 0% to 60% of phase B over 20 min; flow = 1 mL/min. The surface area of the washed-out R25 peptide was measured and compared to the control. The control was prepared by adding 10 μL of R25 stock solution to 100 μL of Tris buffer, pH 7.5. The surface area of this peak was considered as 100%. Each analysis was repeated 3 times.
4. Conclusions
Screening of random bacteriophage libraries against immobilized alendronic acid and subsequent catalytic selection led us to five peptides with potential hydrolytic properties toward CaPP crystals. Incubation of chemically synthesized peptides singled out one of them—R25—as able to lower the amount of pyrophosphate by approximately 20% in 8 days. R25’s catalysis has been shown to be Mg
2+-dependent, and it is able to utilize soluble species of pyrophosphate with higher than 20% efficiency, which suggests that the hydrolysis is probably stopped by inhibition of calcium released from the crystal or limited access to a crystal’s surface. Similar obstacles have been met before by teams trying to use enzymatic approaches [
15,
16,
34,
35]. Previous attempts at the dissolution of CaPP crystals in vitro with chondrocyte alkaline phosphatase and pyrophosphatase resulted in a similar level of hydrolysis of PPi as shown here (the highest was 15% for chondrocyte alkaline phosphatase [
36]). In vivo attempts to use enzymes, however, were unsuccessful, and this is why currently an enzymatic approach is not used in the treatment of CaPP [
10,
17].
Similarly to other enzymatic approaches, R25 turned out to be inhibited by Ca
2+ ions released during the process of CaPP hydrolysis [
35,
37]. Since the R25 peptide is not fully washed out from the alendronate-functionalized resin, nor can it regain catalytic activity with elevated levels of Mg
2+ after the Ca-induced inhibition, its complex with Ca
2+ must be much stronger than the one with Mg
2+.
Structural studies using the
in-silico approach showed that despite strong binding of PPi and Mg
2+, R25 lacks residues able to generate hydroxyl ions in the proximity of the central phosphorus atom the way known inorganic pyrophosphatases do [
23]. Thus, the catalysis most probably is initiated by the hydroxyl ions from the alkaline solution of the reaction cocktail.
In phage display technology, it is customary to undertake two, three, or more cycles of directed evolution in order to select for more and more optimized phenotypes [
38]. However, in our case, it was not possible to perform more than one round of selection. Our one selection round consisted of two stages: the first was a selection of affinity peptides from a large library of bacteriophages (ca. 6 × 10
11); the second was a catalysis assay performed separately for each selected candidate, whose number, therefore, could not be too high. We did select the bacteriophage presenting peptide R25 for the second passage of directed evolution. However, the high titer of bacteriophages resulting from the second affinity selection (approximately 1 × 10
6) made it impossible to screen through this number of potentially active bacteriophages in the second round of catalytic selection. Additionally, since in the first round of affinity selection we already used the strictest selection possible in order to achieve a manageable number of candidates for catalytic selection, it was not possible to further adjust the affinity pressure, resulting in 1 × 10
6 possible second-round catalytic candidates. We learned the sequences of the peptides displayed by the two bacteriophages from the second passage. The sequences did not differ from the original R25.
Combinatorial biology approaches proved to be very effective in selecting for efficient affinity to a given molecular target. However, obtaining an efficient catalyst for a desired reaction is much more challenging. Basically, each specific reaction needs to be tackled separately, and a complex methodology needs to be established in order to obtain any positive results [
39,
40], and even then, the limit to the number of sequences screened is often orders of magnitudes lower than affinity selection. In our study, we designed and performed one such strategy combining the well-known combinatorial biology of phage display with a custom protocol for catalysis selection, along with chemical peptide synthesis for confirmation of the results.
Regardless of its limits, R25 might be a potential prototype for a drug against chondrocalcinosis in the future. As the directed evolution approach to enhance the abilities of R25 was too abundant in results to screen through, we believe that the continuation of this study will have to be done with rational chemical modifications. This is in fact the path that has been taken by many successful drugs in the past. For instance, the development of a peptidomimetic agent acting as an ATIC homodimerization inhibitor (for tumor growth targeting) was possible by screening peptides with a novel directed evolution strategy—SICLOPPS—by Tavassoli et al. [
41], which took a decade to complete [
42,
43,
44]. From this point of view, we are merely at the beginning of our path.


 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}