Abstract
The search for novel antimycobacterial drugs is a matter of urgency, since tuberculosis is still one of the top ten causes of death from a single infectious agent, killing more than 1.4 million people worldwide each year. Nine Amaryllidaceae alkaloids (AAs) of various structural types have been screened for their antimycobacterial activity. Unfortunately, all were considered inactive, and thus a pilot series of aromatic esters of galanthamine, 3-O-methylpancracine, vittatine and maritidine were synthesized to increase biological activity. The semisynthetic derivatives of AAs were screened for their in vitro antimycobacterial activity against Mycobacterium tuberculosis H37Ra and two other mycobacterial strains (M. aurum, M. smegmatis) using a modified Microplate Alamar Blue Assay. The most active compounds were also studied for their in vitro hepatotoxicity on the hepatocellular carcinoma cell line HepG2. In general, the derivatization of the original AAs was associated with a significant increase in antimycobacterial activity. Several pilot derivatives were identified as compounds with micromolar MICs against M. tuberculosis H37Ra. Two derivatives of galanthamine, 1i and 1r, were selected for further structure optimalization to increase the selectivity index.
1. Introduction
Despite the advances made in the treatment of tuberculosis (TB) in recent years, this disease still remains one of the main public health problems. TB is a devastating infectious disease caused by a tubercle bacillus, Mycobacterium tuberculosis complex. From this group of closely related species, the most common causative agent of TB is represented by Mycobacterium tuberculosis (Mtb). The World Health Organization reports that about one-quarter of the global population has latent TB, and around 10.0 million people developed TB globally in 2019, with an estimated 1.4 million TB deaths among HIV-negative people, including 208,000 deaths among HIV-positive individuals [1]. Current treatment for drug-sensitive TB consists of standard first-line drugs, including isoniazid, rifampicin, ethambutol and pyrazinamide, used for at least six months [2]. However, the increasing prevalence of multi-drug-resistant (MDR) strains of Mtb, with resistance to at least rifampicin and isoniazid, has rendered these first-line antibiotics significantly less effective. Therefore, the search for new effective anti-tuberculosis (anti-TB) drugs, possessing activities against various subpopulations of Mtb, or with novel modes of action, remains a serious challenge for medicinal chemistry.
The poor efficiency of identifying new anti-TB and antibacterial drugs by the screening of pharmaceutical libraries is associated with limited chemical diversity within these collections. Additionally, most anti-TB and antibacterial drugs in general do not follow Lipinski´s rule of 5, which defines the optimal drug-like features, whereas pharmaceutical compounds are biased towards these properties [3].
Thus, the key question is: How do we search for new anti-TB and antibacterial drugs and where do we look for them?
One of the options represents different types of natural products (NPs). The importance of natural products (NPs) in drug discovery has been extensively documented, including their contribution to the development of present drugs [4,5]. The chemical diversity of NPs is more closely aligned with drugs than synthetic libraries, thus making them ideal candidates for drug-discovery projects. Moreover, naturally occurring pure compounds from higher and lower forms of plants as well as microorganisms and marine organisms have shown antimycobacterial potential in different studies. Without a doubt, the most studied group of natural products are alkaloids, which are classified into various structural types according to their biosynthetic origin. Recently, several reviews or perspective pieces have been published on this topic [6,7,8,9].
The plant family Amaryllidaceae is known for its structurally unique isoquinoline alkaloids called Amaryllidaceae alkaloids (AAs). Traditionally, plants of this family have been used as folk herbal remedies against various diseases by indigenous people around the world [10]. The biological activity of the plants is connected to the presence of AAs, which demonstrate a wide range of biological activities including antitumor, antibacterial, antioxidant, antiparasitic, antifungal, anti-inflammatory and insect antifeedant effects, as well as acetylcholin- and butyrylcholinesterase inhibitory activities [11,12,13]. The most known compound within the AAs is galanthamine, which is approved for the treatment of Alzheimer´s disease under the commercial names Razadyne© in the USA and Reminyl© in Europe and other countries [14]. Other interesting AAs are lycorine, pancratistatine and haemanthamine, which are intensively studied for their antitumor properties [15,16,17,18].
Given the fact that AAs have been, and are being, intensively studied for a wide range of biological activities, it is surprising that these natural products have not yet been addressed in terms of their antimycobacterial activity. Due to the fact that a number of AAs can be isolated from plants in large quantities, they are also an interesting target for the preparation of semisynthetic derivatives and detailed structure–activity relationship (SAR) studies leading to the optimalization of the structure.
As a part of our ongoing research on AAs and their semisynthetic derivatives as potential drugs, this work reports the antimycobacterial activity of selected AAs and their pilot semisynthetic derivatives.
2. Results and Discussion
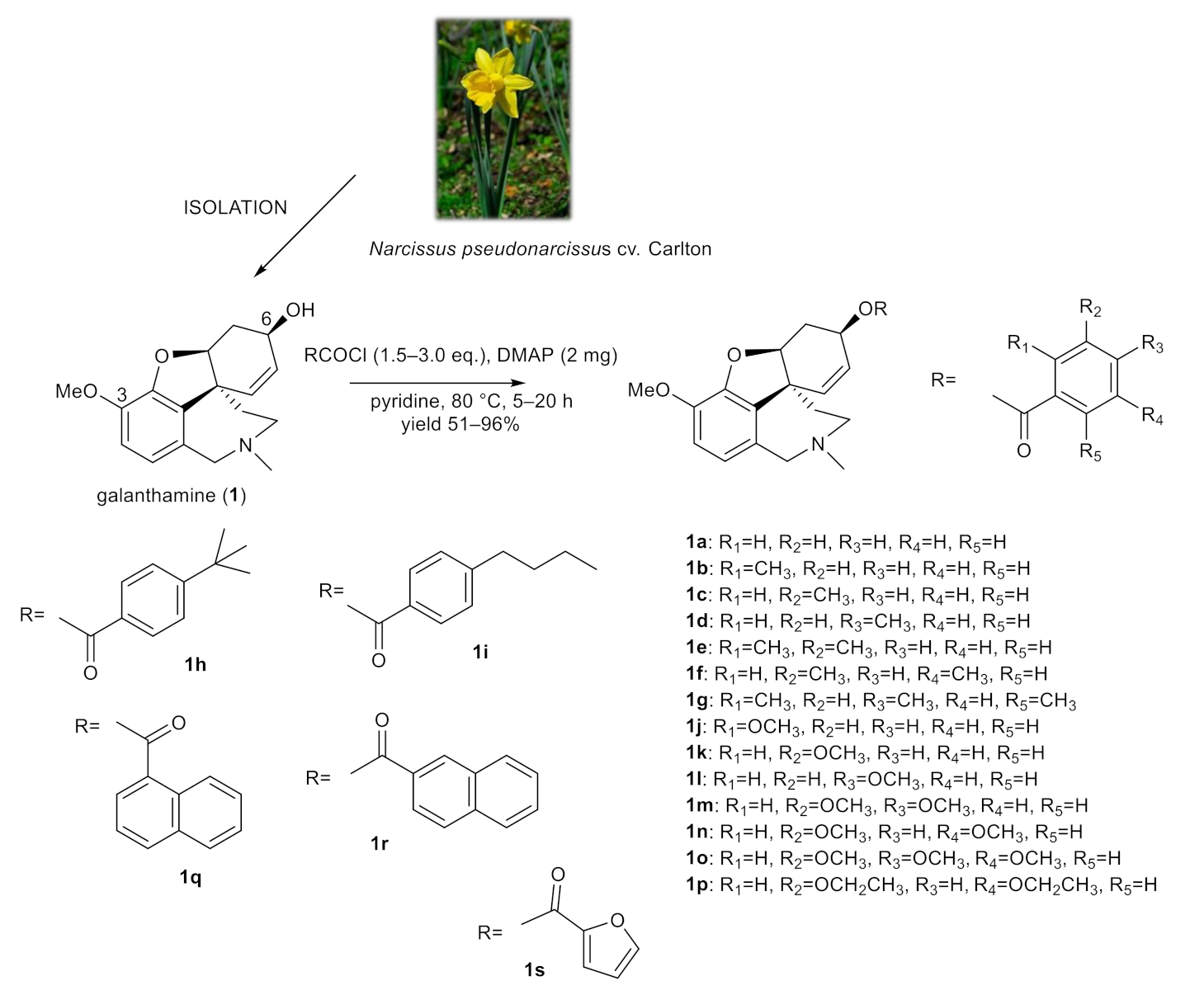
2.1. Synthesis of Galanthamine Derivatives (1a–1s)
Since galanthamine is used as a drug for the treatment of Alzheimer´s disease (AD), the original goal of this study was to develop new galanthamine derivatives, and investigate them as novel chemical entities with improved cholinesterases inhibition profiles. Unfortunately, derivatization of the hydroxy group at position C-6 with differently substituted aromatic moieties was associated with the loss of biological activity. Thus, selected galanthamine derivatives were randomly screened for their antimycobacterial activity. Surprisingly, a few of them demonstrated promising antimycobacterial activity, and thus the remaining derivatives were also screened.
The structural aspects of galanthamine (1) allowed us to prepare novel chemical entities by derivatization of the free hydroxyl group at position C-6 and to inspect the structure–activity relationship of the novel derivatives. Chemical modification of galanthamine was feasible because of its previous large-scale isolation from Narcissus pseudonarcissus cv. Carlton. Modification of the structure was inspired by previous structural changes of haemanthamine, ambelline and vittatine [12,19,20,21]. Using the same method described in our previous reports, the hydroxy group at C-6 was acylated with differently substituted benzoyl chlorides, 1- and 2-naphthoyl chlorides and 2-furoyl chloride affording the corresponding esters 1a–1s (Figure 1). Derivatives (except 1g–1i) were converted to their hydrochlorides to increase their solubility. The structures of the newly synthesized compounds were determined by MS, HRMS and 1D- and 2D-NMR spectroscopic techniques (see Supplementary Materials). The data were fully consistent with the proposed structures. The yield of all reactions was >52% (the yield for each reaction is specified in the experimental part).
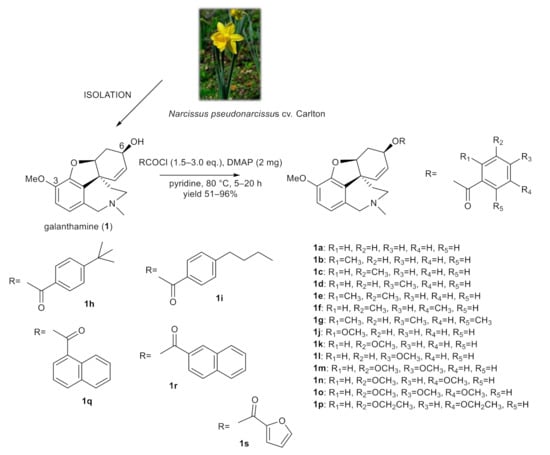
Figure 1.
Chemical procedures affording galanthamine derivatives (1a–1s).
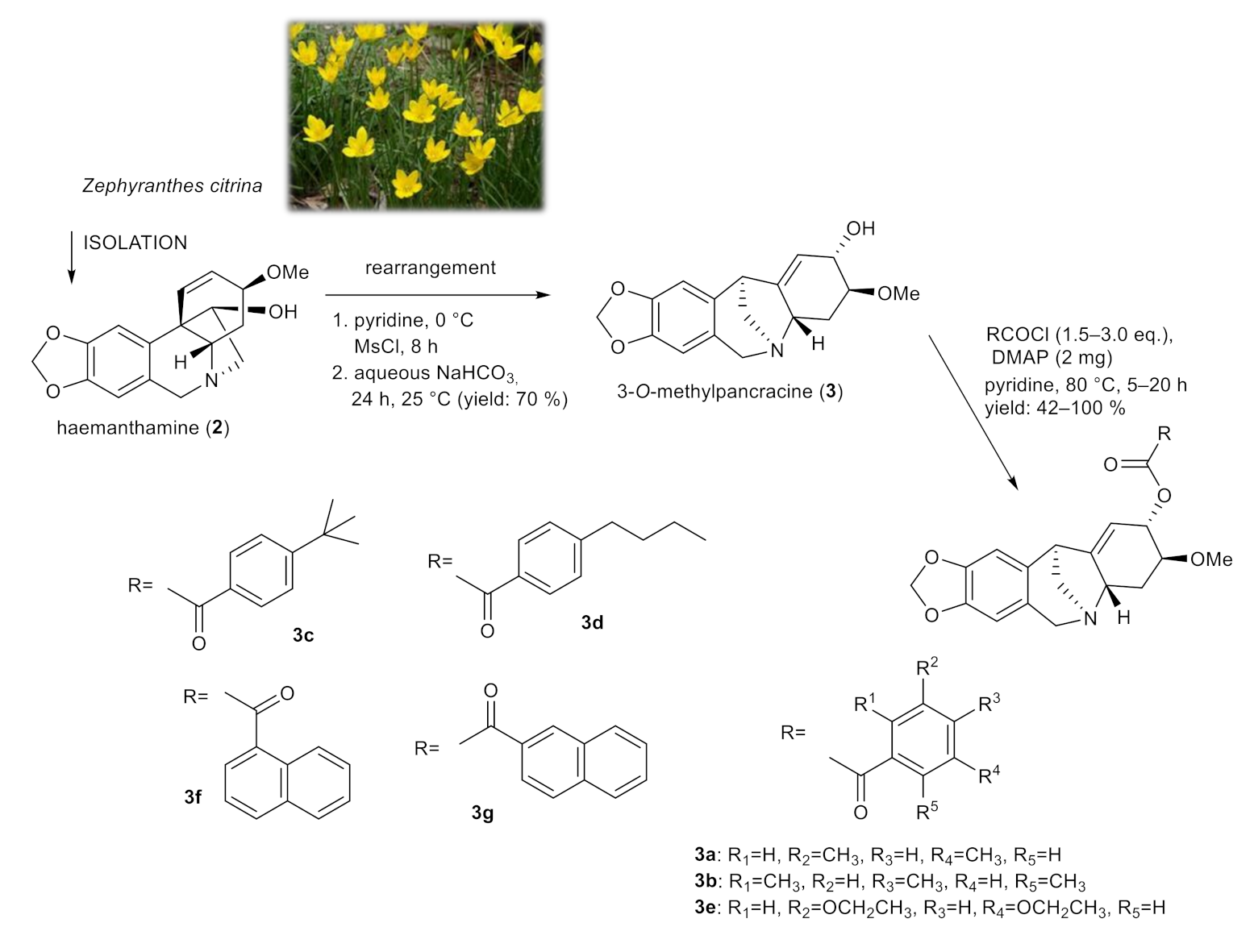
2.2. Synthesis of 3-O-methylpancracine (3a–3g), Vittatine (4a, 4b) and Maritidine Derivatives (5a, 5b)
Derivatives of 3-O-methylpancracine were originally synthesized to study the cytotoxic potential of montanine-type Amaryllidaceae alkaloids. The promising antiproliferative properties of some representatives of this structural type of AA, and the limited presence of these alkaloids in plant material, initiated the total synthesis and production of synthetic analogues of these alkaloids [22,23]. Among these, the montanine-type structure was successfully achieved with intramolecular rearrangement of haemanthamine, which is considered a convenient semi-synthetic method for achieving the montanine-type skeleton due to the ready availability of haemanthamine from plants of the Amaryllidaceae family [23]. Unfortunately, similar to galanthamine derivatives, the structural modification of the montanine skeleton was associated with the loss of cytotoxic activity, and thus we decided to screen selected derivatives of 3-O-methylpancracine for antimycobacterial activity.
Using a skeletal rearrangement of haemanthamine (2) and subsequent acylation, different substituted esters of 3-O-methylpancracine (3) were synthesized. The structural modifications of 3 are displayed in Figure 2.
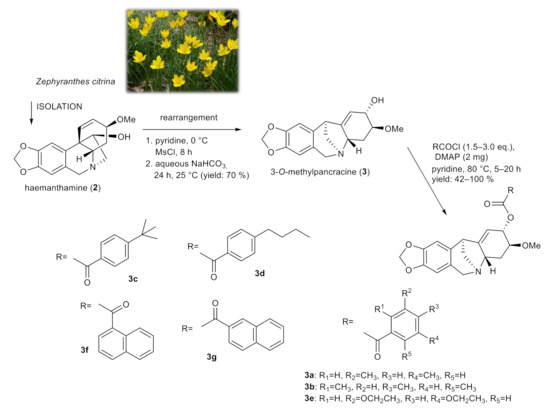
Figure 2.
Chemical procedures affording 3-O-methylpancracine derivatives (3a–3g).
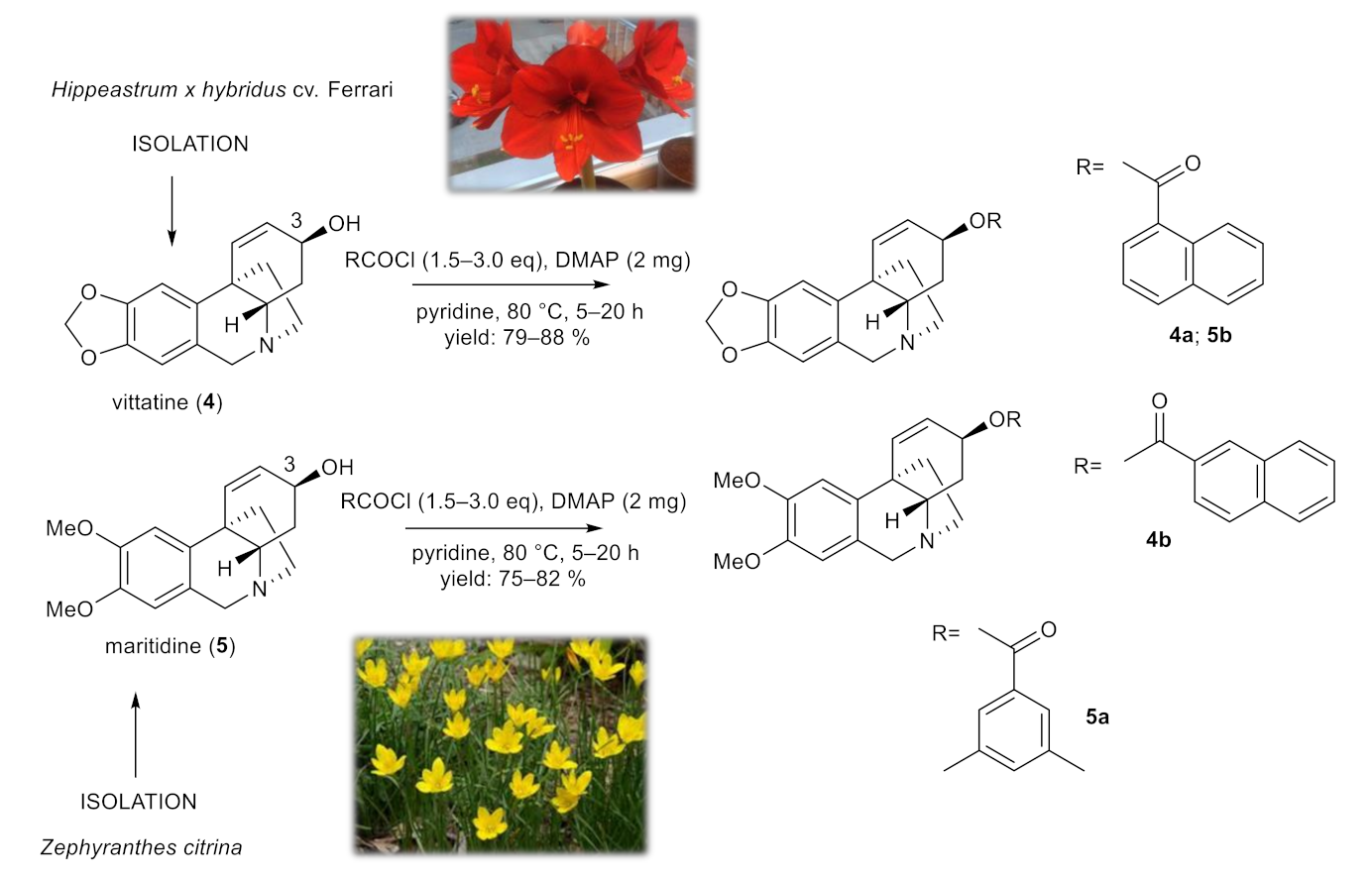
To study the role of the structural type of AAs in the structure of the tested esters, pilot derivatives of vittatine (4) and maritidine (5), corresponding to the active galanthamine analogues, were also synthesized for more detailed SAR studies (Figure 3). The amount of both alkaloids isolated was limited and allowed the preparation of only two pilot derivatives from each alkaloid (4a–4b; 5a–5b). The structural modifications of 4 and 5 are displayed in Figure 3. The structures were determined by HRMS and 1D- and 2D-NMRspectroscopic techniques (see Supplementary Materials). The yield of all reactions was >42% (the yield for each reaction is specified in the experimental part). All synthesized derivatives were converted to their hydrochlorides to improve the solubility for antimycobacterial assay.
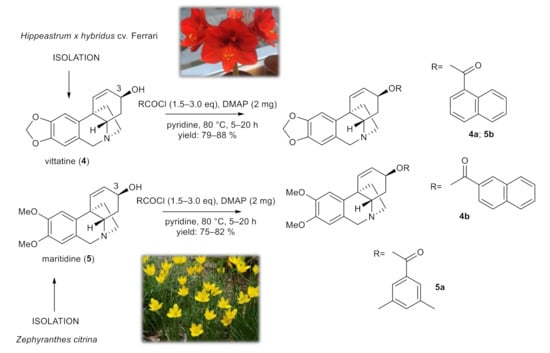
Figure 3.
Chemical procedures affording vittatine (4a,4b) and maritidine derivatives (5a,5b).
2.3. Antimycobacterial Activity of Selected Amaryllidaceae Alkaloids (1–9) and Their Synthesized Derivatives (1a–1s, 3a–3g, 4a–4b and 5a–5b)
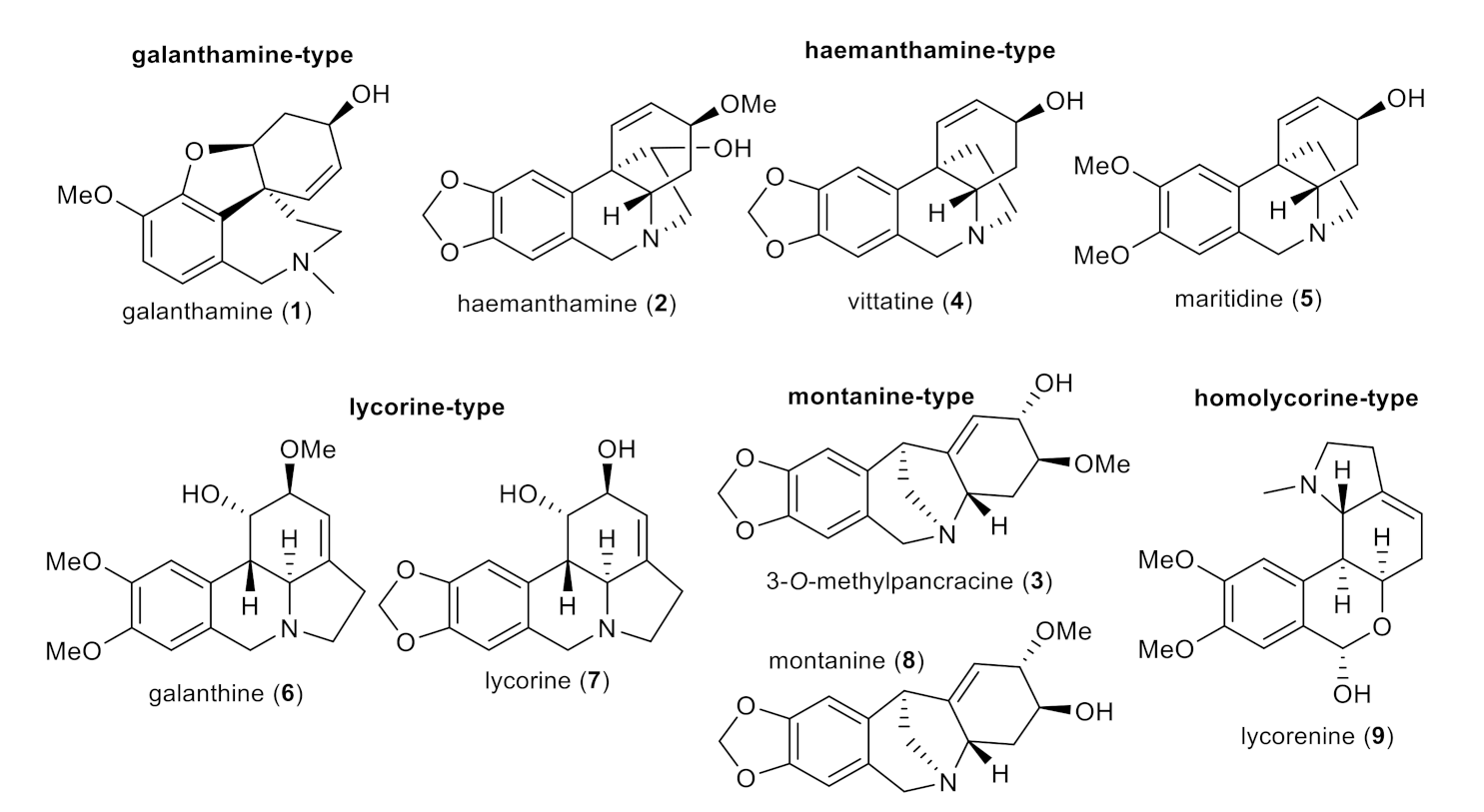
In the current study, nine AAs of various structural types isolated previously from different Amaryllidaceae plants (except 3-O-methylpancracine, which has been synthesized within the current study; Figure 4) [19,20,24], along with 30 newly synthesized derivatives of galanthamine (1a–1s), 3-O-methylpancracine (3a–3g), vittatine (4a–4b) and maritidine (5a–5b) have been screened for their antimycobacterial activity against three different mycobacterial strains: M. tuberculosis H37Ra, M. aurum and M. smegmatis. The last two are fast-growing strains that do not cause tuberculosis in humans, but are much safer to handle. Their phenotype and genotype are close to those of Mtb, which allows their use as a surrogate model of Mtb [25]. Antimycobacterial activity is expressed as the minimum inhibitory concentration (MIC).
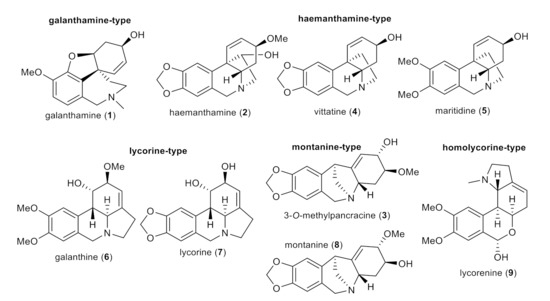
Figure 4.
Structures of tested Amaryllidaceae alkaloids of different structural types.
Although alkaloids of different structural types and biosynthetic origin have been reported as compounds with promising antimycobacterial activities, all screened AAs lacked any antimycobacterial activity (Table 1). These results where surprising to us, especially in view of the fact that these natural products demonstrate a wide range of biological activities.

Table 1.
In vitro antimycobacterial activity against Mtb H37Ra, Mycobacterium aurum and M. smegmatis (MIC), cytotoxicity (IC50), selectivity index (SI) and calculated lipophilicity (logP, ClogP) of Amaryllidaceae alkaloids and their derivatives.
As already mentioned above, the analogues of galanthamine were originally intended to study their cholinesterases inhibition potency, but all derivatives were considered inactive. Thus, selected derivatives were initially screened for antimycobacterial activity, and, surprisingly, some of them showed strong activity, and thus the remaining derivatives were also tested. Interestingly, derivatization of the C-6 hydroxy group was in all cases associated with a significant increase in antimycobacterial activity against all studied Mycobacterium strains (MIC = 1.56–62.5 µg/mL), with a few exceptions (Table 1). Among the substituted benzoyl-derivatives of galanthamine, the strongest activity was related to the presence of either an isobutyl- (1h) or butyl-chain (1i) in the para position on the benzene ring. Both derivatives displayed impressive activity against all the studied strains (MICs = 3.125–7.81 µg/mL and MICs = 1.56–7.81 µg/mL, respectively). The most sensitive strain was M. tuberculosis H37Ra with an MIC = 3.125 µg/mL (6.9 µM) for 6-O-(4-tert-butylbenzoyl)galanthamine (1h) and MIC = 1.56 µg/mL (3.5 µM) for 6-O-(4-butylbenzoyl)galanthamine (1i). The presence of a longer chain on the aromatic ring seems to be responsible for a dramatic increase in antimycobacterial activity. These findings can be deduced by comparing the activities of derivatives 1d and 1i, which differ only in the size of the substituent on the aromatic ring in the para position. When there is a methyl group, as in 6-O-(4-methylbenzoyl)galanthamine (1d), the analogue shows up to ten times lower activity than the substance with a butyl-substitution, as in 6-O-(4-butylbenzoyl)galanthamine (1i) (Table 1). The presence of two or three substituents (methyl-, methoxy- or ethoxy- group) on the benzene ring (like in 1e, 1f, 1g and 1p) was also associated with an interesting increase in antimycobacterial activity. Within these compounds, 6-O-(3,5-diethoxybenzoyl)galanthamine (1p) showed promising antimycobacterial activity, with MICs = 3.91–15.625 µg/mL (7.6–30.2 µM). Interesting activity was also demonstrated by the naphthoyl-derivatives of galanthamine (1q and 1r). Among these, 6-O-(2-naphthoyl)galanthamine (1r) showed up to three times stronger activity, with MICs = 1.98–7.81 µg/mL, in comparison with the galanthamine analogue, for which the 1-naphthoyl chloride was used for esterification (1q, MICs = 6.25–7.81 µg/mL).
These findings encouraged us to study the antimycobacterial activity of further Amaryllidaceae alkaloids (previously isolated or synthesized in our laboratory) with the same derivatization as the most active galanthamine analogues. Thus, the pilot series was tested on analogues of 3-O-methylpancracine (3a–3g), corresponding to the most active galanthamine-derivatives. In general, the derivatives of 3-O-methylpancracine demonstrated either comparable or slightly lower/higher antimycobacterial activity. Interestingly, among the naphthoyl derivatives of 3, the stronger antimycobacterial potency was associated with the presence of a 1-naphthoyl-substituent (3f, MICs = 3.91 µg/mL for all studied strains), instead of a 2-naphthoyl-substituent, like in the galanthamine analogue (1r). To study this phenomenon, two naphthoyl-analogues of vittatine (4) were synthesized and tested (4a–4b). In this case, the same antimycobacterial activity was obtained for both analogues (MICs = 3.91–7.81 µg/mL). The limited amount of maritidine (5) isolated allowed preparation of only two derivatives (5a, 5b), both of which also showed interesting activity (MICs = 3.91–15.625 µg/mL; Table 1).
2.4. Lipophilicity Versus Activity
Lipophilicity is one of the most important physicochemical properties of a compound, which seems to be an important factor related to cell transmembrane transport. The mycobacterial cell wall is rich in mycolic acids, which efficiently prevent the penetration of drugs. Drugs with higher lipophilicity may exert better activity against M. tuberculosis [26], and thus we calculated logP/ClogP values using the ChemBioDraw Ultra program (ver 18.1). The ClogP value is correlated directly to the molecular hydrophobicity, and thereby, to the diffusion through the biological membranes, i.e., through the mycobacterial cell wall. The low antimycobacterial activity of the tested alkaloids is in good agreement with their values for logP/ClogP (logP ≤ 1.45; ClogP ≤ 1.08, Table 1). Actually, the novel active compounds in this study (MIC ≤ 7.81 µg/mL) reached logP values ranging between 3.72 and 5.27 and ClogP 4.20–5.99 (ClogP), indicating their potential for development as antimycobacterial drugs.
2.5. Cytotoxicity of Selected Derivatives of Amaryllidaceae Alkaloids
The most active derivatives were further evaluated for in vitro cytotoxicity on hepatocellular carcinoma cells (HepG2) using an MTT assay. The HepG2 cell line serves as an in vitro model for hepatotoxicity for early drug screening. Moreover, the hepatocellular model was chosen since antitubercular drugs are known to carry the risk of hepatotoxicity and represent the most likely target for chronic toxicity of antitubercular drugs, which often complicates the six-months-long therapy for TB [27].
The evaluation of cytotoxicity allows the calculation of selectivity indexes (SI) as the ratio of IC50,HepG2 to MIC of Mtb H37Ra (Table 1). In general, values of SI higher than 10 indicate more acceptable toxicity (analogous to the therapeutic index). The active compounds were screened at an initial concentration of 50 µM, and the IC50 values were subsequently determined. The effective cytotoxic concentrations of all tested compounds are listed in Table 1. Among these compounds, 6-O-(2,4,6-trimethylbenzoyl)galanthamine (1g) exhibited the highest cytotoxicity with an IC50 value = 13.9 ± 0.8 µM. The most active derivatives in the antimycobacterial assay (1i and 1r) showed cytotoxicity with IC50 values of 14.7 ± 1.6 µM, and 21.2 ± 3.8 µM, reaching an SI of 4.20 for 1i and 5.17 for 1r, which indicates the potential risk of hepatotoxicity. In subsequent structure optimalization of the active antitubercular derivatives, the SI must be increased to reduce the hepatotoxicity of the developed compounds.
3. Materials and Methods
3.1. General Experimental Procedures
All solvents were treated using standard techniques before use. All reagents and catalysts were purchased from Sigma Aldrich, Czech Republic, and used without purification. NMR spectra were recorded in CDCl3 on either a VNMR S500 (Varian) spectrometer operating at 500 MHz for 1H and 125.7 MHz for 13C or a Jeol JNM-ECZ600R operating at 600 MHz for 1H and 151 MHz for 13C at an ambient temperature. The residual signal of CHCl3 (δ 7.26 ppm) was a reference for 1H NMR spectra, and the central signal of the CDCl3 signals (δ 77.0 ppm) was used as a reference for proton-decoupled 13C NMR spectra. The coupling constant (J) is given in Hz, and the chemical shifts are reported in ppm. For unambiguous assignment of 1H and 13C NMR signals, ESI-HRMS were obtained with a Waters Synapt G2-Si hybrid mass analyzer of a quadrupole-time-of-flight (Q-TOF) type, coupled to a Waters Acquity I-Class UHPLC system. EI-MS were obtained on an Agilent 7890A GC 5975 inert MSD operating in EI mode at 70 eV (Agilent Technologies, Santa Clara, CA, USA). A DB-5 column (30 m × 0.25 mm × 0.25 μm, Agilent Technologies, USA) was used with a temperature program: 100–180 °C at 15°C/min, 1 min hold at 180 °C, and 180–300 °C at 5 °C/min and 5 min hold at 300 °C; and a detection range of m/z 40–600. The injector temperature was 280 °C. The flow-rate of the carrier gas (helium) was 0.8 mL/min. A split ratio of 1:15 was used. UV and ECD spectra were recorded on a JASCO J-815 CD spectrometer. Compounds on TLC plates were observed under UV light (254 and 366 nm) and visualized by spraying with Dragendorff’s reagent.
3.2. Amaryllidaceae Alkaloids
Alkaloids 1 and 9 were isolated from Narcissus pseudonarcissus cv. Carlton, alkaloids 2 and 5–7 from Zephyranthes citrina and alkaloids 4 and 8 from Hippeastrum x hybridum cv. Ferrari, as previously reported in our studies [12,20,28]. The purity of all compounds, verified by NMR, was 95%.
3.3. Preparation of Galanthamine (1), Vittatine (4) and Maritidine (5) Derivatives
The same procedure as described previously was used to afford the corresponding esters 1a–1s, 4a–4b and 5a–5b [20,29].
3.3.1. 6-O-benzoylgalanthamine (1a)
Yield 64 mg (96%); white amorphous solid; = −25.3° (c 0.19, MeOH); 1H NMR (500 MHz, CDCl3) δ: 8.08–8.04 (m, 2H), 7.55–7.47 (m, 1H), 7.41–7.34 (m, 2H), 6.69 (d, J = 8.1 Hz, 1H), 6.59 (d, J = 8.1 Hz, 1H), 6.37 (d, J = 10.3 Hz, 1H), 6.05 (dd, J = 10.3 Hz, J = 5.2 Hz, 1H), 5.63–5.56 (m, 1H), 4.69–4.65 (m, 1H), 4.16 (d, J = 15.2 Hz, 1H), 3.90 (s, 3H), 3.69 (d, J = 15.2 Hz, 1H), 3.36 (t, J = 13.8 Hz, 1H), 3.14–3.05 (m, 1H), 2.84–2.75 (m, 1H), 2.42 (s, 3H), 2.25–2.12 (m, 2H), 1.68–1.60 (m, 1H) 13C NMR (126 MHz, CDCl3) δ: 166.2, 146.7, 144.0, 132.7, 132.0, 131.0, 130.6, 129.9, 129.3, 128.1, 122.8, 121.2, 111.4, 86.3, 63.6, 60.3, 56.0, 53.7, 48.2, 41.7, 34.2, 28.0; ESI-HRMS m/z calcd for C24H26NO4 [M + H]+ 392.1856 found 392.1857.
3.3.2. 6-O-(2-methylbenzoyl)galanthamine (1b)
Yield 60 mg (85%); white amorphous solid; = −13.3° (c 0.12, MeOH); 1H NMR (500 MHz, CDCl3) δ: 8.03 (dd, J = 7.9 Hz, J = 1.5 Hz, 1H), 7.35 (td, J = 7.9 Hz, J = 1.5 Hz, 1H), 7.27–7.14 (m, 2H), 6.68 (d, J = 8.1 Hz, 1H), 6.59 (d, J = 8.1 Hz, 1H), 6.35 (d, J = 10.3 Hz, 1H), 6.04 (dd, J = 10.3 Hz, J = 5.1 Hz, 1H), 5.60–5.53 (m, 1H), 4.65–4.63 (m, 1H), 4.15 (d, J = 15.2 Hz, 1H), 3.87 (s, 3H), 3.70 (d, J = 15.2 Hz, 1H), 3.35 (t, J = 12.8 Hz, 1H), 3.15–3.05 (m, 1H), 2.85–2.78 (m, 1H), 2.59 (s, 3H), 2.42 (s, 3H), 2.24–2.13 (m, 2H), 1.69–1.60 (m, 1H); 13C NMR (126 MHz, CDCl3) δ: 167.2, 146.7, 144.0, 140.4, 132.0, 131.8, 131.5, 131.4, 130.9, 129.7, 129.2, 125.6, 122.9, 121.3, 111.5, 86.3, 63.4, 60.4, 56.0, 53.8, 48.1, 41.8, 34.3, 27.9, 21.7; ESI-HRMS m/z calcd for C25H28NO4 [M + H]+ 406.2013 found 406.2013.
3.3.3. 6-O-(3-methylbenzoyl)galanthamine (1c)
Yield 41 mg (58%); white amorphous solid; = −17.0° (c 0.16, MeOH); 1H NMR (500 MHz, CDCl3) δ: 7.89–7.84 (m, 2H), 7.34–7.30 (m, 1H), 7.29–7.23 (m, 1H), 6.69 (d, J = 8.1 Hz, 1H), 6.59 (d, J = 8.1 Hz, 1H), 6.36 (d, J = 10.3 Hz, 1H), 6.03 (dd, J = 10.3 Hz, J = 5.2 Hz, 1H), 5.62–5.57 (m, 1H), 4.68–4.63 (m, 1H), 4.14 (d, J = 15.2 Hz, 1H), 3.88 (s, 3H), 3.68 (d, J = 15.2 Hz, 1H), 3.34 (t, J = 13.7 Hz, 1H), 3.12–3.03 (m, 1H), 2.81–2.73 (m, 1H), 2.41 (s, 3H), 2.37 (s, 3H), 2.23–2.11 (m, 2H), 1.66–1.59 (m, 1H); 13C NMR (126 MHz, CDCl3) δ: 166.3, 146.7, 144.0, 137.8, 133.4, 132.0, 130.9, 130.4, 130.3, 129.3, 127.9, 127.0, 122.8, 121.2, 111.4, 86.3, 63.4, 60.3, 55.9, 53.7, 48.1, 41.8, 34.2, 27.9, 21.1; ESI-HRMS m/z calcd for C25H28NO4 [M + H]+ 406.2013 found 406.2015.
3.3.4. 6-O-(4-methylbenzoyl)galanthamine (1d)
Yield 38 mg (51%); white amorphous solid; = −27.4° (c 0.35, MeOH); 1H NMR (500 MHz, CDCl3) δ: 7.98–7.90 (m, 2H, AA′BB′), 7.20–7.14 (m, 2H, AA′BB′), 6.68 (d, J = 8.1 Hz, 1H), 6.58 (d, J = 8.1 Hz, 1H), 6.35 (d, J = 10.3 Hz, 1H), 6.04 (dd, J = 10.3 Hz, J = 5.2 Hz, 1H), 5.60–5.54 (m, 1H), 4.68–4.62 (m, 1H), 4.14 (d, J = 15.1 Hz, 1H), 3.89 (s, 3H), 3.68 (d, J = 15.1 Hz, 1H), 3.34 (t, J = 12.8 Hz, 1H), 3.12–3.03 (m, 1H), 2.82–2.73 (m, 1H), 2.41 (s, 3H), 2.38 (s, 3H), 2.23–2.11 (m, 2H), 1.66–1.55 (m, 1H); 13C NMR (126 MHz, CDCl3) δ: 166.2, 146.7, 143.9, 143.2, 132.0, 130.8, 129.9, 129.3, 128.8, 127.8, 122.9, 121.1, 111.5, 86.3, 63.4, 60.3, 56.0, 53.7, 48.1, 41.7, 34.2, 28.0, 21.6; ESI-HRMS m/z calcd for C25H28NO4 [M + H]+ 406.2013 found 406.2019.
3.3.5. 6-O-(2,3-dimethylbenzoyl)galanthamine (1e)
Yield 45 mg (61%); white amorphous solid; = −21.4° (c 0.17, MeOH); 1H NMR (600 MHz, CDCl3) δ: 7.74 (d, J = 8.0 Hz, 1H), 7.22 (d, J = 8.0 Hz, 1H), 7.04 (t, J = 8.0 Hz, 1H), 6.66 (d, J = 8.1 Hz, 1H), 6.57 (d, J = 8.1 Hz, 1H), 6.33 (d, J = 10.3 Hz, 1H), 6.02 (dd, J = 10.3 Hz, J = 5.2 Hz, 1H), 5.55–5.50 (m, 1H), 4.64–4.60 (m, 1H), 4.14 (d, J = 15.2 Hz, 1H), 3.84 (s, 3H), 3.68 (d, J = 15.2 Hz, 1H), 3.34 (t, J = 13.5 Hz, 1H), 3.10–3.05 (m, 1H), 2.81–2.75 (m, 1H), 2.43 (s, 3H), 2.40 (s, 3H), 2.27 (s, 3H), 2.20–2.11 (m, 2H), 1.66–1.60 (m, 1H); 13C NMR (151 MHz, CDCl3) δ: 168.2, 146.7, 144.1, 137.9, 137.5, 133.0, 132.0, 130.9, 130.9, 128.8, 125.0, 123.0, 121.3, 111.5, 86.3, 63.6, 60.4, 56.0, 53.7, 48.1, 41.7, 34.2, 27.9, 20.5, 16.5; ESI-HRMS m/z calcd for C26H30NO4 [M + H]+ 420.2169 found 420.2179.
3.3.6. 6-O-(3,5-dimethylbenzoyl)galanthamine (1f)
Yield 41 mg (56%); white amorphous solid; = −15.3° (c 0.23, MeOH); 1H NMR (500 MHz, CDCl3) δ: 7.67 (s, 2H), 7.14 (s, 1H), 6.69 (d, J = 8.1 Hz, 1H), 6.59 (d, J = 8.1 Hz, 1H), 6.35 (d, J = 10.3 Hz, 1H), 6.02 (dd, J = 10.3 Hz, J = 5.1 Hz, 1H), 5.64–5.52 (m, 1H), 4.67–4.62 (m, 1H), 4.14 (d, J = 15.1 Hz, 1H), 3.87 (s, 3H), 3.69 (d, J = 15.1 Hz, 1H), 3.34 (t, J = 13.6 Hz, 1H), 3.12–3.00 (m, 1H), 2.82–2.70 (m, 1H), 2.41 (s, 3H), 2.33 (s, 6H), 2.22–2.06 (m, 2H), 1.66–1.59 (m, 1H); 13C NMR (126 MHz, CDCl3) δ: 166.5, 146.7, 144.0, 137.6, 134.3, 132.0, 130.7, 130.3, 129.2, 127.5, 122.8, 121.2, 111.4, 86.3, 63.2, 60.3, 55.9, 53.7, 48.0, 41.7, 34.2, 27.9, 21.0; ESI-HRMS m/z calcd for C26H30NO4 [M + H]+ 420.2169 found 420.2174.
3.3.7. 6-O-(2,4,6-trimethylbenzoyl)galanthamine (1g)
Yield 50 mg (66%); yellow amorphous solid; = −12.6° (c 0.19, MeOH); 1H NMR (600 MHz, CDCl3) δ: 6.77 (s, 2H), 6.61 (d, J = 8.1 Hz, 1H), 6.54 (d, J = 8.1 Hz, 1H), 6.28 (d, J = 10.3 Hz, 1H), 6.02 (dd, J = 10.3 Hz, J = 4.9 Hz, 1H), 5.57–5.48 (m, 1H), 4.61–4.53 (m, 1H), 4.10 (d, J = 15.1 Hz, 1H), 3.72 (s, 3H), 3.66 (d, J = 15.1 Hz, 1H), 3.29 (t, J = 13.9 Hz, 1H), 3.11–2.99 (m, 1H), 2.83–2.75 (m, 1H), 2.38 (s, 3H), 2.25 (s, 6H), 2.23 (s, 3H), 2.21–2.12 (m, 2H), 1.64–1.58 (m, 1H); 13C NMR (151 MHz, CDCl3) δ: 169.9, 146.8, 144.0, 139.0, 135.6, 131.9, 130.9, 130.6, 129.0, 128.3, 122.7, 121.3, 112.0, 85.9, 64.2, 60.4, 56.1, 53.7, 48.0, 41.9, 34.4, 28.0, 21.1, 19.8. ESI-HRMS m/z calcd for C27H32NO4 [M + H]+ 434.2326 found 434.2332.
3.3.8. 6-O-(4-tert-butylbenzoyl)galanthamine (1h)
Yield 70 mg (89%); yellow amorphous solid; = −25.5° (c 0.44, MeOH); 1H NMR (600 MHz, CDCl3) δ: 7.98–7.93 (m, 2H, AA′BB′), 7.38– 7.34 (m, 2H, AA′BB′), 6.67 (d, J = 8.1 Hz, 1H), 6.57 (d, J = 8.1 Hz, 1H), 6.33 (d, J = 10.2 Hz, 1H), 6.02 (dd, J = 10.2 Hz, J = 5.2 Hz, 1H), 5.57–5.52 (m, 1H), 4.65–4.62 (m, 1H), 4.14 (d, J = 15.1 Hz, 1H), 3.88 (s, 3H), 3.67 (d, J = 15.1 Hz, 1H), 3.34 (t, J = 13.3 Hz, 1H), 3.11–3.03 (m, 1H), 2.78–2.70 (m, 1H), 2.39 (s, 3H), 2.21–2.10 (m, 2H), 1.64–1.58 (m, 1H), 1.30 (s, 9H); 13C NMR (151 MHz, CDCl3) δ: 166.3, 156.2, 146.8, 144.0, 132.1, 130.9 129.8, 129.2, 127.8, 125.0, 123.0, 121.2, 111.5, 86.3, 63.4, 60.3, 56.0, 53.7, 48.2, 41.6, 34.9, 34.2, 31.1, 28.0; ESI-HRMS m/z calcd for C28H34NO4 [M + H]+ 448.2482 found 448.2494.
3.3.9. 6-O-(4-butylbenzoyl)galanthamine (1i)
Yield 75 mg (96%); yellow amorphous solid; = −20.4° (c 0.45, MeOH); 1H NMR (600 MHz, CDCl3) δ: 7.96–7.92 (m, 2H, AA′BB′), 7.17–7.12 (m, 2H, AA′BB′) , 6.67 (d, J = 8.1 Hz, 1H), 6.57 (d, J = 8.1 Hz, 1H), 6.33 (d, J = 10.3 Hz, 1H), 6.02 (dd, J = 10.3 Hz, J = 5.2 Hz, 1H), 5.58–5.52 (m, 1H), 4.68–4.60 (m, 1H), 4.13 (d, J = 15.1 Hz, 1H), 3.88 (s, 3H), 3.67 (d, J = 15.1 Hz, 1H), 3.33 (t, J = 13.8 Hz, 1H), 3.09–3.03 (m, 1H), 2.78–2.73 (m, 1H), 2.62 (t, J = 7.4 Hz, 2H), 2.39 (s, 3H), 2.20–2.10 (m, 2H), 1.66–1.51 (m, 3H), 1.32 (dq, J = 14.8 Hz, J = 7.4 Hz, 2H), 0.90 (t, J = 7.4 Hz, 3H); 13C NMR (151 MHz, CDCl3) δ: 166.3, 148.2, 146.8, 144.0, 132.1, 130.9, 130.0, 129.3, 128.2, 128.0, 123.0, 121.2, 111.5, 86.3, 63.4, 60.3, 56.1, 53.7, 48.2, 41.7, 35.7, 34.2, 33.3, 28.0, 22.2, 13.8; ESI-HRMS m/z calcd for C28H34NO4 [M + H]+ 448.2482 found 448.2493.
3.3.10. 6-O-(2-methoxybenzoyl)galanthamine (1j)
Yield 58 mg (81%); white amorphous solid; = −26.0° (c 0.20, MeOH); 1H NMR (600 MHz, CDCl3) δ: 7.93 (dd, J = 7.7 Hz, J = 1.9 Hz, 1H), 7.46–7.36 (m, 1H), 6.97–6.83 (m, 2H), 6.65 (d, J = 8.0 Hz, 1H), 6.57 (d, J = 8.0 Hz, 1H), 6.32 (d, J = 10.1 Hz, 1H), 6.01 (dd, J = 10.1 Hz, J = 5.3 Hz, 1H), 5.57–5.53 (m, 1H), 4.66–4.58 (m, 1H), 4.13 (d, J = 15.1 Hz, 1H), 3.87 (s, 3H), 3.85 (s, 3H), 3.67 (d, J = 15.1 Hz, 1H), 3.33 (t, J = 13.8 Hz, 1H), 3.10–3.04 (m, 1H), 2.82–2.72 (m, 1H), 2.40 (s, 3H), 2.23–2.08 (m, 2H), 1.65–1.56 (m, 1H); 13C NMR (151 MHz, CDCl3) δ: 165.1, 159.6, 146.7, 144.0, 133.4, 132.7, 132.1, 130.8, 129.0, 123.1, 121.3, 120.0, 119.9, 111.7, 111.5, 86.4, 63.3, 60.3, 56.0, 55.9, 53.7, 48.1, 41.6, 34.2, 27.9; ESI-HRMS m/z calcd for C25H28NO5 [M + H]+ 422.1962 found 422.1970.
3.3.11. 6-O-(3-methoxybenzoyl)galanthamine (1k)
Yield 64 mg (90%); white amorphous solid; = −19.4° (c 0.35, MeOH); 1H NMR (500 MHz, CDCl3) δ: 7.67 (d, J = 7.6 Hz, 1H), 7.59–7.55 (m, 1H), 7.28 (t, J = 7.6 Hz, 1H), 7.06 (dd, J = 7.6 Hz, J = 2.7 Hz, 1H), 6.69 (d, J = 8.1 Hz, 1H), 6.60 (d, J = 8.1 Hz, 1H), 6.35 (d, J = 10.3 Hz, 1H), 6.05 (dd, J = 10.3 Hz, J = 5.1 Hz, 1H), 5.65–5.55 (m, 1H), 4.68–4.63 (m, 1H), 4.18 (d, J = 15.1 Hz, 1H), 3.86 (s, 3H), 3.84 (s, 3H), 3.72 (d, J = 15.1 Hz, 1H), 3.38 (t, J = 13.6 Hz, 1H), 3.17–3.05 (m, 1H), 2.79 (d, J = 16.2 Hz, 1H), 2.43 (s, 3H), 2.24–2.13 (m, 2H), 1.73–1.61 (m, 1H); 13C NMR (126 MHz, CDCl3) δ: 166.0, 159.3, 146.7, 144.0, 131.9, 131.8, 130.9, 129.1, 129.0, 122.7, 122.4, 121.2, 119.4, 113.9, 111.3, 86.3, 63.6, 60.3, 55.8, 55.2, 53.6, 48.1, 41.7, 34.2, 27.8; ESI-HRMS m/z calcd for C25H28NO5 [M + H]+ 422.1962 found 422.1972.
3.3.12. 6-O-(4-methoxybenzoyl)galanthamine (1l)
Yield 45 mg (63%); white amorphous solid; = −73.6° (c 0.12, MeOH); 1H NMR (500 MHz, CDCl3) δ: 8.06–7.91 (m, 2H, AA’BB‘), 6.90–6.78 (m, 2H, AA’BB‘), 6.67 (d, J = 8.1 Hz, 1H), 6.57 (d, J = 8.1 Hz, 1H), 6.34 (d, J = 10.4 Hz, 1H), 6.03 (dd, J = 10.4 Hz, J = 5.2 Hz, 1H), 5.58–5.49 (m, 1H), 4.67–4.62 (m, 1H), 4.14 (d, J = 15.3 Hz, 1H), 3.88 (s, 3H), 3.82 (s, 3H), 3.67 (d, J = 15.3 Hz, 1H), 3.33 (t, J = 13.6 Hz, 1H), 3.11–3.00 (m, 1H), 2.81–2.67 (m, 1H), 2.40 (s, 3H), 2.25–2.01 (m, 2H), 1.68–1.56 (m, 1H); 13C NMR (126 MHz, CDCl3) δ: 165.9, 163.1, 146.7, 143.9, 132.0, 131.9, 130.8, 129.3, 123.0, 123.0, 121.1, 113.3, 111.4, 86.3, 63.2, 60.3, 56.0, 55.2, 53.6, 48.1, 41.7, 34.2, 28.0; ESI-HRMS m/z calcd for C25H28NO5 [M + H]+ 422.1962 found 422.1972.
3.3.13. 6-O-(3,4-dimethoxybenzoyl)galanthamine (1m)
Yield 54 mg (69%); white amorphous solid; = −26.7° (c 0.07, MeOH); 1H NMR (500 MHz, CDCl3) δ: 7.71 (d, J = 8.6 Hz, 1H), 7.55 (s, 1H), 6.83 (d, J = 8.6 Hz, 1H), 6.67 (d, J = 8.1 Hz, 1H), 6.59 (d, J = 8.1 Hz, 1H), 6.33 (d, J = 10.3 Hz, 1H), 6.03 (dd, J = 10.3 Hz, J = 5.0 Hz, 1H), 5.66–5.49 (m, 1H), 4.63 (s, 1H), 4.15 (d, J = 15.1 Hz, 1H), 3.91 (s, 6H), 3.83 (s, 3H), 3.69 (d, J = 15.1 Hz, 1H), 3.34 (t, J = 13.6 Hz, 1H), 3.14–3.02 (m, 1H), 2.84–2.68 (m, 1H), 2.41 (s, 3H), 2.25–2.08 (m, 2H), 1.71–1.52 (m, 1H); 13C NMR (126 MHz, CDCl3) δ: 165.9, 152.8, 148.4, 146.7, 144.0, 132.1, 130.6, 128.9, 124.0, 123.0, 121.3, 112.1, 111.3, 110.0, 86.4, 63.3, 60.3, 55.9, 55.8, 55.7, 53.7, 48.0, 41.7, 34.2, 27.9; ESI-HRMS m/z calcd for C26H30NO6 [M + H]+ 452.2068 found 452.2074.
3.3.14. 6-O-(3,5-dimethoxybenzoyl)galanthamine (1n)
Yield 75 mg (95%); white amorphous solid; = −32.0° (c 0.25, MeOH); 1H NMR (500 MHz, CDCl3) δ: 7.22 (d, J = 2.4 Hz, 2H), 6.68 (d, J = 8.1 Hz, 1H), 6.64–6.58 (m, 2H), 6.34 (d, J = 10.3 Hz, 1H), 6.03 (dd, J = 10.3, 5.3 Hz, 1H), 5.63–5.56 (m, 1H), 4.67–4.62 (m, 1H), 4.17 (d, J = 15.1 Hz, 1H), 3.83 (s, 3H), 3.82 (s, 6H), 3.72 (d, J = 15.1 Hz, 1H), 3.37 (t, J = 13.6 Hz, 1H), 3.17–3.05 (m, 1H), 2.83–2.70 (m, 1H), 2.43 (s, 3H), 2.23–2.09 (m, 2H), 1.71–1.57 (m, 1H); 13C NMR (126 MHz, CDCl3) δ: 166.0, 160.5, 146.8, 144.2, 132.4, 132.0, 130.8, 122.8, 121.4, 111.3, 107.3, 105.9, 86.3, 63.7, 60.2, 55.8, 55.4, 53.6, 48.0, 41.6, 34.1, 27.9; ESI-HRMS m/z calcd for C26H30NO6 [M + H]+ 452.2068 found 452.2075.
3.3.15. 6-O-(3,4,5-trimethoxybenzoyl)galanthamine (1o)
Yield 61 mg (75%); white amorphous solid; = −10.3° (c 0.35, MeOH); 1H NMR (500 MHz, CDCl3) δ: 7.32 (s, 2H), 6.66 (d, J = 8.1 Hz, 1H), 6.60 (d, J = 8.1 Hz, 1H), 6.33 (d, J = 10.4 Hz, 1H), 6.01 (dd, J = 10.4 Hz, J = 5.1 Hz, 1H), 5.64–5.54 (m, 1H), 4.64–4.60 (m, 1H), 4.16 (d, J = 15.0 Hz, 1H), 3.89 (s, 6H), 3.87 (s, 3H), 3.79 (s, 3H), 3.71 (d, J = 15.0 Hz, 1H), 3.35 (t, J = 13.5 Hz, 1H), 3.16–3.05 (m, 1H), 2.80–2.70 (m, 1H), 2.42 (s, 3H), 2.23–2.11 (m, 2H), 1.70–1.57 (m, 1H); 13C NMR (126 MHz, CDCl3) δ: 165.7, 152.7, 146.7, 144.0, 142.0, 132.1, 130.6, 128.4, 125.4, 122.8, 121.5, 111.0, 107.0, 86.4, 63.5, 60.8, 60.2, 56.0, 55.6, 53.6, 47.9, 41.7, 34.2, 27.8; ESI-HRMS m/z calcd for C27H32NO7 [M + H]+ 482.2173 found 482.2174.
3.3.16. 6-O-(3,5-diethoxybenzoyl)galanthamine (1p)
Yield 53 mg (64%); white amorphous solid; = −17.0° (c 0.16, MeOH); 1H NMR (500 MHz, CDCl3) δ: 7.17 (s, 2H), 6.67 (d, J = 8.1 Hz, 1H), 6.62–6.53 (m, 2H), 6.33 (d, J = 10.3 Hz, 1H), 6.00 (dd, J = 10.3 Hz, J = 5.0 Hz, 1H), 5.62–5.48 (m, 1H), 4.67–4.55 (m, 1H), 4.14 (d, J = 15.1 Hz, 1H), 4.02 (q, J = 7.0 Hz, 4H), 3.84 (s, 3H), 3.69 (d, J = 15.1 Hz, 1H), 3.33 (t, J = 13.6 Hz, 1H), 3.14–3.00 (m, 1H), 2.83–2.67 (m, 1H), 2.41 (s, 3H), 2.22–2.08 (m, 2H), 1.70–1.53 (m, 1H), 1.39 (t, J = 7.0 Hz, 6H); 13C NMR (126 MHz, CDCl3) δ: 166.0, 159.7, 146.8, 144.0, 132.2, 132.0, 130.8, 129.0, 122.7, 121.2, 111.6, 107.8, 106.4, 86.2, 63.7, 63.5, 60.3, 56.0, 53.7, 48.0, 41.7, 34.2, 27.8, 14.7; ESI-HRMS m/z calcd for C28H34NO6 [M + H]+ 480.2381 found 480.2386.
3.3.17. 6-O-(1-naphthoyl)galanthamine (1q)
Yield 53 mg (70%); white amorphous solid; = −40.0° (c 0.10, MeOH); 1H NMR (500 MHz, CDCl3) δ: 8.98 (d, J = 8.2 Hz, 1H), 8.34 (d, J = 8.2 Hz, 1H), 7.99 (d, J = 8.2 Hz, 1H), 7.86 (d, J = 8.2 Hz, 1H), 7.58 (t, J = 8.2 Hz, 1H), 7.51 (t, J = 8.2 Hz, 1H), 7.45 (t, J = 8.2 Hz, 1H), 6.72 (d, J = 8.1 Hz, 1H), 6.62 (d, J = 8.1 Hz, 1H), 6.40 (d, J = 10.2 Hz, 1H), 6.12 (dd, J = 10.2 Hz, J = 5.1 Hz, 1H), 5.77–5.61 (m, 1H), 4.75–4.63 (m, 1H), 4.19 (d, J = 15.1 Hz, 1H), 3.89 (s, 3H), 3.73 (d, J = 15.1 Hz, 1H), 3.39 (t, J = 13.5 Hz, 1H), 3.22–3.04 (m, 1H), 2.96–2.81 (m, 1H), 2.44 (s, 3H), 2.30–2.12 (m, 2H), 1.75–1.61 (m, 1H); 13C NMR (126 MHz, CDCl3) δ: 167.1, 146.7, 144.1, 133.7, 133.2, 132.1, 131.5, 131.4, 131.0, 128.8, 128.4, 127.6, 127.1, 125.9, 125.9, 124.5, 123.0, 121.5, 111.5, 86.4, 63.7, 60.3, 56.0, 53.7, 48.1, 41.6, 34.1, 27.9; ESI-HRMS m/z calcd for C28H28NO4 [M + H]+ 442.2013 found 442.2020.
3.3.18. 6-O-(2-naphthoyl)galanthamine (1r)
Yield 56 mg (74%); white amorphous solid; = −35.2° (c 0.25, MeOH); 1H NMR (500 MHz, CDCl3) δ: 8.63 (s, 1H), 8.12–8.05 (m, 1H), 7.95 (d, J = 8.3 Hz, 1H), 7.86 (d, J = 8.3 Hz, 1H), 7.83 (d, J = 8.3 Hz, 1H), 7.60–7.47 (m, 2H), 6.73 (d, J = 8.1 Hz, 1H), 6.62 (d, J = 8.1 Hz, 1H), 6.40 (d, J = 10.3 Hz, 1H), 6.11 (dd, J = 10.3 Hz, J = 5.2 Hz, 1H), 5.70–5.64 (m, 1H), 4.73–4.68 (m, 1H), 4.19 (d, J = 15.1 Hz, 1H), 3.92 (s, 3H), 3.73 (d, J = 15.1 Hz, 1H), 3.39 (t, J = 13.6 Hz, 1H), 3.17–3.07 (m, 1H), 2.91–2.80 (m, 1H), 2.44 (s, 3H), 2.28–2.12 (m, 2H), 1.72–1.62 (m, 1H); 13C NMR (126 MHz, CDCl3) δ: 166.4, 146.8, 144.2, 135.5, 132.5, 132.1, 131.5, 131.0, 129.4, 128.0, 127.8, 127.8, 127.7, 126.2, 125.6, 123.0, 121.4, 111.6, 86.4, 63.7, 60.3, 56.1, 53.7, 48.2, 41.6, 34.2, 28.0; ESI-HRMS m/z calcd for C28H28NO4[M + H]+ 442.2013 found 442.2021.
3.3.19. 6-O-(2-furoyl)galanthamine (1s)
Yield 38 mg (57%); white amorphous solid; = −40.0° (c 0.05, MeOH); 1H NMR (500 MHz, CDCl3) δ: 7.54 (s, 1H), 7.15 (d, J = 3.5 Hz, 1H), 6.68 (d, J = 8.0 Hz, 1H), 6.58 (d, J = 8.0 Hz, 1H), 6.49–6.41 (m, 1H), 6.36 (d, J = 10.3 Hz, 1H), 5.99 (dd, J = 10.3 Hz, J = 5.1 Hz, 1H), 5.61–5.52 (m, 1H), 4.65–4.60 (m, 1H), 4.14 (d, J = 15.1 Hz, 1H), 3.86 (s, 3H), 3.69 (d, J = 15.1 Hz, 1H), 3.34 (t, J = 13.7 Hz, 1H), 3.12–3.00 (m, 1H), 2.83–2.70 (m, 1H), 2.41 (s, 3H), 2.22–2.07 (m, 2H), 1.67–1.54 (m, 1H); 13C NMR (126 MHz, CDCl3) δ: 158.4, 146.7, 146.3, 144.8, 144.0, 131.9, 131.3, 129.2, 122.5, 121.3, 118.1, 111.7, 111.6, 86.1, 63.7, 60.3, 56.0, 53.7, 48.1, 41.6, 34.1, 27.9; ESI-HRMS m/z calcd for C22H24NO5 [M + H]+ 382.1649 found 382.1658.
3.3.20. 3-O-(1-naphthoyl)vittatine (4a)
Yield 51 mg (79%); pale yellow amorphous solid; = –115.2° (c 0.12, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 8.86 (d, J = 8.1 Hz, 1H), 8.09 (dd, J = 8.1 Hz, J = 1.0 Hz, 1H), 7.99 (d, J = 8.1 Hz, 1H), 7.86 (d, J = 8.1 Hz, 1H), 7.58–7.50 (m, 2H), 7.45 (t, J = 8.1 Hz, 1H), 6.89 (s, 1H), 6.80 (d, J = 9.8 Hz, 1H), 6.52 (s, 1H), 6.16 (dd, J = 9.8 Hz, J = 5.5 Hz, 1H), 5.91 (d, overlap, J = 11.7 Hz, 1H), 5.90 (d, overlap, J = 11.7 Hz, 1H), 5.75–5.71 (m, 1H), 4.45 (d, J = 16.9 Hz, 1H), 3.82 (d, J = 16.9 Hz, 1H), 3.51 (dd, J = 13.0 Hz, J = 4.4 Hz, 1H), 3.43 (ddd, J = 13.0 Hz, J = 9.8 Hz, J = 5.0 Hz, 1H), 2.96 (ddd, J = 13.0 Hz, J = 9.8 Hz, J = 5.0 Hz, 1H), 2.29–2.20 (m, 2H), 2.06–1.94 (m, 2H); 13C NMR (126 MHz, CDCl3) δ: 167.0, 146.2, 145.8, 138.2, 134.9, 133.8, 133.2, 131.3, 130.1, 128.5, 127.6, 127.4, 126.5, 126.1, 125.8, 124.4, 123.8, 107.0, 102.8, 100.8, 67.3, 63.5, 62.4, 53.6, 44.4, 44.2, 30.1; ESI-HRMS m/z calcd for C27H24NO4 [M + H]+ 427.1700 found 427.1699.
3.3.21. 3-O-(2-naphthoyl)vittatine (4b)
Yield 56 mg (88%); pale yellow amorphous solid; = –75.0° (c 0.11, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 8.56 (bs, 1H), 8.02 (dd, J = 8.4 Hz, J = 1.5 Hz, 1H), 7.94 (d, J = 8.4 Hz, 1H), 7.86 (d, J = 8.0 Hz, 1H), 7.84 (d, J = 8.0 Hz, 1H), 7.58 (t, J = 8.0 Hz, 1H), 7.52 (t, J = 8.0 Hz, 1H), 6.91 (s, 1H), 6.80 (d, J = 9.8 Hz, 1H), 6.56 (s, 1H), 6.11 (dd, J = 9.8 Hz, J = 5.6 Hz, 1H), 5.94–5.92 (m, 1H), 5.91–5.90 (m, 1H), 5.70–5.67 (m, 1H), 4.49 (d, J = 17.1 Hz, 1H), 3.85 (d, J = 17.1 Hz, 1H), 3.54 (dd, J = 12.7 Hz, J = 5.0 Hz, 1H), 3.44 (ddd, J = 12.7 Hz, J = 9.3 Hz, J = 5.0 Hz, 1H), 2.97 (ddd, J = 12.7 Hz, J = 9.3 Hz, J = 5.0 Hz, 1H), 2.56 (ddd, J = 12.7 Hz, J = 9.3 Hz, J = 5.0 Hz, 1H), 2.23–2.17 (m, 1H), 2.05–2.00 (m, 1H), 1.96 (td, J = 12.7 Hz, J = 5.0 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ: 166.2, 146.3 145.9, 138.1, 135.5, 134.7, 132.4, 131.1, 129.3, 128.2, 128.0, 127.7, 127.6, 126.53, 126.50, 125.3, 123.8, 107.0, 102.9, 100.8, 67.3, 63.5, 62.4, 53.6, 44.4, 44.2, 30.1; ESI-HRMS m/z calcd for C27H24NO4 [M + H]+ 427.1700 found 427.1701.
3.3.22. 3-O-(3,5-dimethylbenzoyl)maritidine (5a)
Yield 37 mg (75%); white amorphous solid; = −75.6° (c 0.18, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 7.60 (s, 2H), 7.15 (s, 1H), 6.90 (s, 1H), 6.83 (d, J = 10.0 Hz, 1H), 6.59 (s, 1H), 6.08 (dd, J = 10.0 Hz, J = 5.1 Hz, 1H), 5.63–5.58 (m, 1H), 4.52 (d, J = 16.7 Hz, 1H), 3.93–3.82 (m, overlap, 1H), 3.91 (s, overlap, 3H), 3.85 (s, overlap, 3H), 3.52 (dd, J = 13.5 Hz, J = 4.3 Hz, 1H), 3.49–3.42 (m, 1H), 3.02–2.94 (m, 1H), 2.33 (s, 6H), 2.30–2.22 (m, 1H), 2.21–2.13 (m, 1H), 2.01 (ddd, J = 12.1 Hz, J = 10.3 Hz, J = 5.8 Hz, 1H), 1.94 (td, J = 13.5 Hz, J = 4.3 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ: 166.4, 147.5, 147.4, 137.9, 136.8, 134.5, 134.3, 130.1, 127.3, 125.0, 123.9, 109.9, 105.8, 67.0, 63.5, 62.0, 56.1, 55.9, 53.5, 44.2, 43.9, 29.9, 21.1; ESI-HRMS m/z calcd for C26H30NO4 [M + H]+ 420.2170 found 420.2172.
3.3.23. 3-O-(1-naphthoyl)maritidine (5b)
Yield 35 mg (82%); white amorphous solid; = −92.4° (c 0.19, CHCl3); 1H NMR (600 MHz, CDCl3) δ: 8.84 (d, J = 7.8 Hz, 1H), 8.07 (d, J = 7.8 Hz, 1H), 7.96 (d, J = 7.8 Hz, 1H), 7.84 (d, J = 7.8 Hz, 1H), 7.54–7.46 (m, 2H), 7.42 (t, J = 7.8 Hz, 1H), 6.88–6.83 (m, 2H), 6.54 (s, 1H), 6.16 (dd, J = 10.0 Hz, J = 5.2 Hz, 1H), 5.73–5.69 (m, 1H), 4.47 (d, J = 16.8 Hz, 1H), 3.93–3.77 (m, overlap, 1H), 3.87 (s, overlap, 3H), 3.81 (s, overlap, 3H), 3.53 (dd, J = 13.2 Hz, J = 4.3 Hz, 1H), 3.47–3.40 (m, 1H), 3.00–2.92 (m, 1H), 2.29–2.19 (m, 2H), 2.06–1.93 (m, 2H); 13C NMR (151 MHz, CDCl3) δ: 167.0, 147.5, 147.4, 136.9, 134.7, 133.8, 133.2, 131.3, 130.1, 128.5, 127.6, 127.3, 126.1, 125.7, 125.1, 124.4, 123.8, 110.0, 105.8, 67.3, 63.6, 62.0, 56.1, 55.9, 53.6, 44.3, 44.0, 30.0; ESI-HRMS m/z calcd for C28H28NO4 [M + H]+ 442.2013 found 442.2019.
3.4. Preparation of 3-O-methylpancracine (3)
3-O-methylpancracine (3) was synthesized through the rearrangement of haemanthamine (2), as described by Govindaraju et al. [23]. One hundred milligrams of haemanthamine was dissolved in 3 ml of dry pyridine and treated with 100 µL of methanesulfonyl chloride. After stirring at 0 °C for 8 h, the reaction mixture was poured into a solution of 100 mg of sodium bicarbonate in 10 ml of distilled water. The solution was again stirred overnight, and then extracted with chloroform, 2 times. The solution was treated with anhydrous sodium sulfate, filtered and the solvent was removed under reduced pressure. The residue was purified by preparative TLC (To:Et2NH 9:1), resulting in 70 mg of 3-O-methylpancracine (70%) as an amorphous white solid. The reaction was repeated several times during the study to provide compound 3 for further derivatization.
= –51.8° (c 0.25, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 6.55 (s, 1H), 6.47 (s, 1H), 5.88 (d, overlap, J = 13.5 Hz, 1H), 5.88 (d, overlap, J = 13.5 Hz, 1H), 5.59–5.51 (m, 1H), 4.36 (d, J = 16.6 Hz, 1H), 4.10–4.04 (m, 1H), 3.84 (d, J = 16.6 Hz, 1H), 3.59–3.52 (m, 1H), 3.41–3.34 (m, overlap, 1H), 3.39 (s, overlap, 3H), 3.30 (d, J = 2.6 Hz, 1H), 3.12 (dd, J = 11.2, J = 2.6 Hz, 1H), 3.06 (d, J = 11.2 Hz, 1H), 2.35–2.26 (m, 1H), 1.54 (td, J = 12.5, J = 3.2 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ: 153.7, 146.8, 146.0, 132.1, 124.3, 115.1, 107.4, 106.8, 100.8, 81.3, 67.3, 60.8, 58.8, 57.0, 55.5, 45.5, 28.6; ESI-HRMS m/z calcd for C17H20NO4 [M + H]+ 302.1378 found 302.1386.
3.5. Preparation of 3-O-methylpancracine (3a–3g) Derivatives
The same procedure as that described previously was used to afford the corresponding esters 3a–3g [29].
3.5.1. 2-O-(3,5-dimethylbenzoyl)-3-O-methylpancracine (3a)
Yield 23 mg (58%); white amorphous solid; = +114.3° (c 0.10, CHCl3); 1H NMR (600 MHz, CDCl3) δ: 7.62 (dd, J = 1.8 Hz, J = 0.7 Hz, 2H), 7.21–7.17 (m, 1H), 6.54 (s, 1H), 6.46 (s, 1H), 5.87 (d, overlap, J = 13.8 Hz, 1H), 5.87 (d, overlap, J = 13.8 Hz, 1H), 5.56–5.53 (m, 1H), 5.39–5.35 (m, 1H), 4.36 (d, J = 16.6 Hz, 1H), 3.85 (d, J = 16.6 Hz, 1H), 3.69–3.64 (m, 1H), 3.46–3.42 (m, overlap, 1H), 3.44 (s, overlap, 3H), 3.31 (d, J = 2.3 Hz, 1H), 3.13 (dd, J = 11.3 Hz, J = 2.3 Hz, 1H), 3.07 (d, J = 11.3 Hz, 1H), 2.39–2.32 (m, overlap, 1H), 2.36 (s, overlap, 6H), 1.56 (td, J = 12.4 Hz, J = 2.7 Hz, 1H); 13C NMR (151 MHz, CDCl3) δ: 166.0, 155.4, 146.9, 146.1, 138.1, 134.8, 131.7, 129.9, 127.3, 124.2, 111.6, 107.5, 106.8, 100.8, 78.4, 68.3, 60.9, 58.4, 57.3, 55.5, 45.7, 30.0, 21.1; ESI-HRMS m/z calcd for C26H28NO5 [M + H]+ 434.1962 found 434.1965.
3.5.2. 2-O-(2,4,6-trimethylbenzoyl)-3-O-methylpancracine (3b)
Yield 29 mg (42%); white amorphous solid; = +17.6° (c 0.41, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 6.87 (s, 2H), 6.56 (s, 1H), 6.48 (s, 1H), 5.89 (d, overlap, J = 11.4 Hz, 1H), 5.89 (d, overlap, J = 11.4 Hz, 1H), 5.60–5.57 (m, 1H), 5.46–5.41 (m, 1H), 4.36 (d, J = 16.6 Hz, 1H), 3.86 (d, J = 16.6 Hz, 1H), 3.73–3.67 (m, 1H), 3.53–3.42 (m, overlap, 1H), 3.49 (s, overlap, 3H), 3.33–3.30 (m, 1H), 3.10–3.03 (m, 2H), 2.40–2.33 (m, 1H), 2.32 (s, 6H), 2.29 (s, 3H), 1.53 (td, J = 12.3 Hz, J = 2.7 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ: 169.0, 156.0, 146.9, 146.0, 139.5, 135.0, 131.8, 130.6, 128.5, 124.5, 111.1, 107.5, 106.8, 100.8, 78.5, 68.6, 61.0, 58.2, 57.3, 55.5, 45.8, 29.9, 21.1, 19.8; ESI-HRMS m/z calcd for =C27H30NO5 [M + H]+ 448.2118 found 448.2119.
3.5.3. 2-O-(4-tert-butylbenzoyl)-3-O-methylpancracine (3c)
Yield 28 mg (98%); white amorphous solid; = +40.9° (c 0.90, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 8.01–7.95 (m, 2H, AA′BB′), 7.50–7.44 (m, 2H, AA′BB′), 6.55 (s, 1H), 6.48 (s, 1H), 5.89 (d, overlap, J = 13.4 Hz, 1H), 5.89 (d, overlap, J = 13.4 Hz, 1H), 5.59–5.54 (m, 1H), 5.46–5.41 (m, 1H), 4.37 (d, J = 16.7 Hz, 1H), 3.87 (d, J = 16.7 Hz, 1H), 3.71–3.64 (m, 1H), 3.50–3.42 (m, overlap, 1H), 3.46 (s, overlap, 3H), 3.32 (d, J = 2.1 Hz, 1H), 3.13 (dd, J = 11.3 Hz, J = 2.1 Hz, 1H), 3.09 (d, J = 11.3 Hz, 1H), 2.40–2.32 (m, 1H), 1.56 (td, J = 12.6 Hz, J = 2.9 Hz, 1H), 1.35 (s, 9H); 13C NMR (126 MHz, CDCl3) δ: 165.7, 156.8, 156.1, 146.8, 146.0, 132.0, 129.5, 127.3, 125.3, 124.7, 111.3, 107.5, 106.8, 100.7, 78.6, 68.3, 61.1, 58.1, 57.3, 55.5, 45.8, 35.1, 31.1, 30.2; ESI-HRMS m/z calcd for C28H32NO5 [M + H]+ 462.2275 found 462.2279.
3.5.4. 2-O-(4-butylbenzoyl)-3-O-methylpancracine (3d)
Yield 32 mg (98%); white amorphous solid; = +48.9° (c 0.15, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 7.98–7.92 (m, 2H, AA′BB′), 7.29–7.23 (m, 2H, AA′BB′), 6.55 (s, 1H), 6.48 (s, 1H), 5.89 (d, overlap, J = 13.4 Hz, 1H), 5.89 (d, overlap, J = 13.4 Hz, 1H), 5.59–5.54 (m, 1H), 5.42–5.37 (m, 1H), 4.36 (d, J = 16.7 Hz, 1H), 3.86 (d, J = 16.7 Hz, 1H), 3.71–3.66 (m, 1H), 3.49–3.42 (m, overlap, 1H), 3.46 (s, overlap, 3H), 3.32 (d, J = 2.0 Hz, 1H), 3.11 (dd, J = 11.1 Hz, J = 2.0 Hz, 1H), 3.08 (d, J = 11.1 Hz, 1H), 2.67 (t, J = 8.1 Hz, 2H), 2.39–2.31 (m, 1H), 1.66–1.58 (m, 2H), 1.55 (td, J = 12.0 Hz, J = 2.6 Hz, 1H), 1.36 (dq, J = 14.7 Hz, J = 7.3 Hz, 2H), 0.93 (t, J = 7.3 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ: 165.8, 156.2, 148.8, 146.8, 145.9, 132.0, 129.7, 128.5, 127.5, 124.7, 111.3, 107.5, 106.8, 100.7, 78.6, 68.3, 61.1, 58.1, 57.3, 55.5, 45.8, 35.7, 33.3, 30.2, 22.2, 13.9; ESI-HRMS m/z calcd for C28H32NO5 [M + H]+ 462.2275 found 462.2281.
3.5.5. 2-O-(3,5-diethoxybenzoyl)-3-O-methylpancracine (3e)
Yield 20 mg (48%); white amorphous solid; = +69.0° (c 0.14, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 7.16 (d, J = 2.3 Hz, 2H), 6.65 (t, J = 2.3 Hz, 1H), 6.55 (s, 1H), 6.48 (s, 1H), 5.89 (d, overlap, J = 13.4 Hz, 1H), 5.89 (d, overlap, J = 13.4 Hz, 1H), 5.57–5.52 (m, 1H), 5.40–5.35 (m, 1H), 4.37 (d, J = 16.6 Hz, 1H), 4.06 (q, J = 7.0 Hz, 4H), 3.86 (d, J = 16.6 Hz, 1H), 3.70–3.65 (m, 1H), 3.49–3.41 (m, overlap, 1H), 3.46 (s, overlap, 3H), 3.32 (d, J = 2.6 Hz, 1H), 3.12 (dd, J = 11.3 Hz, J = 2.6 Hz, 1H), 3.09 (d, J = 11.3 Hz, 1H), 2.39–2.31 (m, 1H), 1.55 (td, J = 12.6 Hz, J = 2.4 Hz, 1H), 1.43 (t, J = 7.0 Hz, 6H); 13C NMR (126 MHz, CDCl3) δ: 165.6, 159.9, 156.3, 146.8, 145.9, 132.0, 131.9, 124.7, 111.1, 107.9, 107.5, 106.8, 106.2, 100.7, 78.5, 68.7, 63.8, 61.1, 58.1, 57.3, 55.5, 45.8, 30.2, 14.7; ESI-HRMS m/z calcd for C28H32NO7 [M + H]+ 494.2173 found 494.2179.
3.5.6. 2-O-(1-naphthoyl)-3-O-methylpancracine (3f)
Yield 38 mg (100%); white amorphous solid; = +30.8° (c 0.19, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 8.93 (d, J = 8.0 Hz, 1H), 8.18 (d, J = 8.0 Hz, 1H), 8.04 (d, J = 8.0 Hz, 1H), 7.90 (d, J = 8.0 Hz, 1H), 7.64 (t, J = 8.0 Hz, 1H), 7.59–7.48 (m, 2H), 6.58 (s, 1H), 6.49 (s, 1H), 5.90 (d, overlap, J = 12.4 Hz, 1H), 5.90 (d, overlap, J = 12.4 Hz, 1H), 5.68–5.65 (m, 1H), 5.56–5.51 (m, 1H), 4.38 (d, J = 16.6 Hz, 1H), 3.88 (d, J = 16.6 Hz, 1H), 3.81–3.78 (m, 1H), 3.59–3.46 (m, overlap, 1H), 3.53 (s, overlap, 3H), 3.35 (d, J = 2.1 Hz, 1H), 3.14 (dd, J = 11.2 Hz, J = 2.1 Hz, 1H), 3.09 (d, J = 11.2 Hz, 1H), 2.45–2.37 (m, 1H), 1.62 (td, J = 13.0 Hz, J = 2.4 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ: 166.5, 156.4, 146.8, 145.9, 133.8, 133.5, 132.0, 131.3, 130.2, 128.6, 127.8, 127.0, 126.2, 125.6, 124.7, 124.4, 111.2, 107.5, 106.8, 100.7, 78.6, 68.7, 61.1, 58.1, 57.3, 55.6, 45.8, 30.2; ESI-HRMS m/z calcd for C28H26NO5 [M + H]+ 456.1805 found 456.1800.
3.5.7. 2-O-(2-naphthoyl)-3-O-methylpancracine (3g)
Yield 39 mg (100%); white amorphous solid; = +86.0° (c 0.15, CHCl3); 1H NMR (500 MHz, CDCl3) δ: 8.61 (d, J = 1.7 Hz, 1H), 8.06 (dd, J = 8.4 Hz, J = 1.7 Hz, 1H), 7.98 (d, J = 8.4 Hz, 1H), 7.90 (d, J = 8.4 Hz, 2H), 7.65–7.53 (m, 2H), 6.57 (s, 1H), 6.49 (s, 1H), 5.93–5.84 (m, 2H), 5.66–5.61 (m, 1H), 5.51–5.46 (m, 1H), 4.38 (d, J = 16.6 Hz, 1H), 3.88 (d, J = 16.6 Hz, 1H), 3.79–3.74 (m, 1H), 3.53–3.45 (m, overlap, 1H), 3.50 (s, overlap, 3H), 3.36 (d, J = 2.5 Hz, 1H), 3.17 (dd, J = 11.1 Hz, J = 2.5 Hz, 1H), 3.11 (d, J = 11.1 Hz, 1H), 2.44–2.36 (m, 1H), 1.63 (td, J = 12.4, J = 2.7 Hz, 1H);13C NMR (126 MHz, CDCl3) δ: 165.9, 156.4, 146.8, 146.0, 135.6, 132.5, 132.0, 131.1, 129.3, 128.4, 128.2, 127.8, 127.4, 126.7, 125.2, 124.8, 111.2, 107.5, 106.8, 100.7, 78.6, 68.8, 61.2, 58.2, 57.3, 55.6, 45.8, 30.3; ESI-HRMS m/z calcd for C28H26NO5 [M + H]+ 456.1805 found 456.1808.
3.6. Conversion of Derivatives to Hydrochlorides
The synthesized derivatives were dissolved in ethereal HCl and converted to the corresponding hydrochlorides.
3.7. Antimycobacterial Screening
The antimycobacterial assay was performed with fast-growing Mycobacterium smegmatis DSM 43465 (ATCC 607), Mycobacterium aurum DSM 43999 (ATCC 23366) from the German Collection of Microorganisms and Cell Cultures (Braunschweig, Germany) and an avirulent strain of Mycobacterium tuberculosis H37Ra ITM-M006710 (ATCC 9431) from Belgian Co-ordinated Collections of Micro-organisms (Antwerp, Belgium). The technique used for activity determination was the microdilution broth panel method using 96-well microtitration plates. The culture medium was Middlebrook 7H9 broth (MB) (Sigma-Aldrich, Steinheim, Germany) enriched with 0.4% glycerol (Sigma-Aldrich, Steinheim, Germany) and 10% Middlebrook OADC growth supplement (Himedia, Mumbai, India).
The mycobacterial strains were cultured on Middlebrook 7H9 agar and suspensions were prepared in Middlebrook 7H9 broth. The final density was adjusted to 1.0 on the McFarland scale and diluted in z ratio of either 1:20 (for fast-growing mycobacteria) or 1:10 (for M. tuberculosis) with broth.
The test compounds were dissolved in DMSO (Sigma-Aldrich, Steinheim, Germany), then MB broth was added to obtain a concentration of 2000 µg/mL. The standards used for activity determination were isoniazid (INH), rifampicin (RIF) and ciprofloxacin (CPX) (Sigma-Aldrich, Steinheim, Germany). Final concentrations were reached by binary dilution and the addition of mycobacterial suspension, and were set as 500, 250, 125, 62.5, 31.25, 15.625, 7.81 and 3.91 µg/mL. Isoniazid was diluted in the range of 500-3.91 µg/mL for screening against fast-growing mycobacteria and 1-0.0078 µg/mL for screening against M. tuberculosis. Rifampicin final concentrations ranged from 50 to 0.39 µg/mL for fast-growing mycobacteria and from 1 to 0.0078 µg/mL for M. tuberculosis. Ciprofloxacin was used for screening antimycobacterial activity with final concentrations of 1, 0.5, 0.25, 0.125, 0.0625, 0.0313, 0.0156 and 0.0078 µg/mL. The final concentration of DMSO did not exceed 2.5% (v/v) and did not affect the growth of M. smegmatis, M. aurum and M. tuberculosis. Positive (broth, DMSO, bacteria) and negative (broth, DMSO) growth controls were included.
Plates were sealed with polyester adhesive film and incubated in the dark at 37 °C without agitation. The addition of a 0.01% solution of resazurin sodium salt followed after 48 h of incubation for M. smegmatis, after 72 h for M. aurum and after 120 h for M. tuberculosis. Microtitration panels were then incubated for a further 2.5 h for the determination of activity against M. smegmatis, 4 hours for M. aurum and 18 hours for M. tuberculosis.
The antimycobacterial activity was expressed as the minimal inhibition concentration (MIC) and the value was read on the basis of stain color change (blue color—active compound; pink color—inactive compound). MIC values for standards were in the range of 7.81–15.625 µg/mL for INH, 12.5–25 µg/mL for RIF and 0.0625–0.125 µg/mL for CPX against M. smegmatis, 1.95–3.91 µg/mL for INH, 0.39–0.78 µg/mL for RIF and 0.0078–0.0156 µg/mL for CPX against M. aurum and 0.125–0.25 µg/mL for INH, 0.0039–0.0078 µg/mL for RIF and 0.125–0.25 for CPX against M. tuberculosis. All experiments were conducted in duplicate.
3.8. Cytotoxicity Assay
Human hepatocellular carcinoma HepG2 cells (ATCC HB-8065; passage 20–25) purchased from Health Protection Agency Culture Collections (ECACC, Salisbury, UK) were cultured in the Minimum Essential Eagle Medium supplemented with 10% fetal bovine serum and 1% l-glutamine solution (Sigma-Aldrich, St. Louis, MO, USA) at 37 °C in a humidified atmosphere containing 5% CO2. For passaging, the cells were treated with trypsin/EDTA (Sigma-Aldrich, St. Louis, MO, USA) at 37 °C and then harvested. For the cytotoxicity evaluation, the cells treated with the test substances were used while untreated HepG2 cells served as the control group; no cell control was included. The cells were seeded in a 96-well plate at a density of 50,000 cells per well and incubated for 24 h. The following day, the cells were treated with each of the test compounds dissolved in DMSO (the highest DMSO concentration used was 0.5% v/v). The test substances were prepared at different concentrations in triplicate according to their solubility and incubated for 24 h in a humidified atmosphere containing 5% CO2 at 37 °C. After incubation, a solution of thiazolyl blue tetrazolium bromide (Sigma-Aldrich, St. Louis, MO, USA) in RPMI 1640 without phenol red (BioTech) was added and incubated for 30 min in a humidified atmosphere containing 5% CO2 at 37 °C. Then, the formazan crystals formed were dissolved in DMSO and the absorbance of the samples was recorded at 570 nm (BioTek, Synergy Neo2 Multi-Mode Reader NEO2SMALPHAB). IC50 values were calculated by nonlinear regression from a semi-logarithmic plot of incubation concentration versus the percentage of absorbance relative to untreated controls using GraphPad Prism software (version 9; GraphPad Software, Inc., La Jolla, CA, USA). The obtained results of the experiments are presented as the concentration that reduces the viability of cells from the maximal viability to 50% (IC50).
4. Conclusions
Amaryllidaceae alkaloids and some of their derivatives have demonstrated a wide range of biological activities, including antimicrobial potential. On the other hand, no information about the antimycobacterial activity of AAs was available in the literature, thus the selected AAs, previously isolated from various Amaryllidaceae plants, have been initially screened for antimycobacterial activity. Moreover, pilot semisynthetic derivatives of galanthamine, 3-O-methyl pancracine, vittatine and maritidine have been synthesized and tested. Interestingly, the natural AAs lacked any antimycobacterial activity, but on the other hand, several semisynthetic derivatives showed great antimycobacterial potential. The most active derivatives were also evaluated for their cytotoxic activity on HepG2 cells, and thus the SI was also determined. Acylchlorides (namely 4-tert-butylbenzoyl chloride, 4-butylbenzoyl chloride, 3,5-diethoxybenzoyl chloride, 1-naphthoylchloride and 2-naphthoylchloride), which were used for the preparation of active derivatives in this study, will be used for the preparation of esters of other AAs previously isolated in our laboratory (e.g., haemanthamine, lycorine, galanthine) in order to study the effect of the structural type of AAs on antimycobacterial and cytotoxic activity. After additional isolation of vittatine, maritidine and other AAs, this pilot study will continue with the further structure modification of selected alkaloids. The second series of galanthamine derivatives, especially aromatic ethers and amines, will be synthesized for more detailed SAR studies. Based on the obtained results, compounds 1i and 1r were selected for further structure optimalization to improve the antimycobacterial potential and selectivity index by reducing the cytotoxicity. The selected compounds, showing both great antitubercular activity and low cytotoxicity, will be further subjected to searching of partner compounds from a portfolio of anti-TB drugs by checkerboard studies, and an evaluation of in vivo toxicity and in vivo treatment assays by the establishment of the Galleria mellonella animal model. This pilot study indicated the potential of semisynthetic derivatives of AAs for further structure optimalization as promising antimycobacterial compounds.
Supplementary Materials
The following are available online at, Figure S1. ESI-HRMS spectrum of 6-O-benzoylgalanthamine (1a); Figure S2. 1H NMR spectrum of 6-O-benzoylgalanthamine (1a) in CDCl3; Figure S3. 13C NMR spectrum of 6-O-benzoylgalanthamine (1a) in CDCl3; Figure S4. ESI-HRMS spectrum of 6-O-(2-methylbenzoyl)galanthamine (1b); Figure S5. 1H NMR spectrum of 6-O-(2-methylbenzoyl)galanthamine (1b) in CDCl3; Figure S6. 13C NMR spectrum of 6-O-(2-methylbenzoyl)galanthamine (1b) in CDCl3; Figure S7. ESI-HRMS spectrum of 6-O-(3-methylbenzoyl)galanthamine (1c); Figure S8. 1H NMR spectrum of 6-O-(3-methylbenzoyl)galanthamine (1c) in CDCl3; Figure S9. 13C NMR spectrum of 6-O-(3-methylbenzoyl)galanthamine (1c) in CDCl3; Figure S10. ESI-HRMS spectrum of 6-O-(4-methylbenzoyl)galanthamine (1d); Figure S11. 1H NMR spectrum of 6-O-(4-methylbenzoyl)galanthamine (1d) in CDCl3; Figure S12. 13C NMR spectrum of 6-O-(4-methylbenzoyl)galanthamine (1d) in CDCl3; Figure S13. ESI-HRMS spectrum of 6-O-(2,3-dimethylbenzoyl)galanthamine (1e); Figure S14. 1H NMR spectrum of 6-O-(2,3-dimethylbenzoyl)galanthamine (1e) in CDCl3; Figure S15. 13C NMR spectrum of 6-O-(2,3-dimethylbenzoyl)galanthamine (1e) in CDCl3; Figure S16. ESI-HRMS spectrum of 6-O-(3,5-dimethylbenzoyl)galanthamine (1f); Figure S17. 1H NMR spectrum of 6-O-(3,5-dimethylbenzoyl)galanthamine (1f) in CDCl3; Figure S18. 13C NMR spectrum of 6-O-(3,5-dimethylbenzoyl)galanthamine (1f) in CDCl3; Figure S19. ESI-HRMS spectrum of 6-O-(2,4,6-trimethylbenzoyl)galanthamine (1g); Figure S20. 1H NMR spectrum of 6-O-(2,4,6-trimethylbenzoyl)galanthamine (1g) in CDCl3; Figure S21. 13C NMR spectrum of 6-O-(2,4,6-trimethylbenzoyl)galanthamine (1g) in CDCl3; Figure S22. ESI-HRMS spectrum of 6-O-(4-tert-butylbenzoyl)galanthamine (1h); Figure S23. 1H NMR spectrum of 6-O-(4-tert-butylbenzoyl)galanthamine (1h) in CDCl3; Figure S24. 13C NMR spectrum of 6-O-(4-tert-butylbenzoyl)galanthamine (1h) in CDCl3; Figure S25. ESI-HRMS spectrum of 6-O-(4-butylbenzoyl)galanthamine (1i); Figure S26. 1H NMR spectrum of 6-O-(4-butylbenzoyl)galanthamine (1i) in CDCl3; Figure S27. 13C NMR spectrum of 6-O-(4-butylbenzoyl)galanthamine (1i) in CDCl3; Figure S28. ESI-HRMS spectrum of 6-O-(2-methoxybenzoyl)galanthamine (1j); Figure S29. 1H NMR spectrum of 6-O-(2-methoxybenzoyl)galanthamine (1j) in CDCl3; Figure S30. 13C NMR spectrum of 6-O-(2-methoxybenzoyl)galanthamine (1j) in CDCl3; Figure S31. ESI-HRMS spectrum of 6-O-(3-methoxybenzoyl)galanthamine (1k); Figure S32. 1H NMR spectrum of 6-O-(3-methoxybenzoyl)galanthamine (1k) in CDCl3; Figure S33. 13C NMR spectrum of 6-O-(3-methoxybenzoyl)galanthamine (1k) in CDCl3; Figure S34. ESI-HRMS spectrum of 6-O-(4-methoxybenzoyl)galanthamine (1l); Figure S35. 1H NMR spectrum of 6-O-(4-methoxybenzoyl)galanthamine (1l) in CDCl3; Figure S36. 13C NMR spectrum of 6-O-(4-methoxybenzoyl)galanthamine (1l) in CDCl3; Figure S37. ESI-HRMS spectrum of 6-O-(3,4-dimethoxybenzoyl)galanthamine (1m); Figure S38. 1H NMR spectrum of 6-O-(3,4-dimethoxybenzoyl)galanthamine (1m) in CDCl3; Figure S39. 13C NMR spectrum of 6-O-(3,4-dimethoxybenzoyl)galanthamine (1m) in CDCl3; Figure S40. ESI-HRMS spectrum of 6-O-(3,5-dimethoxybenzoyl)galanthamine (1n); Figure S41. 1H NMR spectrum of 6-O-(3,5-dimethoxybenzoyl)galanthamine (1n) in CDCl3; Figure S42. 13C NMR spectrum of 6-O-(3,5-dimethoxybenzoyl)galanthamine (1n) in CDCl3; Figure S43. ESI-HRMS spectrum of 6-O-(3,4,5-trimethoxybenzoyl)galanthamine (1o); Figure S44. 1H NMR spectrum of 6-O-(3,4,5-trimethoxybenzoyl)galanthamine (1o) in CDCl3; Figure S45. 13C NMR spectrum of 6-O-(3,4,5-trimethoxybenzoyl)galanthamine (1o) in CDCl3; Figure S46. ESI-HRMS spectrum of 6-O-(3,5-diethoxybenzoyl)galanthamine (1p); Figure S47. 1H NMR spectrum of 6-O-(3,5-diethoxybenzoyl)galanthamine (1p) in CDCl3; Figure S48. 13C NMR spectrum of 6-O-(3,5-diethoxybenzoyl)galanthamine (1p) in CDCl3; Figure S49. ESI-HRMS spectrum of 6-O-(1-naphtoyl)galanthamine (1q); Figure S50. 1H NMR spectrum of 6-O-(1-naphtoyl)galanthamine (1q) in CDCl3; Figure S51. 13C NMR spectrum of 6-O-(1-naphtoyl)galanthamine (1q) in CDCl3; Figure S52. ESI-HRMS spectrum of 6-O-(2-naphtoyl)galanthamine (1r); Figure S53. 1H NMR spectrum of 6-O-(2-naphtoyl)galanthamine (1r) in CDCl3; Figure S54. 13C NMR spectrum of 6-O-(2-naphtoyl)galanthamine (1r) in CDCl3; Figure S55. ESI-HRMS spectrum of 6-O-(2-furoyl)galanthamine (1s); Figure S56. 1H NMR spectrum of 6-O-(2-furoyl)galanthamine (1s) in CDCl3; Figure S57. 13C NMR spectrum of 6-O-(2-furoyl)galanthamine (1s) in CDCl3; Figure S58. ESI-HRMS spectrum of 3-O-methylpancracine (3); Figure S59. 1H NMR spectrum of 3-O-methylpancracine (3) in CDCl3; Figure S60. 13C NMR spectrum of 3-O-methylpancracine (3) in CDCl3; Figure S61. ESI-HRMS spectrum of 2-O-(3,5-dimethylbenzoyl)-3-O-methylpancracine (3a); Figure S62. 1H NMR spectrum of 2-O-(3,5-dimethylbenzoyl)-3-O-methylpancracine (3a) in CDCl3; Figure S63. 13C NMR spectrum of 2-O-(3,5-dimethylbenzoyl)-3-O-methylpancracine (3a) in CDCl3; Figure S64. ESI-HRMS spectrum of 2-O-(2,4,6-trimethylbenzoyl)-3-O-methylpancracine (3b); Figure S65. 1H NMR spectrum of 2-O-(2,4,6-trimethylbenzoyl)-3-O-methylpancracine (3b) in CDCl3; Figure S66. 13C NMR spectrum of 2-O-(2,4,6-trimethylbenzoyl)-3-O-methylpancracine (3b) in CDCl3; Figure S67. ESI-HRMS spectrum of 2-O-(4-tert-butylbenzoyl)-3-O-methylpancracine (3c); Figure S68. 1H NMR spectrum of 2-O-(4-tert-butylbenzoyl)-3-O-methylpancracine (3c) in CDCl3; Figure S69. 13C NMR spectrum of 2-O-(4-tert-butylbenzoyl)-3-O-methylpancracine (3c) in CDCl3; Figure S70. ESI-HRMS spectrum of 2-O-(4-butylbenzoyl)-3-O-methylpancracine (3d); Figure S71. 1H NMR spectrum of 2-O-(4-butylbenzoyl)-3-O-methylpancracine (3d) in CDCl3; Figure S72. 13C NMR spectrum of 2-O-(4-butylbenzoyl)-3-O-methylpancracine (3d) in CDCl3; Figure S73. ESI-HRMS spectrum of 2-O-(3,5-diethoxybenzoyl)-3-O-methylpancracine (3e); Figure S74. 1H NMR spectrum of 2-O-(3,5-diethoxybenzoyl)-3-O-methylpancracine (3e) in CDCl3; Figure S75. 13C NMR spectrum of 2-O-(3,5-diethoxybenzoyl)-3-O-methylpancracine (3e) in CDCl3; Figure S76. ESI-HRMS spectrum of 2-O-(1-naphtoyl)-3-O-methylpancracine (3f); Figure S77. 1H NMR spectrum of 2-O-(1-naphtoyl)-3-O-methylpancracine (3f) in CDCl3; Figure S78. 13C NMR spectrum of 2-O-(1-naphtoyl)-3-O-methylpancracine (3f) in CDCl3; Figure S79. ESI-HRMS spectrum of 2-O-(2-naphtoyl)-3-O-methylpancracine (3g); Figure S80. 1H NMR spectrum of 2-O-(2-naphtoyl)-3-O-methylpancracine (3g) in CDCl3; Figure S81. 13C NMR spectrum of 2-O-(2-naphtoyl)-3-O-methylpancracine (3g) in CDCl3; Figure S82. ESI-HRMS spectrum of 3-O-(1-naphtoyl)vittatine (4a); Figure S83. 1H NMR spectrum of 3-O-(1-naphtoyl)vittatine (4a) in CDCl3; Figure S84. 13C NMR spectrum of 3-O-(1-naphtoyl)vittatine (4a) in CDCl3; Figure S85. ESI-HRMS spectrum of 3-O-(2-naphtoyl)vittatine (4b); Figure S86. 1H NMR spectrum of 3-O-(2-naphtoyl)vittatine (4b) in CDCl3; Figure S87. 13C NMR spectrum of 3-O-(2-naphtoyl)vittatine (4b) in CDCl3; Figure S88. ESI-HRMS spectrum of 3-O-(3,5-dimethylbenzoyl)maritidine (5a); Figure S89. 1H NMR spectrum of 3-O-(3,5-dimethylbenzoyl)maritidine (5a) in CDCl3; Figure S90. 13C NMR spectrum of 3-O-(3,5-dimethylbenzoyl)maritidine (5a) in CDCl3; Figure S91. ESI-HRMS spectrum of 3-O-(1-naphtoyl)maritidine (5b); Figure S92. 1H NMR spectrum of 3-O-(1-naphtoyl)maritidine (5b) in CDCl3; Figure S93. 13C NMR spectrum of 3-O-(1-naphtoyl)maritidine (5b) in CDCl3.
Author Contributions
N.M., A.A.M., D.H. and E.K. contributed to the synthesis of compounds; A.H., M.Š. and L.C. contributed to the isolation of Amaryllidaceae alkaloids; J.M., J.K. and L.N. contributed to the measurement and interpretation of MS, HRMS, NMR and optical rotatory data. O.J., K.B., A.D., K.K. and D.K. contributed to the measurement of various biological activities of the isolated alkaloids and synthetic derivatives. L.C. and O.J. designed the study, supervised the laboratory work, and wrote the manuscript. All authors read the final manuscript and approved the submission.
Funding
This project was supported by Charles University grants (SVV UK 260 548; SVV 260 547), and by pre-application research into the innovative medicines and medical technologies project co-funded by the European Union (reg. No. CZ.02.1.01/0.0/0.0/18_069/0010046).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available within the article or Supplementary Materials.
Acknowledgments
The authors wish to thank Gerald Blunden for critical reading of the manuscript and corrections of English, and Jana Vacková and Ida Dufková for assistance with antimycobacterial screening.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are available from the authors.
References
- Kanabalan, R.D.; Lee, L.J.; Lee, T.Y.; Chong, P.P.; Hassan, L.; Ismail, R.; Chin, V.K. Human tuberculosis and Mycobacterium tuberculosis complex: A review on genetic diversity, pathogenesis and omics approaches in host biomarkers discovery. Microbiol. Res. 2021, 246, 126674. [Google Scholar] [CrossRef]
- Tiberi, S.; Muñoz-Torrico, M.; Duarte, R.; Dalcolmo, M.; D’Ambrosio, L.; Migliori, G.B. New drugs and perspectives for new anti-tuberculosis regimens. Pulmonology 2018, 24, 86–98. [Google Scholar] [CrossRef]
- Koul, A.; Arnoult, E.; Lounis, N.; Guillemont, J.; Andries, K. The challenge of new drug discovery for tuberculosis. Nature 2011, 469, 483–490. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patridge, E.; Gareiss, P.; Kinch, M.S.; Hoyer, D. An analysis of FDA-approved drugs: Natural products and their derivatives. Drug Discov. Today 2016, 21, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Okunade, A.L.; Elvin-Lewis, M.P.; Lewis, W.H. Natural antimycobacterial metabolites: Current status. Phytochemistry 2004, 65, 1017–1032. [Google Scholar] [CrossRef] [PubMed]
- Copp, B.R.; Pearce, A.N. Natural product growth inhibitors of Mycobacterium tuberculosis. Nat. Prod. Rep. 2007, 24, 278–297. [Google Scholar] [CrossRef]
- Kishore, N.; Mishra, B.B.; Tripathi, V.; Tiwari, V.K. Alkaloids as potential anti-tubercular agents. Fitoterapia 2009, 80, 149–163. [Google Scholar] [CrossRef]
- Mishra, S.K.; Tripathi, G.; Kishore, N.; Singh, R.K.; Singh, A.; Tiwari, V.K. Drug development against tuberculosis: Impact of alkaloids. Eur. J. Med. Chem. 2017, 137, 504–544. [Google Scholar] [CrossRef]
- Jin, Z.; Yao, G. Amaryllidaceae and Sceletium alkaloids. Nat. Prod. Rep. 2019, 36, 1462–1488. [Google Scholar] [CrossRef]
- Nair, J.J.; Wilhelm, A.; Bonnet, S.L.; van Staden, J. Antibacterial constituents of the plant family Amaryllidaceae. Bioorg. Med. Chem. Lett. 2017, 27, 4943–4951. [Google Scholar] [CrossRef]
- Al Mamun, A.; Maříková, J.; Hulcová, D.; Janoušek, J.; Šafratová, M.; Nováková, L.; Kučera, T.; Hrabinová, M.; Kuneš, J.; Korábečný, J.; et al. Amaryllidaceae Alkaloids of Belladine-Type from Narcissus pseudonarcissus cv. Carlton as New Selective Inhibitors of Butyrylcholinesterase. Biomolecules 2020, 10, 800. [Google Scholar] [CrossRef] [PubMed]
- Kornienko, A.; Evidente, A. Chemistry, Biology, and Medicinal Potential of Narciclasine and its Congeners. Chem. Rev. 2008, 108, 1982–2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinrich, M.; Lee Teoh, H. Galanthamine from snowdrop--the development of a modern drug against Alzheimer’s disease from local Caucasian knowledge. J. Ethnopharmacol. 2004, 92, 147–162. [Google Scholar] [CrossRef] [PubMed]
- Cahlíková, L.; Kawano, I.; Řezáčová, M.; Blunden, G.; Hulcová, D.; Havelek, R. The Amaryllidaceae alkaloids haemanthamine, haemanthidine and their semisynthetic derivatives as potential drugs. Phytochem. Rev. 2021, 20, 303–323. [Google Scholar] [CrossRef]
- McLachlan, A.; Kekre, N.; McNulty, J.; Pandey, S. Pancratistatin: A natural anti-cancer compound that targets mitochondria specifically in cancer cells to induce apoptosis. Apoptosis 2005, 10, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Griffin, C.; Karnik, A.; McNulty, J.; Pandey, S. Pancratistatin selectively targets cancer cell mitochondria and reduces growth of human colon tumor xenografts. Mol. Cancer Ther. 2011, 10, 57–68. [Google Scholar] [CrossRef] [Green Version]
- Roy, M.; Liang, L.; Xiao, X.; Feng, P.; Ye, M.; Liu, J. Lycorine: A prospective natural lead for anticancer drug discovery. Biomed. Pharmacother. 2018, 107, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Peřinová, R.; Maafi, N.; Korábečný, J.; Kohelová, E.; De Simone, A.; Mamun, A.A.; Hulcová, D.; Marková, J.; Kučera, T.; Jun, D.; et al. Functionalized aromatic esters of the Amaryllidaceae alkaloid haemanthamine and their in vitro and in silico biological activity connected to Alzheimer’s disease. Bioorg. Chem. 2020, 100, 103928. [Google Scholar] [CrossRef]
- Al Shammari, L.; Hulcová, D.; Maříková, J.; Kučera, T.; Šafratová, M.; Nováková, L.; Schmidt, M.; Pulkrábková, L.; Janoušek, J.; Soukup, O.; et al. Amaryllidaceae alkaloids from Hippeastrum X hybridum cv. Ferrari, and preparation of vittatine derivatives as potential ligands for Alzheimer´s disease. South Afr. J. Bot. 2021, 136, 137–146. [Google Scholar] [CrossRef]
- Maříková, J.; Ritomská, A.; Korábečný, J.; Peřinová, R.; Al Mamun, A.; Kučera, T.; Kohelová, E.; Hulcová, D.; Kobrlová, T.; Kuneš, J.; et al. Aromatic Esters of the Crinane Amaryllidaceae Alkaloid Ambelline as Selective Inhibitors of Butyrylcholinesterase. J. Nat. Prod. 2020, 83, 1359–1367. [Google Scholar] [CrossRef]
- Koutová, D.; Maafi, N.; Havelek, R.; Opletal, L.; Blunden, G.; Řezáčová, M.; Cahlíková, L. Chemical and Biological Aspects of Montanine-Type Alkaloids Isolated from Plants of the Amaryllidaceae Family. Molecules 2020, 25, 2337. [Google Scholar] [CrossRef]
- Govindaraju, K.; Ingels, A.; Hasan, M.N.; Sun, D.; Mathieu, V.; Masi, M.; Evidente, A.; Kornienko, A. Synthetic analogues of the montanine-type alkaloids with activity against apoptosis-resistant cancer cells. Bioorg. Med. Chem. Lett. 2018, 28, 589–593. [Google Scholar] [CrossRef]
- Safratova, M.; Hostalkova, A.; Hulcova, D.; Breiterova, K.; Hrabcova, V.; Machado, M.; Fontinha, D.; Prudencio, M.; Kunes, J.; Chlebek, J.; et al. Alkaloids from Narcissus poeticus cv. Pink Parasol of various structural types and their biological activity. Arch. Pharmacal Res. 2018, 41, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Phelan, J.; Maitra, A.; McNerney, R.; Nair, M.; Gupta, A.; Coll, F.; Pain, A.; Bhakta, S.; Clark, T.G. The draft genome of Mycobacterium aurum, a potential model organism for investigating drugs against Mycobacterium tuberculosis and Mycobacterium leprae. Int. J. Mycobacteriology 2015, 4, 207–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piccaro, G.; Poce, G.; Biava, M.; Giannoni, F.; Fattorini, L. Activity of lipophilic and hydrophilic drugs against dormant and replicating Mycobacterium tuberculosis. J. Antibiot. 2015, 68, 711–714. [Google Scholar] [CrossRef] [Green Version]
- Senousy, B.E.; Belal, S.I.; Draganov, P.V. Hepatotoxic effects of therapies for tuberculosis. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Kohelová, E.; Maříková, J.; Korábečný, J.; Hulcová, D.; Kučera, T.; Jun, D.; Chlebek, J.; Jenčo, J.; Šafratová, M.; Hrabinová, M.; et al. Alkaloids of Zephyranthes citrina (Amaryllidaceae) and their implication to Alzheimer’s disease: Isolation, structural elucidation and biological activity. Bioorganic Chem. 2021, 107, 104567. [Google Scholar] [CrossRef]
- Kohelová, E.; Peřinová, R.; Maafi, N.; Korábečný, J.; Hulcová, D.; Maříková, J.; Kučera, T.; Martínez González, L.; Hrabinova, M.; Vorčáková, K.; et al. Derivatives of the β-Crinane Amaryllidaceae Alkaloid Haemanthamine as Multi-Target Directed Ligands for Alzheimer’s Disease. Molecules 2019, 24, 1307. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).