
Aspergillus oryzae-Fermented Wheat Peptone Enhances the Potential of Proliferation and Hydration of Human Keratinocytes through Activation of p44/42 MAPK


 ,
,  ,
, 
Abstract
:1. Introduction
2. Results
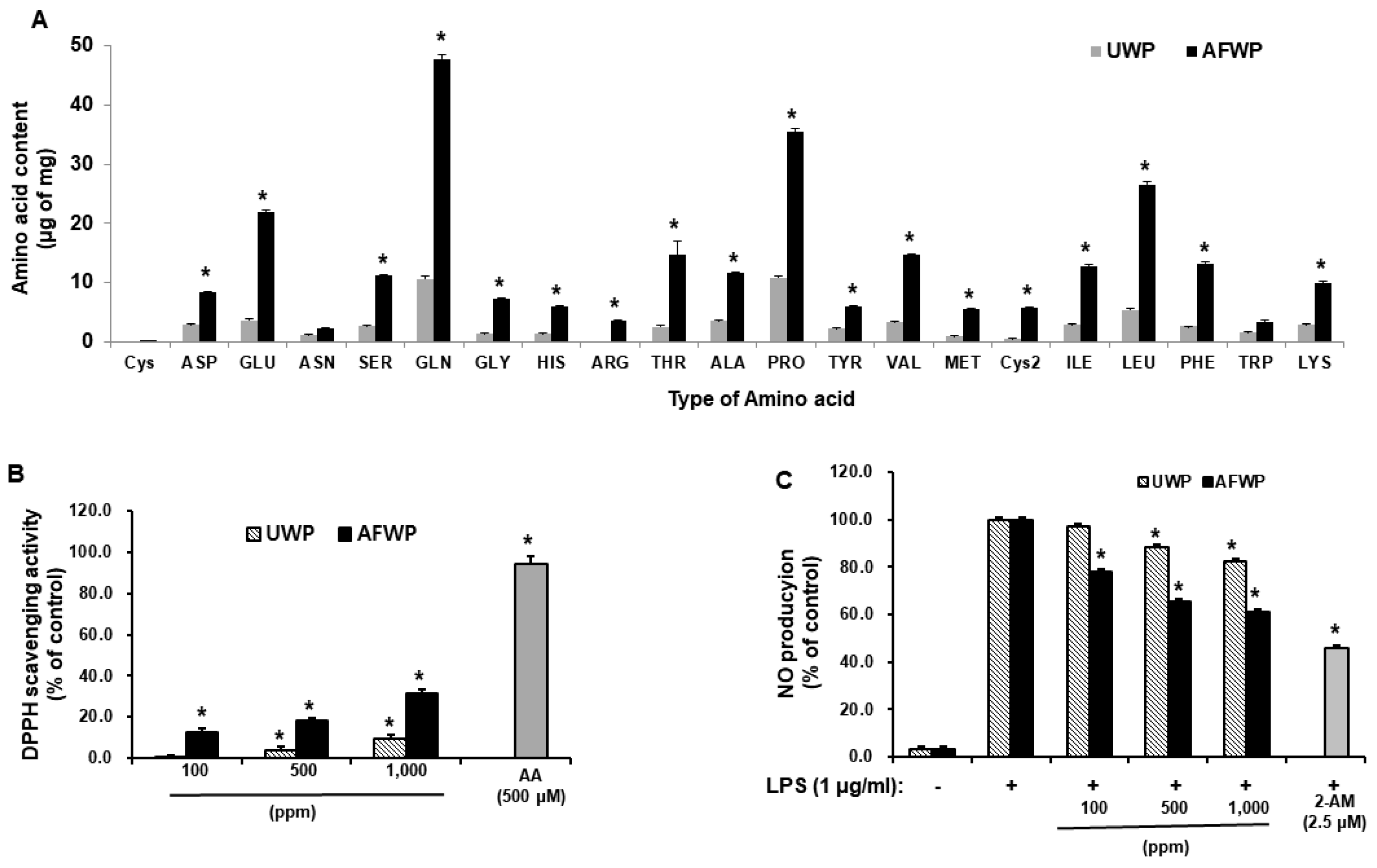
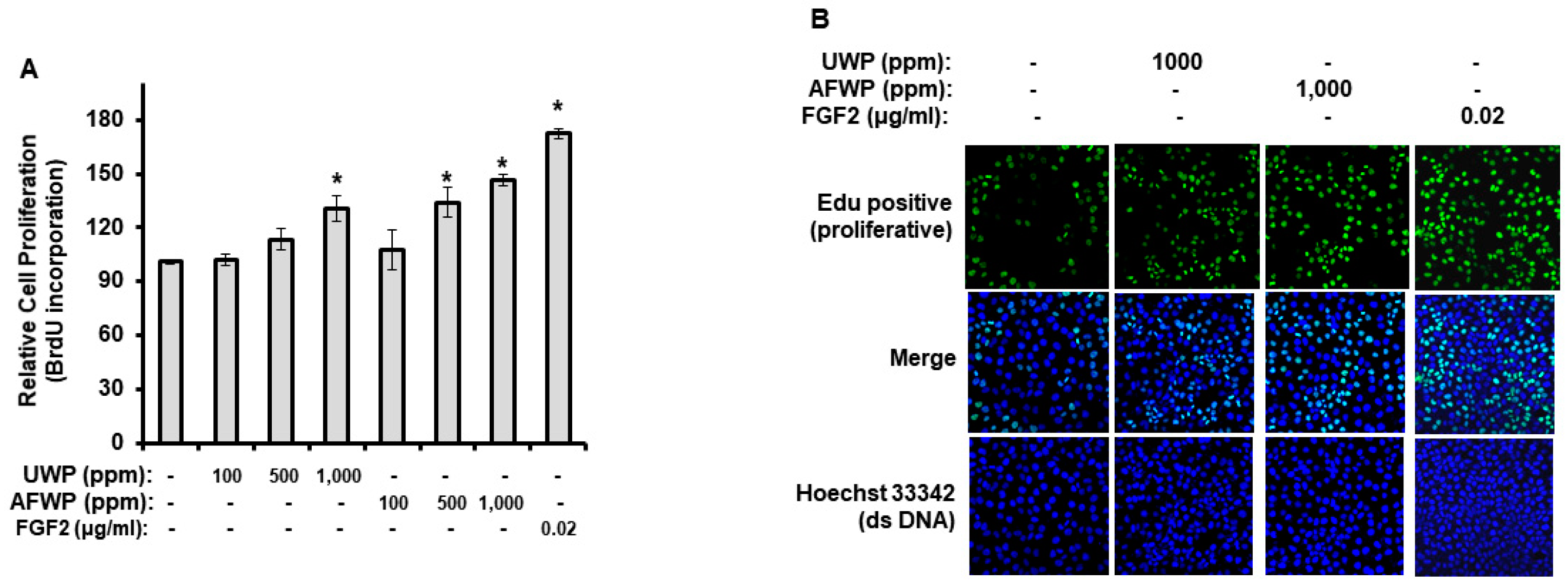
2.1. AFWP Exerted Increased Antioxidant and Proliferation Activities
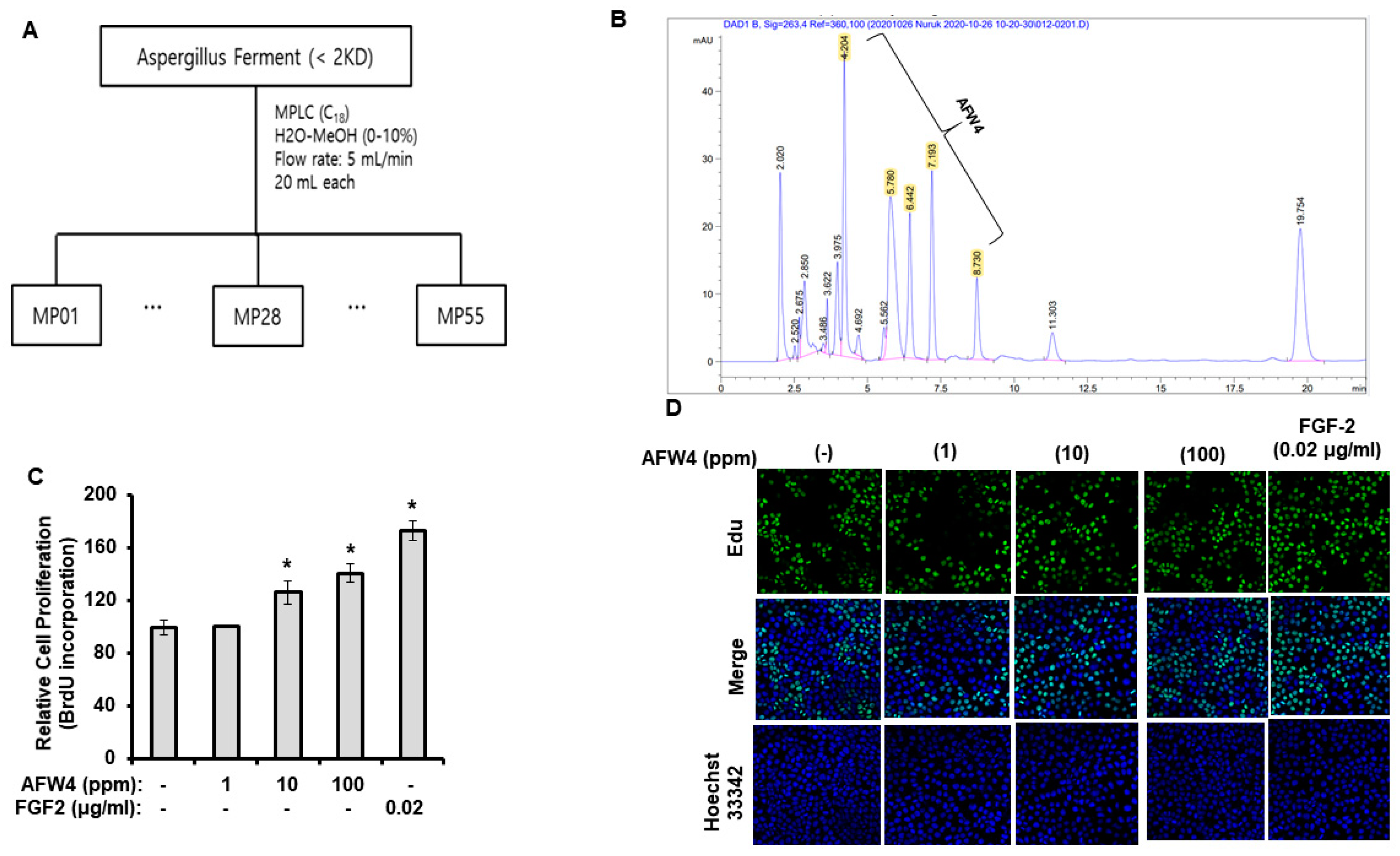
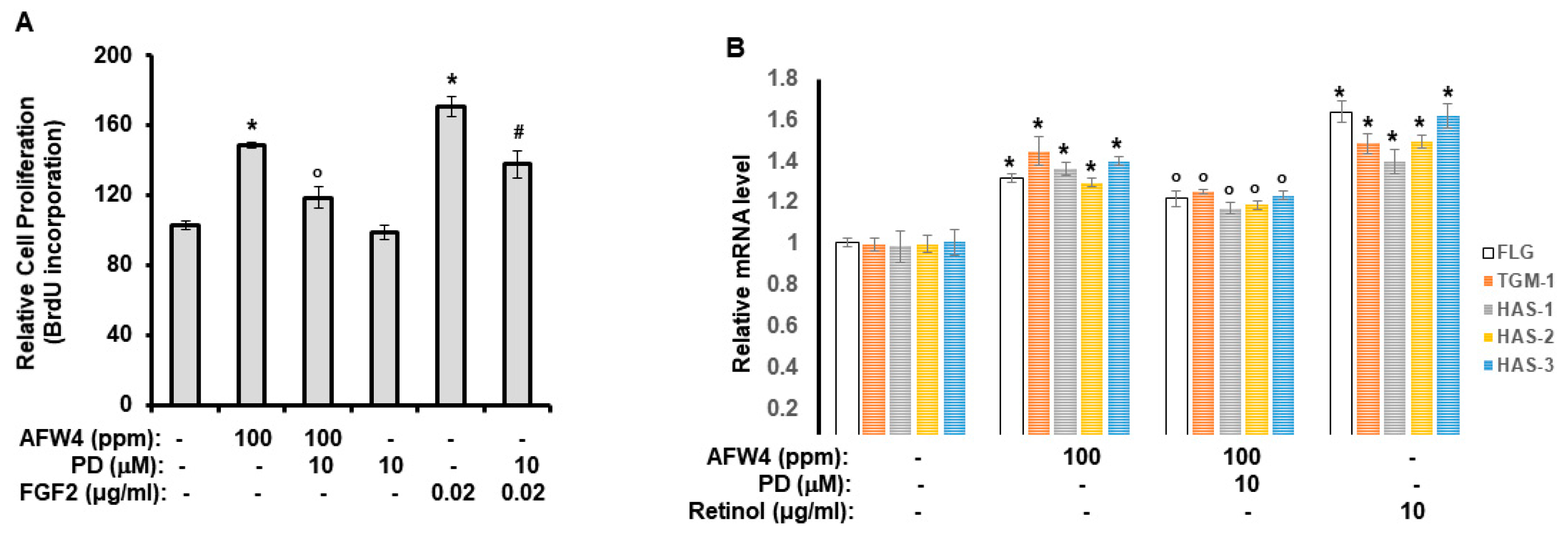
2.2. AFW4 Is the Main Component of AFWP Contributing to Cell Proliferation Potential
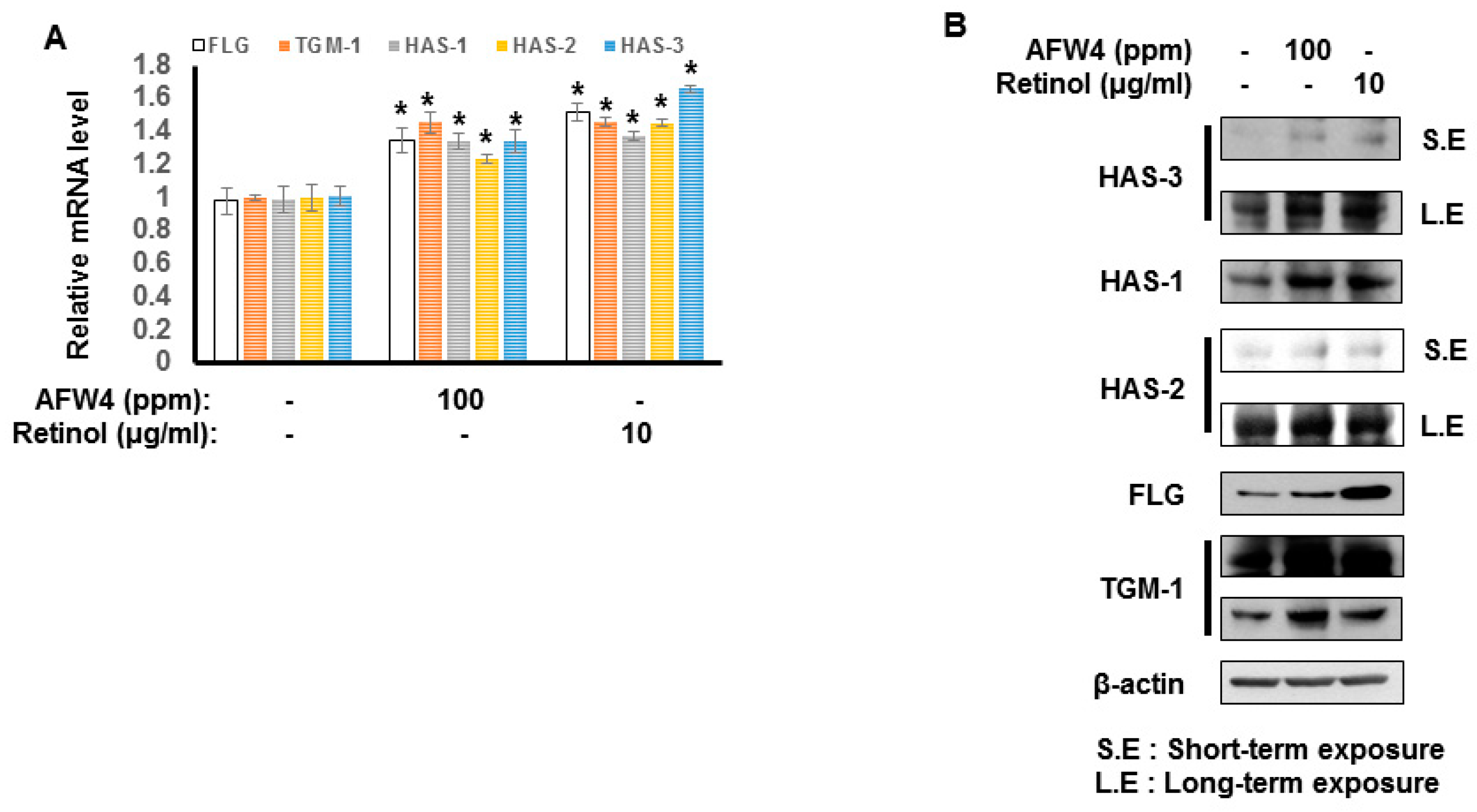
2.3. AFW4 Contributes to Skin Hydration through the Upregulation of NMF Levels
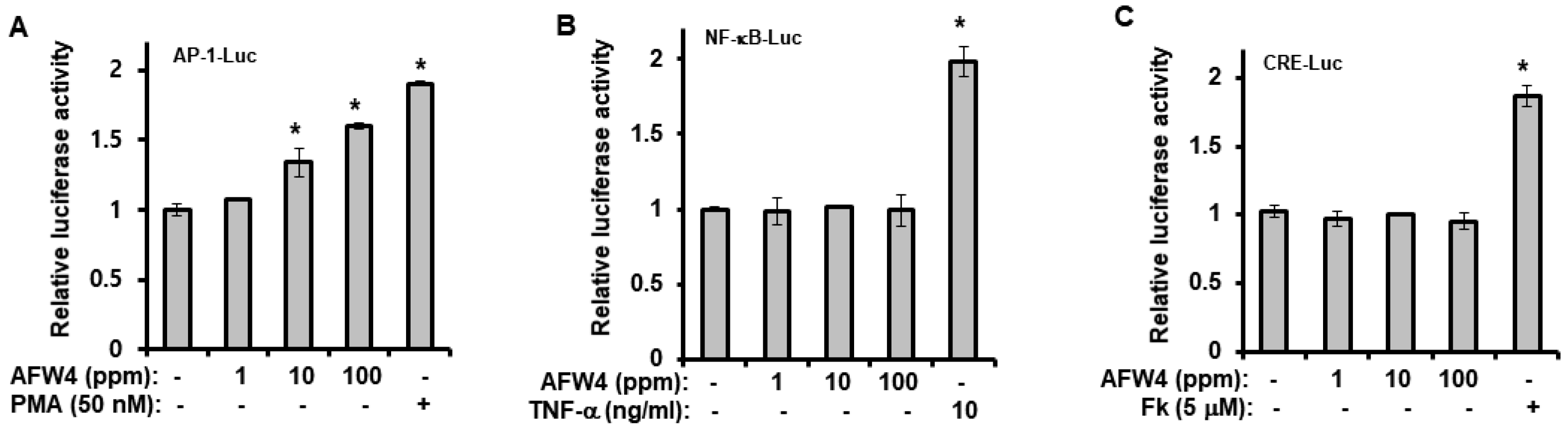
2.4. AFW4 Induces Cell Proliferation and Expression of NMF-Related Genes through p44/42 MAPK Activation
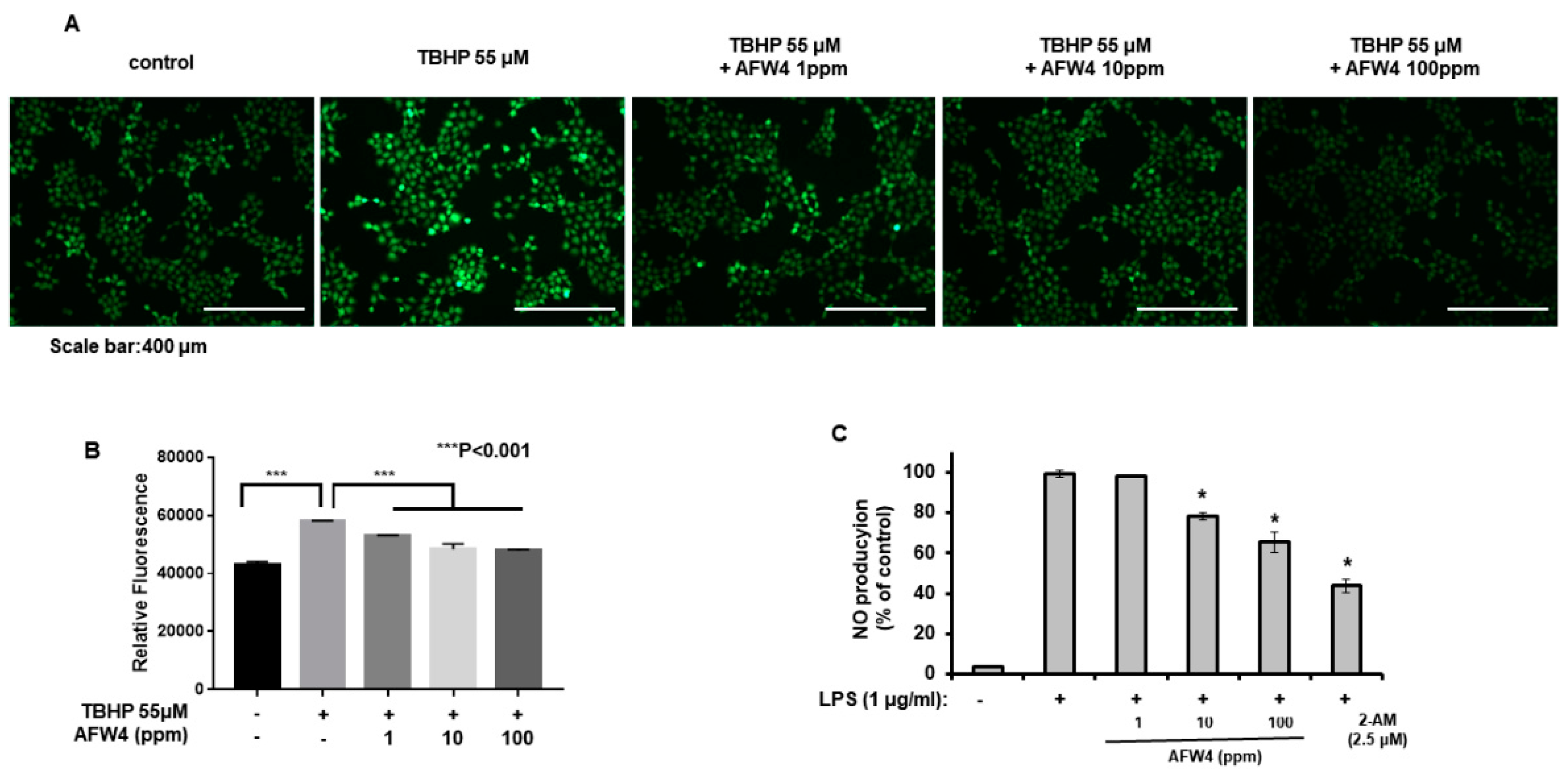
2.5. AFW4 Exerts Antioxidant Activity
2.6. AFW4 Improves Skin Hydration and TEWL
2.6.1. Moisture Content
2.6.2. Trans-Epidermal Water Loss (TEWL)
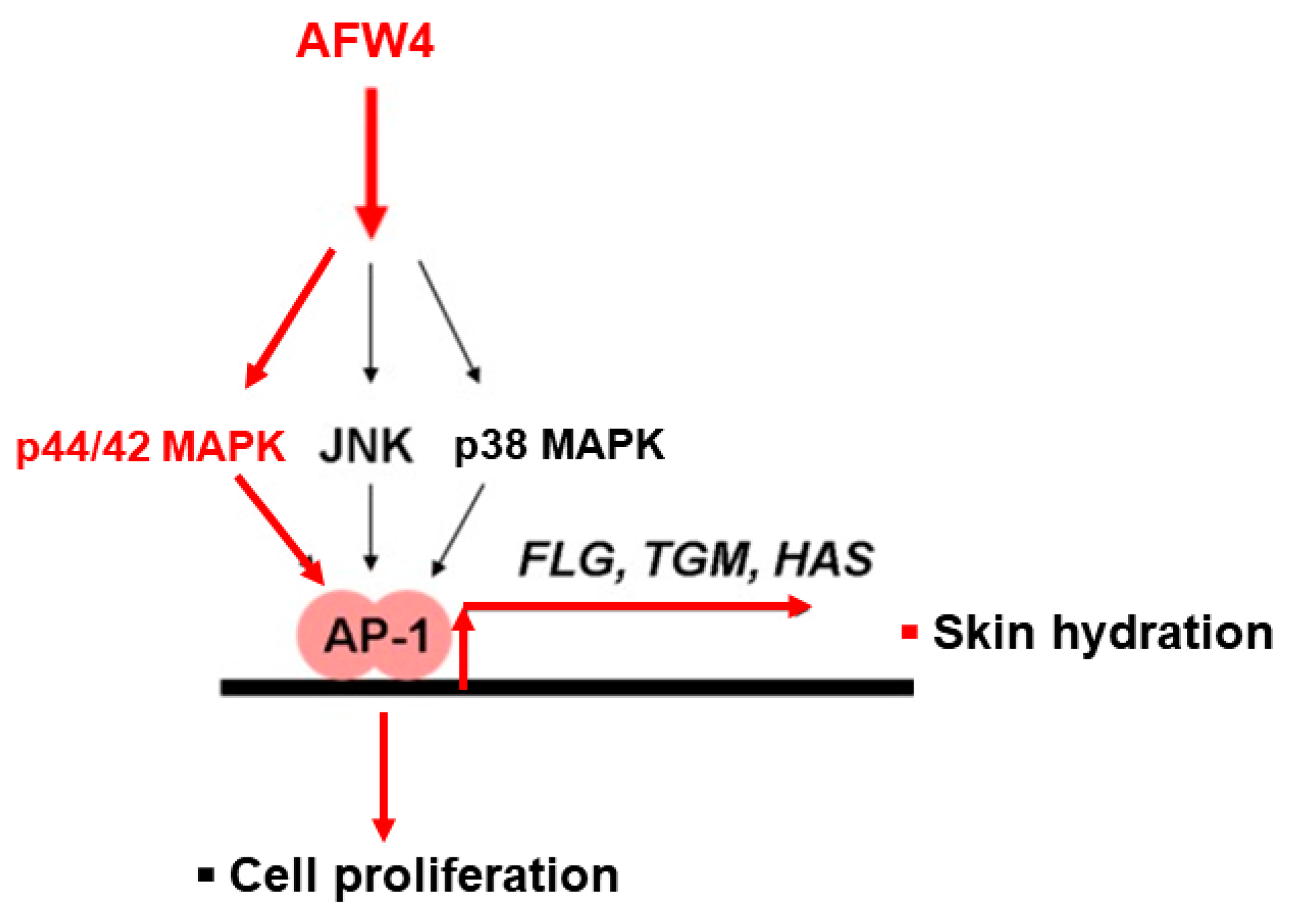
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Sample Preparation
4.2.1. Preparation of AFWP
4.2.2. Preparation of Low Molecular Weight Wheat Peptone
4.3. Gel Permeation Chromatography (GPC) Analysis
4.4. Amino Acid Composition Analysis
4.5. Medium-Pressure Liquid Chromatography (MPLC) Analysis
4.6. Peptide-Sequencing Analysis
4.7. 2,2′-Diphenyl-1-picrylhydrazyl (DPPH) Radical Scavenging Activity
4.8. Cell Culture
4.9. Cell Proliferation
4.10. Quantification of Nitric Oxide Levels
4.11. Western Blotting Analysis
4.12. MAPK-Phosphorylation Analysis
4.13. Analysis of mRNA Levels Using Real-Time Quantitative Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
4.14. Luciferase Reporter Assay
4.15. Cellular Reactive Oxygen Species (ROS) Detection Assay
4.16. Clinical Evaluation and Study Design
4.16.1. Skin Hydration
4.16.2. TEWL
4.16.3. Statistical Analyses of Clinical Data
4.17. Statistical Analyses of Experimental Data
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Igaki, M.; Higashi, T.; Hamamoto, S.; Kodama, S.; Naito, S.; Tokuhara, S. A study of the behavior and mechanism of thermal conduction in the skin under moist and dry heat conditions. Ski. Res. Technol. 2013, 20, 43–49. [Google Scholar] [CrossRef]
- Kim, H.-Y.; Agrahari, G.; Lee, M.; Tak, L.-J.; Ham, W.-K.; Kim, T.-Y. Low-Temperature Argon Plasma Regulates Skin Moisturizing and Melanogenesis-Regulating Markers Through Yes-Associated Protein. Int. J. Mol. Sci. 2021, 22, 1895. [Google Scholar] [CrossRef]
- Farage, M.A.; Miller, K.W.; Elsner, P.; Maibach, H.I. Intrinsic and extrinsic factors in skin ageing: A review. Int. J. Cosmet. Sci. 2008, 30, 87–95. [Google Scholar] [CrossRef]
- Wang, A.S.; Dreesen, O. Biomarkers of Cellular Senescence and Skin Aging. Front. Genet. 2018, 9, 247. [Google Scholar] [CrossRef]
- Ryu, Y.S.; Kang, K.A.; Piao, M.J.; Ahn, M.J.; Yi, J.M.; Bossis, G.; Hyun, Y.-M.; Park, C.O.; Hyun, J.W. Particulate matter-induced senescence of skin keratinocytes involves oxidative stress-dependent epigenetic modifications. Exp. Mol. Med. 2019, 51, 1–14. [Google Scholar] [CrossRef]
- Rittié, L.; Fisher, G.J. Natural and Sun-Induced Aging of Human Skin. Cold Spring Harb. Perspect. Med. 2015, 5, a015370. [Google Scholar] [CrossRef] [PubMed]
- Papakonstantinou, E.; Roth, M.; Karakiulakis, G. Hyaluronic acid: A key molecule in skin aging. Derm.-Endocrinol. 2012, 4, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Tammi, R.; Ripellino, J.A.; Margolis, R.U.; Maibach, H.I.; Tammi, M. Hyaluronate Accumulation in Human Epidermis Treated with Retinoic Acid in Skin Organ Culture. J. Investig. Dermatol. 1989, 92, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Dicker, K.T.; Gurski, L.A.; Pradhan-Bhatt, S.; Witt, R.L.; Farach-Carson, M.; Jia, X. Hyaluronan: A simple polysaccharide with diverse biological functions. Acta Biomater. 2014, 10, 1558–1570. [Google Scholar] [CrossRef]
- Hansen, L.A.; Brown, D.; Virador, V.; Tanaka, T.; Andreola, F.; Strain, K.; Dancheck, B.; Riley, R.; Arbeit, J.M.; De Luca, L.M.; et al. A PMLRARA transgene results in a retinoid-deficient phenotype associated with enhanced susceptibility to skin tumor-igenesis. Cancer Res. 2003, 63, 5257–5265. [Google Scholar]
- Kobayashi, T.; Chanmee, T.; Itano, N. Hyaluronan: Metabolism and Function. Biomolecules 2020, 10, 1525. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, A.V.; Harding, C.R. Moisturization and skin barrier function. Dermatol. Ther. 2004, 17, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Bow, J.R.; Sonoki, Y.; Uchiyama, M.; Shimizu, E.; Tanaka, K.; Dauskardt, R.H. Lipid Loss Increases Stratum Corneum Stress and Drying Rates. Ski. Pharmacol. Physiol. 2020, 33, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Son, E.D.; Kim, Y.; Joo, K.M.; Kim, H.J.; Lee, E.; Nam, G.W.; Cho, E.G.; Noh, M.; Chung, J.H.; Byun, S.Y.; et al. Skin dryness in apparently healthy human skin is associated with decreased expression of bleomycin hydrolase in the stratum corneum. Clin. Exp. Dermatol. 2014, 40, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Aluko, R.E. Identification and Inhibitory Properties of Multifunctional Peptides from Pea Protein Hydrolysate. J. Agric. Food Chem. 2010, 58, 11471–11476. [Google Scholar] [CrossRef] [PubMed]
- Franěk, F.; Hohenwarter, O.; Katinger, H. Plant Protein Hydrolysates: Preparation of Defined Peptide Fractions Promoting Growth and Production in Animal Cells Cultures. Biotechnol. Prog. 2000, 16, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Jan, D.C.-H.; Jones, S.J.; Emery, A.N.; Al-Rubeai, M. Peptone, a low-cost growth-promoting nutrient for intensive animal cell culture. Cytotechnology 1994, 16, 17–26. [Google Scholar] [CrossRef]
- Jung, E.; Cho, J.Y.; Park, D.; Kim, M.H.; Park, B.; Lee, S.Y.; Lee, J. Vegetable peptones increase production of type I collagen in human fibroblasts by inducing the RSK-CCAAT/enhancer binding protein-β phosphorylation pathway. Nutr. Res. 2015, 35, 127–135. [Google Scholar] [CrossRef]
- Pettit, R.K. Mixed fermentation for natural product drug discovery. Appl. Microbiol. Biotechnol. 2009, 83, 19–25. [Google Scholar] [CrossRef]
- Song, S.H. Analysis of Microflora Profile in Korean Traditional Nuruk. J. Microbiol. Biotechnol. 2013, 23, 40–46. [Google Scholar] [CrossRef]
- Bal, J.; Yun, S.-H.; Song, H.-Y.; Yeo, S.-H.; Kim, J.H.; Kim, J.-M.; Kim, D.-H. Mycoflora dynamics analysis of Korean traditional wheat-based nuruk. J. Microbiol. 2014, 52, 1025–1029. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Choi, S.J.; Kwak, J.; Kim, K.; Seo, M.; Moon, T.W.; Lee, Y.-W. Aspergillus oryzae strains isolated from traditional Korean Nuruk: Fermentation properties and influence on rice wine quality. Food Sci. Biotechnol. 2013, 22, 425–432. [Google Scholar] [CrossRef]
- Papaspyridi, L.-M.; Aligiannis, N.; Topakas, E.; Christakopoulos, P.; Skaltsounis, A.-L.; Fokialakis, N. Submerged Fermentation of the Edible Mushroom Pleurotus ostreatus in a Batch Stirred Tank Bioreactor as a Promising Alternative for the Effective Production of Bioactive Metabolites. Molecules 2012, 17, 2714–2724. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, N.; Zhang, T.; Rahl, P.B.; Abraham, B.J.; Reddy, J.; Ficarro, S.B.; Dastur, A.; Amzallag, A.; Ramaswamy, S.; Tesar, B.; et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 2014, 511, 616–620. [Google Scholar] [CrossRef]
- Blois, M.S. Antioxidant Determinations by the Use of a Stable Free Radical. Nat. Cell Biol. 1958, 181, 1199–1200. [Google Scholar] [CrossRef]
- Yoo, J.A.; Yu, E.; Park, S.-H.; Oh, S.W.; Kwon, K.; Park, S.J.; Kim, H.; Yang, S.; Park, J.Y.; Cho, J.Y.; et al. Blue Light Irradiation Induces Human Keratinocyte Cell Damage via Transient Receptor Potential Vanilloid 1 (TRPV1) Regulation. Oxidative Med. Cell. Longev. 2020, 2020, 1–14. [Google Scholar] [CrossRef]
- Lee, S.E.; Park, S.-H.; Yoo, J.A.; Kwon, K.; Kim, J.W.; Oh, S.W.; Park, S.J.; Kim, J.; Yu, E.; Han, B.S.; et al. Antagonizing Effects of Clematis apiifolia DC. Extract against Benzo[a]pyrene-Induced Damage to Human Keratinocytes. Oxidative Med. Cell. Longev. 2019, 2019, 2386163. [Google Scholar] [CrossRef]
- Smale, S.T. β-Galactosidase Assay. Cold Spring Harb. Protoc. 2010, 2010, 5423. [Google Scholar] [CrossRef]
- Hashimoto-Kumasaka, K.; Takahashi, K.; Tagami, H. Electrical measurement of the water content of the stratum corneum in vivo and in vitro under various conditions: Comparison between skin surface hygrometer and corneometer in evaluation of the skin surface hydration state. Acta Derm.-Venereol. 1993, 73, 335–339. [Google Scholar]
- Meng, H.; Lin, W.; Dong, Y.; Li, L.; Yi, F.; Meng, Q.; Li, Y.; He, Y. Statistical analysis of age-related skin parameters. Technol. Health Care 2021, 29, 65–76. [Google Scholar] [CrossRef]
- Lodén, M. Role of Topical Emollients and Moisturizers in the Treatment of Dry Skin Barrier Disorders. Am. J. Clin. Dermatol. 2003, 4, 771–788. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, A.V.; Matts, P.J. Stratum Corneum Moisturization at the Molecular Level: An Update in Relation to the Dry Skin Cycle. J. Investig. Dermatol. 2005, 124, 1099–1110. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, D.R.; Kroll, L.M.; Basehoar, A.; Reece, B.; Cunningham, C.T.; Koenig, D.W. Immediate and extended effects of abrasion on stratum corneum natural moisturizing factor. Ski. Res. Technol. 2015, 21, 366–372. [Google Scholar] [CrossRef] [PubMed]
- Chiou, Y.B.; Blume-Peytavi, U. Stratum Corneum Maturation. Ski. Pharmacol. Physiol. 2004, 17, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Nolan, K.; Marmur, E. Moisturizers: Reality and the skin benefits. Dermatol. Ther. 2012, 25, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Zhang, J.; Li, H. Selenium, aging and aging-related diseases. Aging Clin. Exp. Res. 2018, 31, 1035–1047. [Google Scholar] [CrossRef]
- Gilchrest, B.A. Skin aging and photoaging: An overview. J. Am. Acad. Dermatol. 1989, 21, 610–613. [Google Scholar] [CrossRef]
- Panich, U.; Sittithumcharee, G.; Rathviboon, N.; Jirawatnotai, S. Ultraviolet Radiation-Induced Skin Aging: The Role of DNA Damage and Oxidative Stress in Epidermal Stem Cell Damage Mediated Skin Aging. Stem Cells Int. 2016, 2016, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Parrado, C.; Mercado-Saenz, S.; Perez-Davó, A.; Gilaberte, Y.; Gonzalez, S.; Juarranz, A. Environmental Stressors on Skin Aging. Mechanistic Insights. Front. Pharmacol. 2019, 10, 759. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Database | Enzyme Used for Hydrolysis | The Most Suitable Amino Acid Sequence |
|---|---|---|---|
| AFW4 | Uniprot Database: Organism: T. aestivum (Wheat) | Random | MASI |
| TMIT | |||
| Trypsin-Chymotrypsin (K, R, W, H, M, L, Y/X-X) | MLSLF YGSD LPQHSRDSLR AAQPR |
| Sample | Mean ± SEM | p-Value | |||
|---|---|---|---|---|---|
| 0 Week | 2 Weeks | 4 Weeks | 0 vs. 2 Weeks | 0 vs. 4 Weeks | |
| AFWP4 | 23.3 ± 1.32 | 28.1 ± 1.68 | 31.8 ± 1.45 | 0.003 ** | 0.003 ** |
| AFWP CON | 24.1 ± 1.61 | 26.3 ± 1.68 | 29.5 ± 1.24 | 0.000 ### | 0.000 ### |
| Weeks | Mean ± SEM | Title 4 | |
|---|---|---|---|
| AFWP4 | AFWP CON | AFWP4 vs. AFWP CON | |
| 2 | 20.6 ± 2.15 | 9.7 ± 1.89 | 0.003 ** |
| 4 | 38.3 ± 4.27 | 25.7 ± 5.33 | 0.017 * |
| Sample | Mean ± SEM | p-Value | |||
|---|---|---|---|---|---|
| 0 Week | 2 Weeks | 4 Weeks | 0 vs. 2 Weeks | 0 vs. 4 Weeks | |
| AFWP4 | 18.8 ± 1.43 | 17.4 ± 1.30 | 15.9 ± 1.14 | 0.010 * | 0.003 ** |
| AFWP CON | 18.9 ± 1.15 | 18.3 ± 1.05 | 17.3 ± 1.07 | 0.167 | 0.008 ** |
| Weeks | Mean ± SEM | Title 4 | |
|---|---|---|---|
| AFWP4 | AFWP CON | AFWP4 vs. AFWP CON | |
| 2 | 7.3 ± 2.08 | 2.6 ± 1.76 | 0.103 |
| 4 | 14.8 ± 2.25 | 8.2 ± 2.04 | 0.042 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hahm, K.M.; Park, S.-H.; Oh, S.W.; Kim, J.H.; Yeom, H.S.; Lee, H.J.; Yang, S.; Cho, J.Y.; Park, J.O.; Lee, J. Aspergillus oryzae-Fermented Wheat Peptone Enhances the Potential of Proliferation and Hydration of Human Keratinocytes through Activation of p44/42 MAPK. Molecules 2021, 26, 6074. https://doi.org/10.3390/molecules26196074
Hahm KM, Park S-H, Oh SW, Kim JH, Yeom HS, Lee HJ, Yang S, Cho JY, Park JO, Lee J. Aspergillus oryzae-Fermented Wheat Peptone Enhances the Potential of Proliferation and Hydration of Human Keratinocytes through Activation of p44/42 MAPK. Molecules. 2021; 26(19):6074. https://doi.org/10.3390/molecules26196074
Chicago/Turabian StyleHahm, Kyung Man, See-Hyoung Park, Sae Woong Oh, Ji Hye Kim, Hyun Sook Yeom, Hye Ja Lee, Seoyeon Yang, Jae Youl Cho, Jin Oh Park, and Jongsung Lee. 2021. "Aspergillus oryzae-Fermented Wheat Peptone Enhances the Potential of Proliferation and Hydration of Human Keratinocytes through Activation of p44/42 MAPK" Molecules 26, no. 19: 6074. https://doi.org/10.3390/molecules26196074
APA StyleHahm, K. M., Park, S.-H., Oh, S. W., Kim, J. H., Yeom, H. S., Lee, H. J., Yang, S., Cho, J. Y., Park, J. O., & Lee, J. (2021). Aspergillus oryzae-Fermented Wheat Peptone Enhances the Potential of Proliferation and Hydration of Human Keratinocytes through Activation of p44/42 MAPK. Molecules, 26(19), 6074. https://doi.org/10.3390/molecules26196074