
LC-MS/MS Determination of 21 Non-Steroidal Anti-Inflammatory Drugs Residues in Animal Milk and Muscles



Abstract
:1. Introduction
2. Results and Discussion
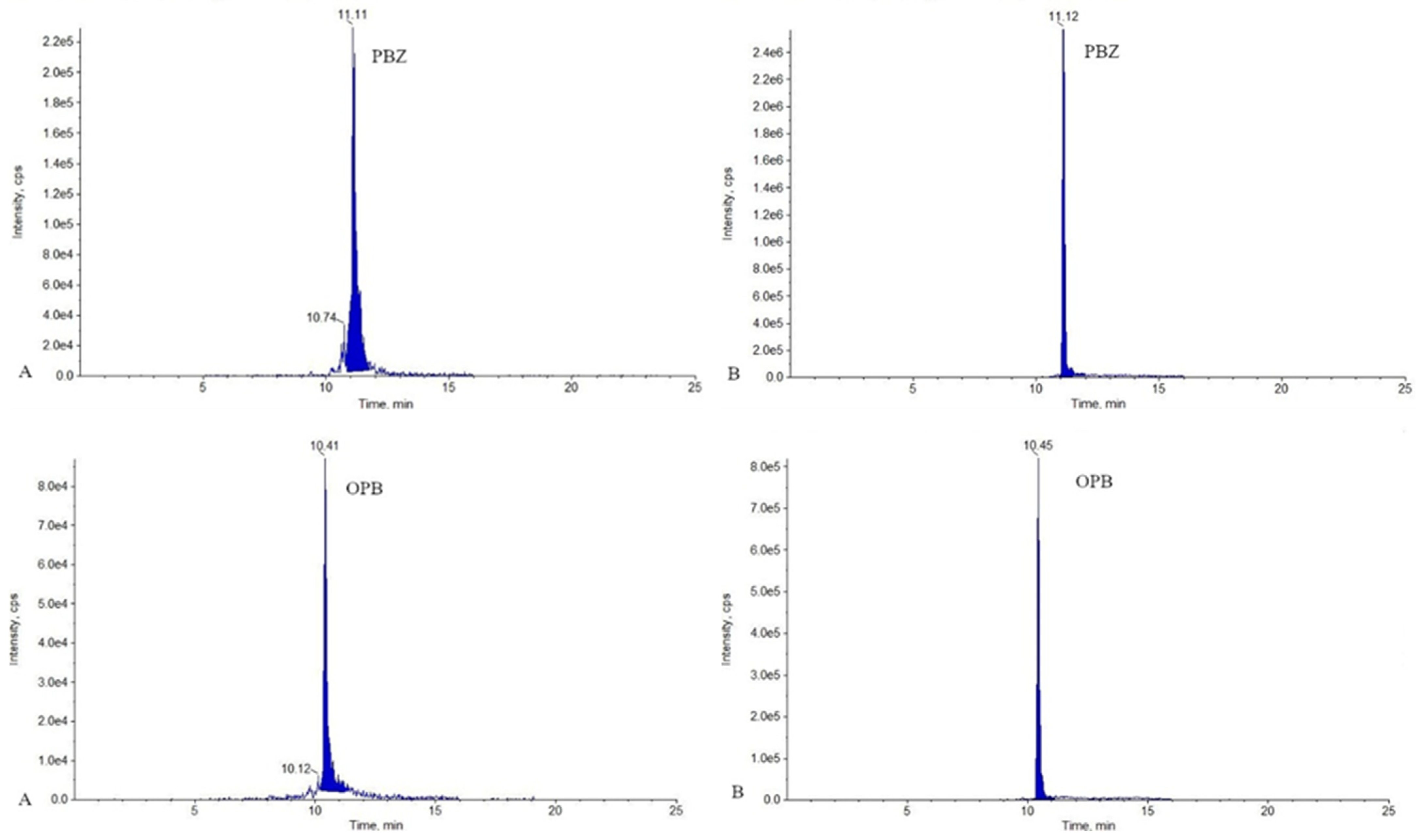
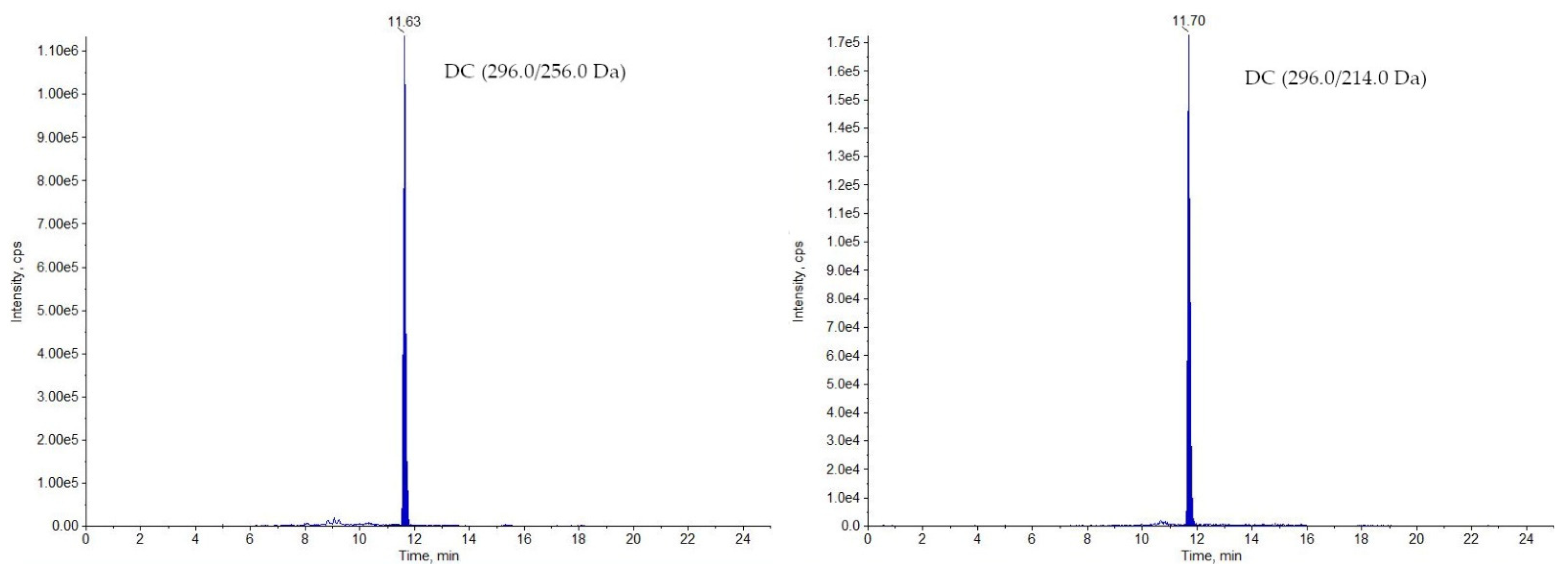
2.1. LC-MS/MS Analysis
2.2. Extraction and Clean-Up
2.3. Method Validation
2.4. Real Samples Analysis
3. Material and Methods
3.1. Reagents and Chemicals
3.2. Standard Solutions, Buffers, and Samples
3.3. Instrumentation
3.4. Optimisation of LC-MS/MS Conditions
3.5. Sample Preparation
3.5.1. Milk
3.5.2. Muscle Tissue
3.6. LC-MS/MS Analysis
3.7. Validation Concept
3.7.1. Selectivity
3.7.2. Recovery and Precision (Repeatability and Within-Laboratory Reproducibility)
3.7.3. The Decision Limit (CCα) and Detection Capability (CCβ)
3.7.4. Working Range
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Wöhrl, S. NSAID hypersensitivity—Recommendations for diagnostic work up and patient management. Allergo J. Int. 2018, 27, 114–121. [Google Scholar] [CrossRef] [Green Version]
- Lanas, A. NSAIDs and aspirin: Recent advances and implications for clinical management. NSAIDs Aspirin Recent Adv. Implic. Clin. Manag. 2016, 1–263. [Google Scholar] [CrossRef]
- Brune, K.; Patrignani, P. New insights into the use of currently available non-steroidal anti-inflammatory drugs. J. Pain Res. 2015, 8, 105–118. [Google Scholar] [CrossRef] [Green Version]
- Tyumina, E.A.; Bazhutin, G.A.; Cartagena Gómez, A.D.P.; Ivshina, I.B. Nonsteroidal Anti-inflammatory Drugs as Emerging Contaminants. Microbiol. (Russ. Fed.) 2020, 89, 148–163. [Google Scholar] [CrossRef]
- Nambirajan, K.; Muralidharan, S.; Roy, A.A.; Manonmani, S. Residues of Diclofenac in Tissues of Vultures in India: A Post-ban Scenario. Arch. Environ. Contam. Toxicol. 2018, 74, 292–297. [Google Scholar] [CrossRef]
- European Parliment and the Council COMMISSION REGULATION No 37/2010 of of 22 December 2009 on pharmacologically active substances and their classification regarding maximum residue limits in foodstuffs of animal origin. Off. J. Eur. Union 2009, 15, 1–80.
- Report for 2018 on the results from the monitoring of veterinary medicinal product residues and other substances in live animals and animal products. EFSA Support. Publ. 2020, 17. [CrossRef]
- Gallo, P.; Fabbrocino, S.; Dowling, G.; Salini, M.; Fiori, M.; Perretta, G.; Serpe, L. Confirmatory analysis of non-steroidal anti-inflammatory drugs in bovine milk by high-performance liquid chromatography with fluorescence detection. J. Chromatogr. A 2010, 1217, 2832–2839. [Google Scholar] [CrossRef] [PubMed]
- Jedziniak, P.; Szprengier-Juszkiewicz, T.; Olejnik, M.; Zmudzki, J. Determination of non-steroidal anti-inflammatory drugs residues in animal muscles by liquid chromatography-tandem mass spectrometry. Anal. Chim. Acta 2010, 672, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Jedziniak, P.; Pietruk, K.; Śledzińska, E.; Olejnik, M.; Szprengier-Juszkiewicz, T.; Zmudzki, J. Rapid method for the determination of metamizole residues in bovine muscle by LC-MS/MS. Food Addit. Contam.—Part A Chem. Anal. Control. Expo. Risk Assess. 2013, 30, 977–982. [Google Scholar] [CrossRef]
- Hu, T.; Peng, T.; Li, X.J.; Chen, D.D.; Dai, H.H.; Deng, X.J.; Yue, Z.F.; Wang, G.M.; Shen, J.Z.; Xia, X.; et al. Simultaneous determination of thirty non-steroidal anti-inflammatory drug residues in swine muscle by ultra-high-performance liquid chromatography with tandem mass spectrometry. J. Chromatogr. A 2012, 1219, 104–113. [Google Scholar] [CrossRef]
- Gentili, A.; Caretti, F.; Bellante, S.; Mainero Rocca, L.; Curini, R.; Venditti, A. Development and validation of two multiresidue liquid chromatography tandem mass spectrometry methods based on a versatile extraction procedure for isolating non-steroidal anti-inflammatory drugs from bovine milk and muscle tissue. Anal. Bioanal. Chem. 2012, 404, 1375–1388. [Google Scholar] [CrossRef]
- Kaufmann, A.; Butcher, P.; Maden, K.; Walker, S.; Widmer, M. Determination of corticosteroids, anabolic steroids, and basic non-steroidal anti-inflammatory drugs in milk and animal tissues. J. AOAC Int. 2014, 97, 263–272. [Google Scholar] [CrossRef]
- Rúbies, A.; Guo, L.; Centrich, F.; Granados, M. Analysis of non-steroidal anti-inflammatory drugs in milk using QuEChERS and liquid chromatography coupled to mass spectrometry: Triple quadrupole versus Q-Orbitrap mass analysers. Anal. Bioanal. Chem. 2016, 408, 5769–5778. [Google Scholar] [CrossRef] [PubMed]
- Britzi, M.; Frieda, S. Development and Validation of a High-Throughput Method for the Determination of Eight Non-Steroidal Anti-Inflammatory Drugs and Chloramphenicol in Milk, using Liquid Chromatography-Tandem Mass Spectroscopy. Int. J. Anal. Bioanal. Methods 2019, 1, 1–12. [Google Scholar]
- van Pamel, E.; Daeseleire, E. A multiresidue liquid chromatographic/tandem mass spectrometric method for the detection and quantitation of 15 non-steroidal anti-inflammatory drugs (NSAIDs) in bovine meat and milk. Anal. Bioanal. Chem. 2015, 407. [Google Scholar] [CrossRef] [PubMed]
- Community Reference Laboratories Residues (CRLs) 20/1/2010 Guidelines for the Validation of Screening Methods for Residues of Veterinary Medicines. 2010. Available online: https://ec.europa.eu/food/system/files/2016-10/cs_vet-med-residues_guideline_validation_screening_en.pdf (accessed on 24 September 2021).
- Jedziniak, P.; Szprengier-Juszkiewicz, T.; Pietruk, K.; Ledziska, E.; Żmudzki, J. Determination of non-steroidal anti-inflammatory drugs and their metabolites in milk by liquid chromatography-tandem mass spectrometry. Anal. Bioanal. Chem. 2012, 403, 2955–2963. [Google Scholar] [CrossRef] [PubMed]
- Dubreil-Chéneau, E.; Pirotais, Y.; Bessiral, M.; Roudaut, B.; Verdon, E. Development and validation of a confirmatory method for the determination of 12 non steroidal anti-inflammatory drugs in milk using liquid chromatography-tandem mass spectrometry. J. Chromatogr. A 2011, 1218, 6292–6301. [Google Scholar] [CrossRef]
- Kim, H.J.; Lee, J.H.; Park, H.J.; Kim, J.Y.; Cho, S.; Kim, W.S. Determination of non-opioid analgesics in adulterated food and dietary supplements by LC-MS/MS. Food Addit. Contam.—Part A Chem. Anal. Control. Expo. Risk Assess. 2014, 31, 973–978. [Google Scholar] [CrossRef]
- Jedziniak, P.; Olejnik, M.; Szprengier-Juszkiewicz, T.; Smulski, S.; Kaczmarowski, M.; Zmudzki, J. Identification of flunixin glucuronide and depletion of flunixin and its marker residue in bovine milk. J. Vet. Pharmacol. Ther. 2013, 36, 571–575. [Google Scholar] [CrossRef]
- Van Hoof, N.; De Wasch, K.; Poelmans, S.; Noppe, H.; De Brabander, H. Multi-residue liquid chromatography/tandem mass spectrometry method for the detection of non-steroidal anti-inflammatory drugs in bovine muscle: Optimisation of ion trap parameters. Rapid Commun. Mass Spectrom. 2004, 18, 2823–2829. [Google Scholar] [CrossRef] [PubMed]
- Jedziniak, P.; Olejnik, M.; Pietruk, K.; Protasiuk, E.; Szprengier-Juszkiewicz, T.; Żmudzki, J. Simultaneous Determination of Residues of Non-Steroidal Anti-Inflammatory Drugs and Glucocorticosteroids in Animal Muscle by Liquid Chromatography-Tandem Mass Spectrometry. Food Anal. Methods 2016, 9, 1837–1848. [Google Scholar] [CrossRef] [Green Version]
- Shimada, H.; Kobayashi, Y.; Tanahashi, S.; Kawase, A.; Ogiso, T.; Iwaki, M. Correlation between glucuronidation and covalent adducts formation with proteins of non-steroidal anti-inflammatory drugs. Eur. J. Pharm. Sci. 2018, 112, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Olejnik, M.; Jedziniak, P.; Szprengier-Juszkiewicz, T.; Zmudzki, J. Influence of matrix effect on the performance of the method for the official residue control of non-steroidal anti-inflammatory drugs in animal muscle. Rapid Commun. Mass Spectrom. 2013, 27, 437–442. [Google Scholar] [CrossRef]
- European Parliament and the Council of the European Union Commission Regulation (EU) No 37/2010. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32017R0201&from=NL (accessed on 24 September 2021).
- Meucci, V.; Minunni, M.; Vanni, M.; Sgorbini, M.; Corazza, M.; Intorre, L. Selective and simultaneous determination of NSAIDs in equine plasma by HPLC with molecularly imprinted solid-phase extraction. Bioanalysis 2014, 6, 2147–2158. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Analyte | Animal Species | MRL/Level * | Matrix |
|---|---|---|---|
| Diclofenac (DC) | Bovine | 5 μg/kg | Muscle |
| 0.1 μg/kg | Milk | ||
| Porcine | 5 μg/kg | Muscle | |
| Firocoxib (FIRO) | Equidae | 10 μg/kg | Muscle |
| Flunixin (FLU) | Bovine | 20 μg/kg | Muscle |
| Porcine | 50 μg/kg | Muscle | |
| Equidae | 10 μg/kg | Muscle | |
| 5-Hydroxyflunixin (5-OH FLU) | Bovine | 40 μg/kg | Milk |
| Tolfenamic acid (TOL) | Bovine, porcine | 50 μg/kg | Muscle |
| Bovine | 50 μg/kg | Milk | |
| Meloxicam (MEL) | Bovine, caprine, porcine, rabbit, Equidae | 20 μg/kg | Muscle |
| Bovine, caprine | 15 μg/kg | Milk | |
| Metamizole (as 4-Methylaminoantipyrin) (4-MAA) | Bovine, porcine, Equidae | 100 μg/kg | Muscle |
| Bovine | 50 μg/kg | Milk | |
| Carprofen (Sum of carprofen and carprofen glucuronide conjugate) (CPF) | Bovine, Equidae | 500 μg/kg | Muscle |
| Phenylbutazone (PBZ) Oxyphenbutazone (OPB) | - | 5 μg/kg * | Muscle, milk |
| Ibuprofen (IBU) Naproxen (NAP) Mefenamic acid (MEF) | - | 10 μg/kg * | Muscle, milk |
| Analyte | Range (µg/kg) | Validation Level (µg/kg) | Recovery (%) | Precision (CV, %) | ccα (µg/kg) | ccβ (µg/kg) |
|---|---|---|---|---|---|---|
| CELE | 1.25–25.0 | 5.00 | 105 | 12.7 | 3.08 | 3.86 |
| CPF | 1.25–25.0 | 5.00 | 104 | 8.83 | 2.64 | 3.09 |
| DC | 0.025–0.50 | 0.10 | 99.7 | 16.2 | 0.15 | 0.22 |
| FIRO | 1.25–25.0 | 5.00 | 103 | 14.6 | 3.58 | 4.99 |
| FLU | 1.25–25.0 | 5.00 | 88.0 | 5.68 | 3.21 | 3.69 |
| FLUF | 1.25–25.0 | 5.00 | 108 | 9.54 | 2.70 | 3.25 |
| IBU | 1.25–25.0 | 5.00 | 95.3 | 11.2 | 3.61 | 4.48 |
| KTP | 1.25–25.0 | 5.00 | 108 | 6.21 | 2.66 | 2.95 |
| MEF | 1.25–25.0 | 5.00 | 108 | 11.7 | 2.67 | 3.11 |
| MEL | 3.75–75.0 | 15.0 | 102 | 8.22 | 16.8 | 18.8 |
| NAP | 1.25–25.0 | 5.00 | 103 | 10.3 | 2.95 | 3.41 |
| NIF | 1.25–25.0 | 5.00 | 105 | 10.9 | 2.55 | 2.92 |
| OPZ | 1.25–25.0 | 5.00 | 106 | 7.72 | 2.89 | 3.31 |
| PBZ | 1.25–25.0 | 5.00 | 106 | 17.0 | 3.26 | 4.32 |
| ROFE | 1.25–25.0 | 5.00 | 94.5 | 15.6 | 3.74 | 5.03 |
| TOLF | 12.5–250 | 50.0 | 95.3 | 13.0 | 56.0 | 70.3 |
| 4-AA | 1.25–25.0 | 5.00 | 105 | 10.9 | 3.03 | 3.96 |
| 4-AcAA | 1.25–25.0 | 5.00 | 101 | 5.51 | 2.87 | 3.21 |
| 4-FAA | 1.25–25.0 | 5.00 | 108 | 12.9 | 2.76 | 3.41 |
| 4-MAA | 12.5–250 | 50.0 | 95.3 | 13.0 | 56.0 | 70.3 |
| 5-OH FLU | 10.0–200 | 40.0 | 86.3 | 13.3 | 45.9 | 59.4 |
| Analyte | Range (µg/kg) | Validation Level (µg/kg) | Recovery (%) | Precision (CV. %) | ccα (µg/kg) | ccβ (µg/kg) |
|---|---|---|---|---|---|---|
| CELE | 1.25–25.0 | 5.00 | 93.8 | 7.14 | 5.65 | 6.46 |
| CPF | 5.00–1000 | 20.0 | 101 | 13.7 | 6.45 | 790 |
| DC | 1.25–25.0 | 5.00 | 102 | 6.71 | 5.59 | 6.28 |
| FIRO | 2.50–50.0 | 10.0 | 85.0 | 7.62 | 10.4 | 12.1 |
| FLU | 2.50–100 | 10.0 | 96.0 | 16.1 | 14.2 | 19.1 |
| FLUF | 1.25–25.0 | 5.00 | 101 | 4.73 | 5.69 | 7.22 |
| IBU | 2.50–50.0 | 10.0 | 85.7 | 13.1 | 11.0 | 13.6 |
| KTP | 1.25–25.0 | 5.00 | 98.6 | 9.75 | 5.67 | 6.45 |
| MEF | 5.00–100 | 20.0 | 105 | 6.77 | 23.6 | 26.9 |
| MEL | 5.00–100 | 20.0 | 99.3 | 7.91 | 22.6 | 24.8 |
| NAP | 2.50–50.0 | 10.0 | 97.7 | 15.7 | 12.9 | 17.7 |
| NIF | 1.25–25.0 | 5.00 | 104 | 7.67 | 5.56 | 4.48 |
| OPB | 1.25–25.0 | 5.00 | 103 | 8.14 | 5.85 | 6.96 |
| PBZ | 1.25–25.0 | 5.00 | 104 | 12.3 | 5.88 | 7.15 |
| ROFE | 1.25–25.0 | 5.00 | 109 | 9.63 | 5.88 | 7.08 |
| TOLF | 12.5–250 | 50.0 | 101 | 6.02 | 57.1 | 65.6 |
| 4-AA | 2.50–50.0 | 10.0 | 92.2 | 15.6 | 12.8 | 18.0 |
| 4-AcAA | 2.50–50.0 | 10.0 | 94.6 | 11.7 | 12.2 | 15.8 |
| 4-FAA | 2.50–50.0 | 10.0 | 101 | 16.1 | 14.2 | 19.1 |
| 4-MAA | 2.50–200 | 10.0 | 105 | 16.6 | 12.3 | 146 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pietruk, M.; Jedziniak, P.; Olejnik, M. LC-MS/MS Determination of 21 Non-Steroidal Anti-Inflammatory Drugs Residues in Animal Milk and Muscles. Molecules 2021, 26, 5892. https://doi.org/10.3390/molecules26195892
Pietruk M, Jedziniak P, Olejnik M. LC-MS/MS Determination of 21 Non-Steroidal Anti-Inflammatory Drugs Residues in Animal Milk and Muscles. Molecules. 2021; 26(19):5892. https://doi.org/10.3390/molecules26195892
Chicago/Turabian StylePietruk, Marta, Piotr Jedziniak, and Małgorzata Olejnik. 2021. "LC-MS/MS Determination of 21 Non-Steroidal Anti-Inflammatory Drugs Residues in Animal Milk and Muscles" Molecules 26, no. 19: 5892. https://doi.org/10.3390/molecules26195892
APA StylePietruk, M., Jedziniak, P., & Olejnik, M. (2021). LC-MS/MS Determination of 21 Non-Steroidal Anti-Inflammatory Drugs Residues in Animal Milk and Muscles. Molecules, 26(19), 5892. https://doi.org/10.3390/molecules26195892