
A QuEChERS-HPLC-MS/MS Method with Matrix Matching Calibration Strategy for Determination of Imidacloprid and Its Metabolites in Procambarus clarkii (Crayfish) Tissues

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
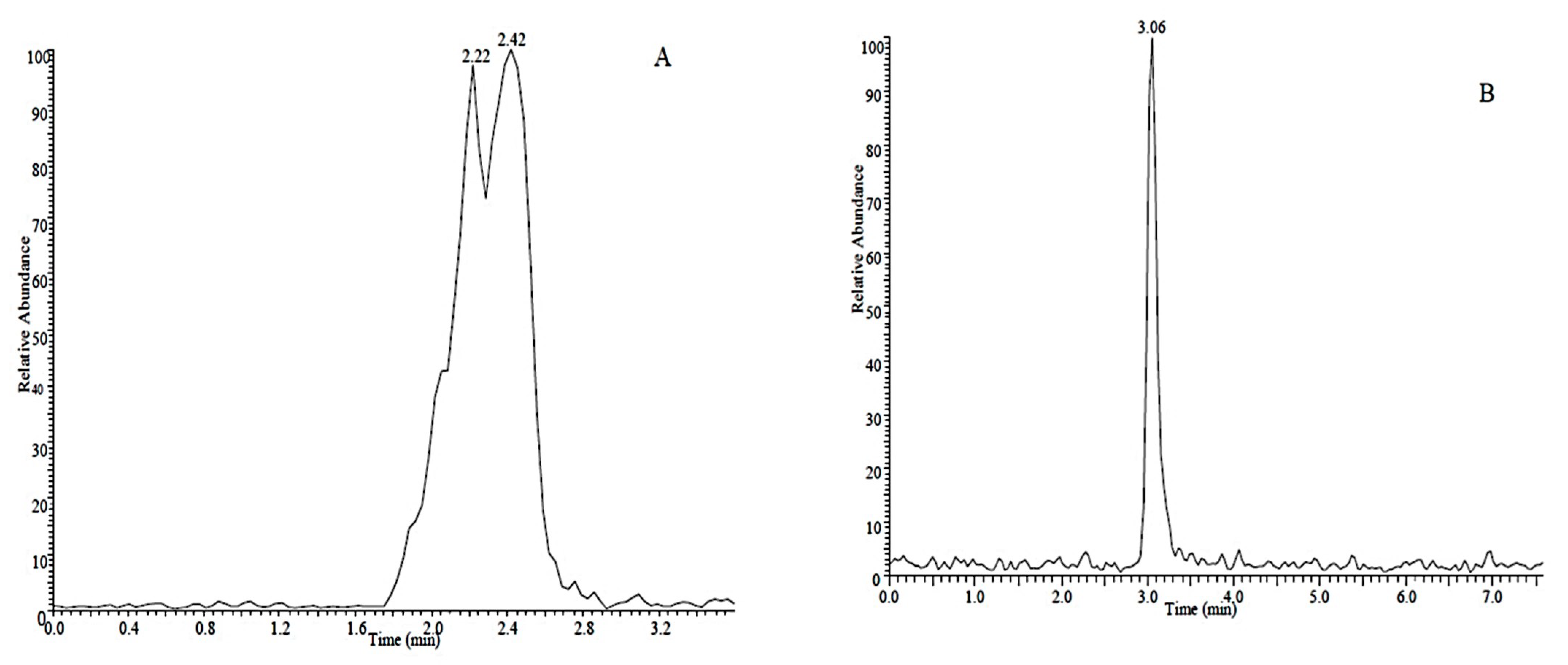
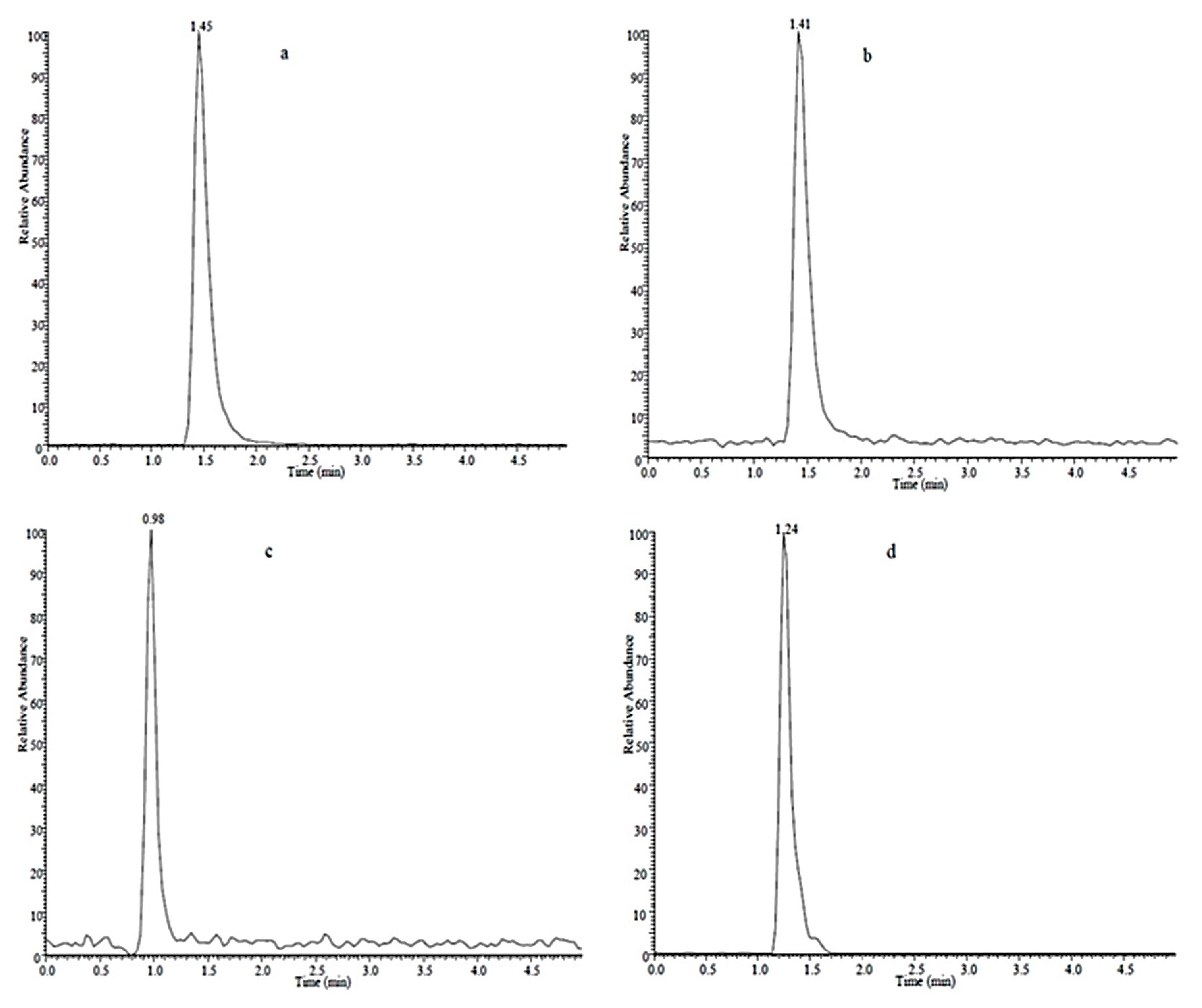
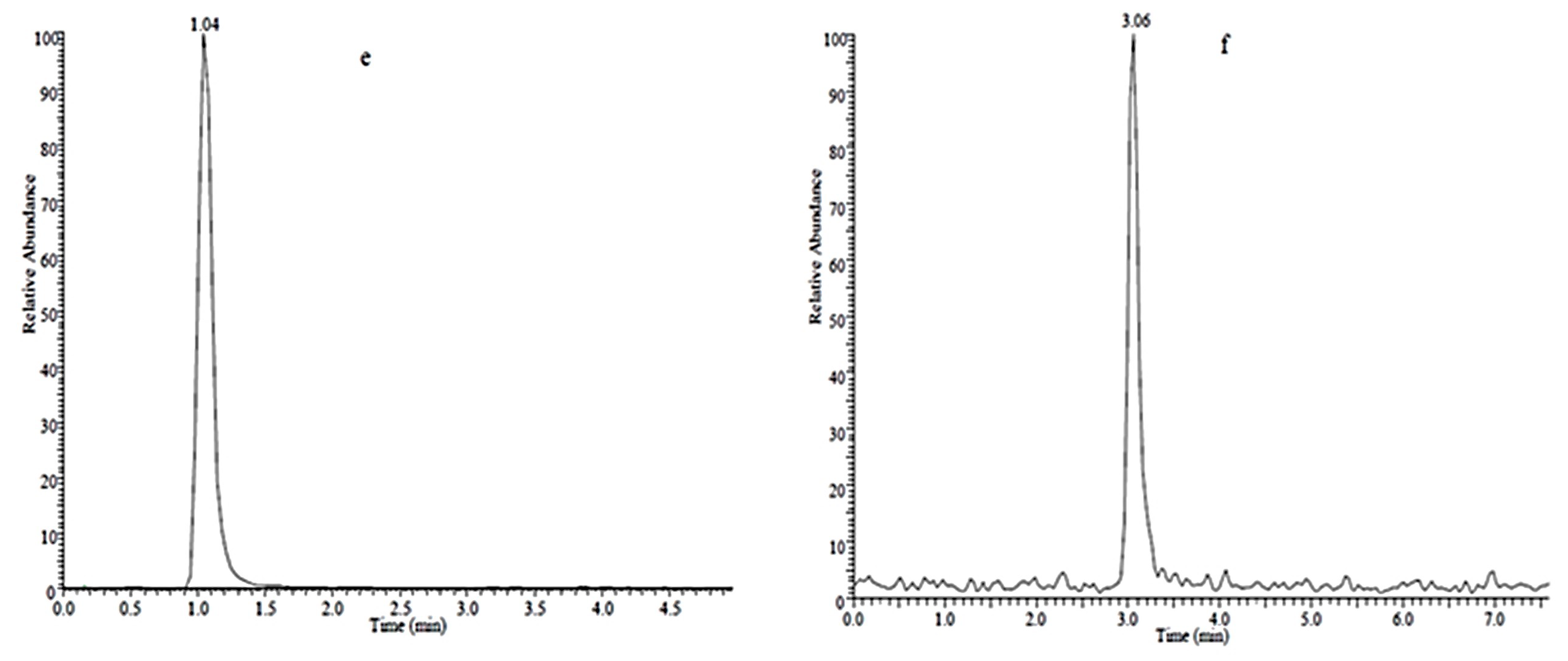
2.1. Analyte Separation and Identification
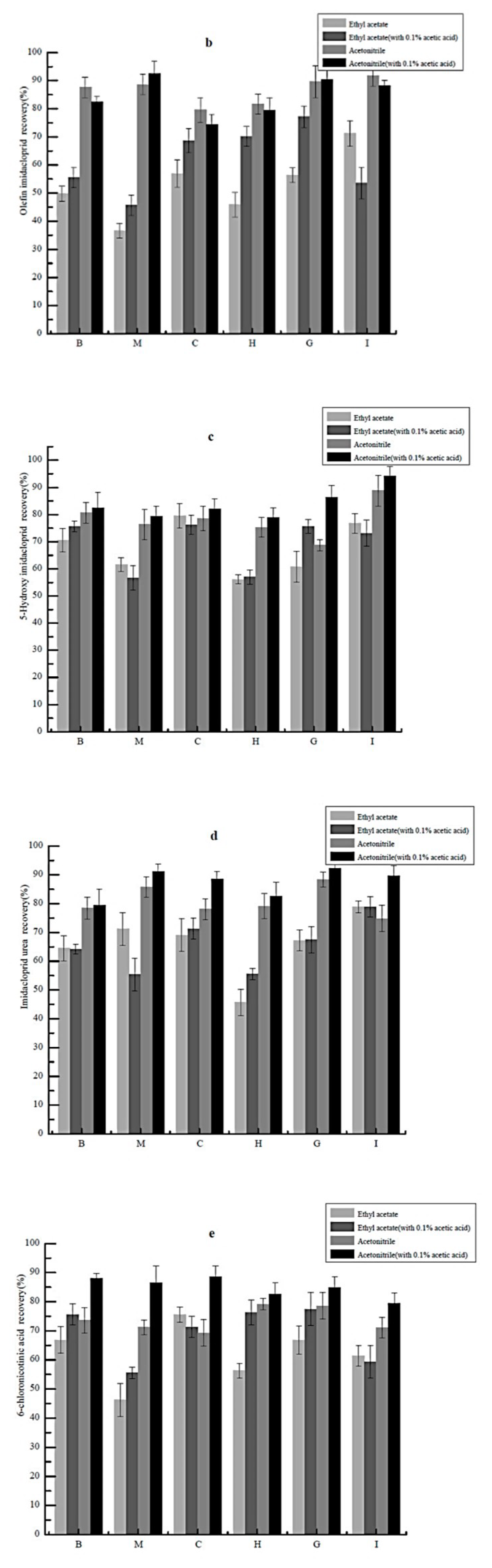
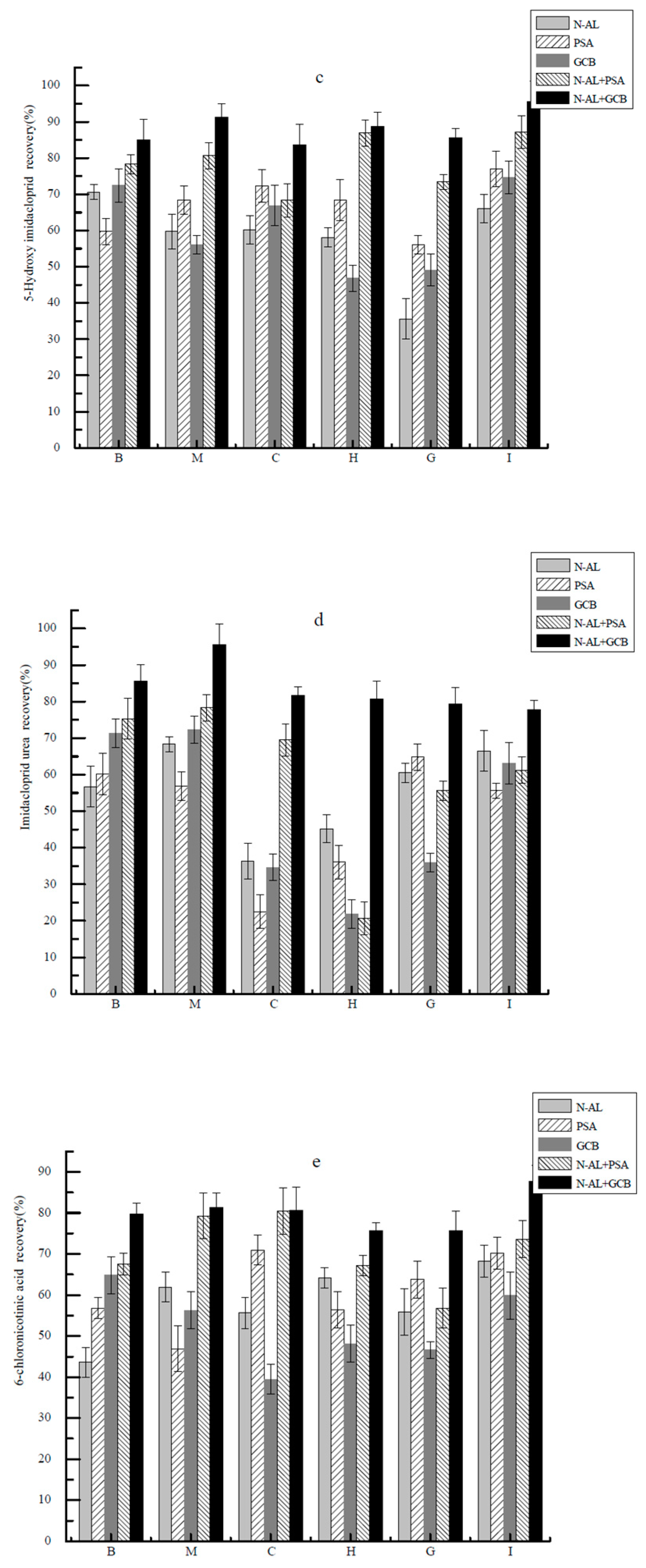
2.2. Selection of Extraction Solvent
2.3. Optimization of Purification Conditions
2.4. Linear Range, Matrix Effects, Detection Limits, Recoveries and RSD
3. Real Sample Analysis
4. Experimental
4.1. Reagents
4.2. Standard Solutions
4.3. Sample Preparation
4.4. Sample Extraction
4.5. Analyte Identification
4.6. Validation of the Analytical Method
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Compliance with Ethical Standards
Conflicts of Interest
Sample Availability
References
- Kang, P.; Mao, B.; Fan, C.; Wang, Y. Transcriptomic information from the ovaries of red swamp crayfish (Procambarus clarkii) provides new insights into development of ovaries and embryos. Aquaculture 2019, 505, 333–343. [Google Scholar] [CrossRef]
- Qian, X.; Zhu, F. Hesperetin protects crayfish Procambarus clarkii against white spot syndrome virus infection. Fish Shellfish Immunol. 2019, 93, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Liu, H.; Li, S.; Xu, N.; Liu, Y.; Ai, X. Determination of imidacloprid residues in water, sediment and Procambarus clarkii samples by high performance liquid chromatography-tandem mass spectrometry. Agrochemicals 2018, 57, 427–430. [Google Scholar]
- Chen, L.; Li, B. Determination of imidacloprid in rice by molecularly imprinted-matrix solid-phase dispersion with liquid chromatography tandem mass spectrometry. J. Chromatogr. B 2012, 897, 32–36. [Google Scholar] [CrossRef]
- Pandiselvi, S.; Sathiyanarayanan, S.; Ramesh, A. Determination of spirotetramat and imidacloprid residues in cotton seed, lint, oil and soil by HPLC UV method and their dissipation in cotton plant. Pestic. Res. J. 2010, 22, 168–173. [Google Scholar]
- Sajid, I.; Riaz, U.; Sumayya, S.; Mubarik, A.; Muhammad, A.; Sahar, A. Extraction, cleanup, and chromatographic determination of imidacloprid residues in wheat. Bull Env. Contam. Toxicol. 2012, 88, 555–558. [Google Scholar]
- Li, Y.; Yang, L.; Yan, H.; Zhang, M.; Ge, J.; Yu, X.i. Uptake, translocation and accumulation of imidacloprid in six leafy vegetables at three growth stages. Ecotoxicol. Environ. Saf. 2018, 164, 690–695. [Google Scholar] [CrossRef]
- Kapoor, U.; Srivastava, M.K.; Srivastava, A.K.; Patel, D.K.; Garg, V.; Srivastava, L.P. Analysis of imidacloprid residues in fruits, vegetables, cereals, fruit juices, and baby foods, and daily intake estimation in and around Lucknow, India. Environ. Toxicol. Chem. 2013, 32, 723–727. [Google Scholar] [CrossRef]
- Stokstad, E. Field research on bees raises concern about low-dose pesticides. Science 2012, 335, 1555. [Google Scholar] [CrossRef] [Green Version]
- Whitehorn, P.R.; O’Connor, S.; Wackers, F.L.; Goulson, D. Neonicotinoid pesticide reduces bumble bee colony growth and queen production. Science 2012, 336, 351–352. [Google Scholar] [CrossRef] [Green Version]
- Michlig, M.P.; Merke, J.; Pacini, A.C.; Orellano, E.M.; Beldoménico, H.R.; Repetti, M.R. Determination of imidacloprid in beehive samples by UHPLC-MS/MS. Microchem. J. 2018, 143, 72–81. [Google Scholar] [CrossRef]
- Faraji, M.; Noorbakhsh, R.; Shafieyan, H.; Ramezani, M. Determination of acetamiprid, imidacloprid, and spirotetramat and their relevant metabolites in pistachio using modified QuEChERS combined with liquid chromatography-tandem mass spectrometry. Food Chem. 2018, 240, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Qin, X.; Luo, X.; Yu, W.; Yang, G.; Zhang, K.; Hu, D. A liquid chromatography with tandem mass spectrometry method to simultaneously determinate chlorpyrifos, imidacloprid and imidacloprid metabolites in wheat. J. Sep. Sci. 2019, 42, 1210–1221. [Google Scholar] [CrossRef] [PubMed]
- Rancan, M.; Sabatini, A.G.; Achilli, G.; Galletti, G.C. Determination of Imidacloprid and metabolites by liquid chromatography with an electrochemical detector and post column photochemical reactor. Anal. Chim. Acta 2006, 555, 20–24. [Google Scholar] [CrossRef]
- Fan, Y.; Shi, X.; Gao, X. Research progresses on the metabolism of neonicotinoids imidacloprid and thiamethoxam. Chin. J. Pestic. Sci. 2012, 14, 587–596. [Google Scholar]
- Liang, X.; Wang, X.; Xue, X.; Qi, P.; Liu, Z.; Liu, Z.; Zhou, L.; Wang, X.; Wang, Q. Determination of imidacloprid and its three metabolites in honey and bee pollen by liquid chromatography-tandem mass spectrometry. Chin. J. Anal. Chem. 2017, 45, 553–559. [Google Scholar]
- Deng, T.; Gao, Y.; Chen, X. Mechanisms for selective action of neonicotinoid insecticides. J. Henan Inst. Sci. Technol. 2010, 38, 23–26. [Google Scholar]
- Xia, Z.; Zhang, J.; Zhou, Y.; Peng, L.; Zheng, D.; Zhang, X.; Hu, X.; Xia, H. Status of the Quality and Safety Standards for Crayfish in China: A Review. Mod. Food Sci. Technol. 2020, 36, 310–318. [Google Scholar]
- Kiljanek, T.; Niewiadowska, A.; Semeniuk, S.; Gaweł, M.; Borzęcka, M.; Posyniak, A. Multi-residue method for the determination of pesticides and pesticide metabolites in honeybees by liquid and gas chromatography coupled with tandem mass spectrometry-Honeybee poisoning incidents. J. Chromatogr. A 2016, 1435, 100–114. [Google Scholar] [CrossRef]
- Ko, A.-Y.; Rahman, M.M.; Abd El-Aty, A.M.; Jang, J.; Park, J.-H.; Cho, S.-K.; Shim, J.-H. Development of a simple extraction and oxidation procedure for the residue analysis of imidacloprid and its metabolites in lettuce using gas chromatography. Food Chem. 2014, 148, 402–409. [Google Scholar] [CrossRef]
- Amelin, V.G.; Bol’shakov, D.S.; Tretiakov, A.V. Identification and determination of synthetic pyrethroids, chlorpyriphos, and neonicotinoids in water by gas and liquid chromatography. J. Anal. Chem. 2012, 67, 354–359. [Google Scholar] [CrossRef]
- Rossi, S.; Sabatini, A.G.; Cenciarini, R.; Ghini, S.; Girotti, S. Use of High-performance liquid chromatography-UV and gas chromatography-mass spectrometry for determination of the imidacloprid content of honeybees, pollen, paper filters, grass, and flowers. Chromatographia 2005, 61, 189–195. [Google Scholar] [CrossRef]
- Huang, Y.; Shi, T.; Luo, X.; Xiong, H.; Min, F.; Chen, Y.; Nie, S.; Xie, M. Determination of multi-pesticide residues in green tea with a modified QuEChERS protocol coupled to HPLC-MS/MS. Food Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Bhattacharyya, A.; Shinde, R.; Dhanshetty, M.; Elliott, C.T.; Banerjee, K. Development and validation of a multiresidue method for pesticides and selected veterinary drugs in animal feed using liquid- and gas chromatography with tandem mass spectrometry. J. Chromatogr. A 2020, 1627, 461416. [Google Scholar] [CrossRef] [PubMed]
- Pagani, A.P.; Ibañez, G.A. Pesticide residues in fruits and vegetables: High-order calibration based on spectrofluorimetric/pH data. Microchem. J. 2019, 149, 104042. [Google Scholar] [CrossRef]
- He, J.; Zhang, B.; Zhang, H.; Hao, L.; Ma, T.; Wang, J.; Han, S. Monitoring of 49 Pesticides and 17 Mycotoxins in Wine by QuEChERS and UHPLC–MS/MS Analysis. J. Food Sci. 2019, 84, 2688–2697. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Kim, M.; Kim, E.; Choe, W. Determination of 66 pesticide residues in livestock products using QuEChERS and GC–MS/MS. Food Sci Biotechnol. 2020, 29, 1573–1586. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Ai, X.; Li, S.; Liu, H.; Liu, Y. Determination of pendimethalin in water, sediment, and Procambarus clarkii by high performance liquid chromatography-triple quadrupole mass spectrometry. Environ. Monit. Assess. 2019, 191, 621. [Google Scholar] [CrossRef]
- Maestroni, B.; Besil, N.; Bojorge, A.; Gérez, N.; Pérez-Parada, A.; Cannavan, A.; Heinzen, H.; Cesio, M.V. Optimization and validation of a single method for the determination of pesticide residues in Peumus boldus Molina leaves using GC-MSD, GC-MS/MS and LC-MS/MS. J. Appl. Res. Med. Aromat. Plants 2020, 18, 100254. [Google Scholar] [CrossRef]
- Shelepchikov, A.A.; Ovcharenko, V.V.; Kozhushkevich, A.I.; Brodskii, E.S.; Komarov, A.A.; Turbabina, K.A.; Kalantaenko, A.M. A new method for purifying fat-containing extracts in the determination of polybrominated diphenyl ethers. J. Anal. Chem. 2019, 74, 574–583. [Google Scholar] [CrossRef]
- Caban, M.; Lis, H.; Kobylis, P.; Stepnowski, P. The triple-sorbents solid-phase extraction for pharmaceuticals and estrogens determination in wastewater samples. Microchem. J. 2019, 149, 103965. [Google Scholar] [CrossRef]
- Jiang, C.; Dai, J.; Han, H.; Wang, C.; Zhu, L.; Lu, C.; Chen, H. Determination of thirteen acidic phytohormones and their analogues in tea (Camellia sinensis) leaves using ultra high performance liquid chromatography tandem mass spectrometry. J. Chromatogr. B 2020, 1149, 122144. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Qiao, C.; Luo, J.; Guo, L.; Pang, T.; Pang, R.; Li, J.; Wang, C.; Wang, R.; Xie, H. Development of a fast multi-residue method for the determination of succinate dehydrogenase inhibitor fungicides in cereals, vegetables and fruits by modified QuEChERS and UHPLC-MS/MS. J. Chromatogr. B 2020, 1152, 122261. [Google Scholar] [CrossRef] [PubMed]
- Hao, C.; Noestheden, M.R.; Zhao, X.; Morse, D. Liquid chromatography tandem mass spectrometry analysis of neonicotinoid pesticides and 6-chloronicotinic acid in environmental water with direct aqueous injection. Anal. Chim. Acta 2016, 925, 43–50. [Google Scholar] [CrossRef]
- Xu, N.; Dong, J.; Yang, Y. Development of a liquid chromatography–tandem mass spectrometry method with modified QuEChERS extraction for the quantification of mebendazole and its metabolites, albendazole and its metabolites, and levamisole in edible tissues of aquatic animals. Food Chem. 2018, 269, 442–449. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Linear Range (μg·L−1) | Regression Equation a | Correlation Coefficient (R2) | LOD b (μg·L−1) | LOQ c (μg·L−1) | RSD (%, N = 3) | |
|---|---|---|---|---|---|---|---|
| Inter-Day | Intraday | ||||||
| IMI | 0.05–2.00 | Y = 0.874X + 0.079 | 0.9922 | 0.02 | 0.05 | 4.3 | 5.7 |
| 5-Hydroxy IMI | 1.00–20.00 | Y = 0.134X + 0.070 | 0.9947 | 0.50 | 1.00 | 6.8 | 4.4 |
| olefin IMI | 2.00–100.00 | Y = 0.298X + 0.034 | 0.9981 | 0.50 | 2.00 | 5.2 | 5.6 |
| IMI urea | 0.10–10.00 | Y = 1.631X + 0.464 | 0.9948 | 0.03 | 0.10 | 7.1 | 6.1 |
| 6-Chloronicotinic acid | 1.00–50.00 | Y = 0.436X − 0.856 | 0.9996 | 0.50 | 1.00 | 3.3 | 7.5 |
| Compound | Spike Level (μg·L−1) | Plasma | Cephalothorax | Hepatopancrea | Gill | Intestine | Muscle | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IS-N MF | CV | IS-N MF | CV | IS-N MF | CV | IS-N MF | CV | IS-N MF | CV | IS-N MF | CV | ||
| IMI † | 0.1 | 1.055 | 0.09 | 1.752 | 0.05 | 0.695 | 0.08 | 1.023 | 0.05 | 1.103 | 0.06 | 0.359 | 0.06 |
| 5.0 | 0.987 | 0.11 | 1.568 | 0.07 | 0.712 | 0.13 | 0.968 | 0.08 | 0.988 | 0.08 | 0.475 | 0.08 | |
| 5-Hydroxy IMI | 2.0 | 0.923 | 0.08 | 0.897 | 0.13 | 0.566 | 0.11 | 0.582 | 0.1 | 0.723 | 0.13 | 1.125 | 0.11 |
| 100.0 | 0.897 | 0.07 | 0.974 | 0.07 | 0.691 | 0.08 | 0.637 | 0.08 | 0.632 | 0.06 | 0.969 | 0.12 | |
| olefin IMI | 5.0 | 0.947 | 0.11 | 0.364 | 0.05 | 1.454 | 0.12 | 0.581 | 0.12 | 0.345 | 0.11 | 0.457 | 0.15 |
| 200.0 | 0.963 | 0.12 | 0.451 | 0.04 | 1.387 | 0.06 | 0.448 | 0.09 | 0.421 | 0.07 | 0.611 | 0.13 | |
| IMI urea | 0.2 | 0.996 | 0.05 | 0.865 | 0.1 | 0.503 | 0.07 | 0.999 | 0.04 | 0.538 | 0.05 | 1.325 | 0.13 |
| 10.0 | 0.897 | 0.09 | 0.905 | 0.12 | 0.487 | 0.11 | 0.896 | 0.09 | 0.476 | 0.08 | 1.451 | 0.14 | |
| 6-CNA † | 2.0 | 0.869 | 0.06 | 1.471 | 0.09 | 1.417 | 0.09 | 0.894 | 0.11 | 1.308 | 0.07 | 1.326 | 0.06 |
| 100.0 | 0.857 | 0.14 | 1.302 | 0.06 | 1.349 | 0.05 | 1.057 | 0.14 | 1.411 | 0.07 | 1.259 | 0.15 | |
| Compound | Index | Plasma | Cephalothorax | Hepatopancrea | Gill | Intestine | Muscle |
|---|---|---|---|---|---|---|---|
| IMI † | Linear range (μg·L−1) | 0.1–5.0 | 0.1–5.0 | 0.1–5.0 | 0.1–5.0 | 0.1–5.0 | 0.1–5.0 |
| Regression equation d | y = 0.937x + 0.395 | y = 0.370x + 0.691 | y = 1.478x − 0.284 | y = 0.633x + 0.121 | y = 0.882x + 0.109 | y = 0.902x + 0.251 | |
| Correlation coefficient (R2) | 0.9931 | 0.9978 | 0.9969 | 0.9935 | 0.9904 | 0.9937 | |
| MOQ e (μg·L−1 or μg·kg−1) | 0.05 | 0.05 | 0.05 | 0.05 | 0.05 | 0.05 | |
| 5-Hydroxy IMI | Linear range (μg·L−1) | 2.0–100.0 | 2.0–100.0 | 2.0–100.0 | 2.0–100.0 | 2.0–100.0 | 2.0–100.0 |
| Regression equation d | y = 0.119x + 0.396 | y = 0.137x − 0.275 | y = 0.116x + 0.703 | y = 0.096x + 0.439 | y = 0.082x + 0.123 | y = 0.090x − 0.096 | |
| Correlation coefficient (R2) | 0.9977 | 0.9916 | 0.9901 | 0.9947 | 0.9996 | 0.9914 | |
| MOQ e (μg·L−1 or μg·kg−1) | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | |
| olefin IMI | Linear range (μg·L−1) | 5.0–200.0 | 5.0–200.0 | 5.0–200.0 | 5.0–200.0 | 5.0–200.0 | 5.0–200.0 |
| Regression equation d | y = 0.272x − 0.259 | y = 0.150x − 0.043 | y = 0.056x − 0.305 | y = 0.400x − 2.774 | y = 0.147x + 0.610 | y = 0.076x + 0.090 | |
| Correlation coefficient (R2) | 0.9946 | 0.9948 | 0.9973 | 0.9942 | 0.9982 | 0.9965 | |
| MOQ e (μg·L−1 or μg·kg−1) | 2.0 | 2.0 | 2.0 | 2.0 | 2.0 | 2.0 | |
| IMI urea | Linear range (μg·L−1) | 0.2–10.0 | 0.2–10.0 | 0.2–10.0 | 0.2–10.0 | 0.2–10.0 | 0.2–10.0 |
| Regression equation d | y = 1.593x + 0.058 | y = 2.058x + 0.194 | y = 1.567x + 0.922 | y = 0.723x + 4.432 | y = 1.570x + 0.956 | y = 0.754x + 0.948 | |
| Correlation coefficient (R2) | 0.9979 | 0.9951 | 0.9920 | 0.9984 | 0.9968 | 0.9927 | |
| MOQ e (μg·L−1 or μg·kg−1) | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | |
| 6-CNA † | Linear range (μg·L−1) | 2.0–100.0 | 2.0–100.0 | 2.0–100.0 | 2.0–100.0 | 2.0–100.0 | 2.0–100.0 |
| Regression equation d | y = 0.225x + 1.364 | y = 0.528x + 0.576 | y = 0.571x + 0.143 | y = 0.236x + 1.269 | y = 0.417x + 2.456 | y = 0.542x + 0.954 | |
| Correlation coefficient (R2) | 0.9934 | 0.9947 | 0.9963 | 0.9981 | 0.9976 | 0.9935 | |
| MOQ e (μg·L−1 or μg·kg−1) | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 |
| Compound | Spike level (μg kg−1) | Plasma | Cephalothorax | Hepatopancrea | Gill | Intestine | Muscle | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Recovery (%) | Intraday RSD | Inter-days RSD | Recovery (%) | Intraday RSD | Inter-days RSD | Recovery (%) | Intraday RSD | Inter-days RSD | Recovery (%) | Intraday RSD | Inter-days RSD | Recovery (%) | Intraday RSD | Inter-days RSD | Recovery (%) | Intraday RSD | Inter-days RSD | ||
| IMI | 0.1 | 89.6 | 7.2 | 5.6 | 96.3 | 6.3 | 6.8 | 102.2 | 9.6 | 6.5 | 91.6 | 9.8 | 6.4 | 100.5 | 8.1 | 10.5 | 106.3 | 4.6 | 8.0 |
| 1 | 97.2 | 9.5 | 6.9 | 87.4 | 5.5 | 7.8 | 97.6 | 7.3 | 5.9 | 86.4 | 7.7 | 7.5 | 96.8 | 5.9 | 8.6 | 110.4 | 8.6 | 7.7 | |
| 5 | 102.3 | 4.3 | 8.2 | 106.7 | 9.8 | 9.4 | 98.3 | 5.2 | 8.1 | 88.9 | 6.1 | 8.6 | 94.1 | 4.2 | 9.4 | 112.7 | 9.1 | 6.4 | |
| 5-Hydroxy IMI | 2.0 | 89.6 | 6.0 | 5.4 | 107.6 | 8.1 | 10.4 | 89.6 | 6.8 | 6.3 | 97.6 | 5.0 | 9.1 | 103.8 | 6.8 | 7.9 | 94.6 | 7.6 | 5.2 |
| 20 | 85.1 | 8.5 | 6.3 | 99.8 | 9.5 | 12.3 | 83.4 | 6.1 | 8.4 | 96.3 | 9.1 | 11.0 | 85.6 | 9.9 | 5.7 | 84.1 | 5.4 | 9.3 | |
| 100 | 97.7 | 9.7 | 8.0 | 110.7 | 11.5 | 8.4 | 91.2 | 7.8 | 9.5 | 102.6 | 6.5 | 7.3 | 107.6 | 10.5 | 6.1 | 86.9 | 6.9 | 4.6 | |
| olefin IMI | 5.0 | 88.7 | 10.6 | 4.6 | 97.2 | 7.6 | 7.6 | 85.2 | 9.5 | 7.3 | 110.1 | 4.6 | 6.9 | 96.8 | 8.6 | 8.9 | 88.7 | 7.6 | 8.2 |
| 50.0 | 90.3 | 5.8 | 7.6 | 88.7 | 6.8 | 9.4 | 86.8 | 6.4 | 6.9 | 95.4 | 8.6 | 8..8 | 94.6 | 7.4 | 9.9 | 94.2 | 9.3 | 9.4 | |
| 200.0 | 100.5 | 6.4 | 6.8 | 95.4 | 8.3 | 8.3 | 91.6 | 3.7 | 9.5 | 86.3 | 9.2 | 9.1 | 82.6 | 9.9 | 6.8 | 90.5 | 8.2 | 7.6 | |
| IMI urea | 0.2 | 98.6 | 7.2 | 7.7 | 86.7 | 9.4 | 9.4 | 86.7 | 7.9 | 10.9 | 91.5 | 7.7 | 10.6 | 86.4 | 5.8 | 12.5 | 101.4 | 8.6 | 9.5 |
| 2.0 | 91.8 | 9.1 | 8.0 | 89.8 | 6.6 | 6.3 | 81.1 | 5.1 | 12.6 | 86.8 | 8.5 | 8.3 | 89.1 | 6.8 | 7.8 | 90.6 | 7.2 | 4.3 | |
| 10.0 | 81.5 | 8.8 | 9.6 | 92.5 | 11.8 | 8.5 | 80.6 | 5.2 | 9.4 | 97.1 | 4.9 | 9.1 | 102.2 | 7.1 | 9.3 | 80.4 | 6.8 | 9.8 | |
| 6-CNA † | 2.0 | 87.1 | 6.9 | 5.1 | 103.3 | 10.6 | 10.5 | 97.6 | 9.8 | 8.2 | 95.6 | 8.2 | 7.6 | 87.6 | 8.6 | 8.6 | 99.6 | 9.4 | 8.6 |
| 20.0 | 80.9 | 8.3 | 8.5 | 88.6 | 9.1 | 11.9 | 99.7 | 8.8 | 7.3 | 85.6 | 5.8 | 12.1 | 91.7 | 9.8 | 10.0 | 89.4 | 12.6 | 5.5 | |
| 100.0 | 89.7 | 11.2 | 5.8 | 97.4 | 8.3 | 10.6 | 105.9 | 7.3 | 10.6 | 97.7 | 11.9 | 5.5 | 90.5 | 12.2 | 9.3 | 100.4 | 11.3 | 6.7 | |
| Analyte | Ionization Mode | Precursor Ion (m/z) | Product Ion (m/z) | Collision Energy (eV) |
|---|---|---|---|---|
| Imidacloprid | positive | 256.0 | 208.8/174.9 | 16/19 |
| Imidacloprid-D4 | positive | 260 | 179/213 | 19/19 |
| 5-Hydroxy Imidacloprid | positive | 272.0 | 225/226.1 | 14/9 |
| Olefin imidacloprid | negative | 251.9 | 204.9/81.1 | 14/10 |
| Imidacloprid urea | positive | 212.0 | 126/128 | 24/18 |
| 6-Chloronicotinic acid | negative | 156.1 | 112/35.1 | 13/26 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Q.; Ai, X.; Dong, J.; Liu, Y.; Zhou, S.; Yang, Y.; Xu, N. A QuEChERS-HPLC-MS/MS Method with Matrix Matching Calibration Strategy for Determination of Imidacloprid and Its Metabolites in Procambarus clarkii (Crayfish) Tissues. Molecules 2021, 26, 274. https://doi.org/10.3390/molecules26020274
Yang Q, Ai X, Dong J, Liu Y, Zhou S, Yang Y, Xu N. A QuEChERS-HPLC-MS/MS Method with Matrix Matching Calibration Strategy for Determination of Imidacloprid and Its Metabolites in Procambarus clarkii (Crayfish) Tissues. Molecules. 2021; 26(2):274. https://doi.org/10.3390/molecules26020274
Chicago/Turabian StyleYang, Qiuhong, Xiaohui Ai, Jing Dong, Yongtao Liu, Shun Zhou, Yibin Yang, and Ning Xu. 2021. "A QuEChERS-HPLC-MS/MS Method with Matrix Matching Calibration Strategy for Determination of Imidacloprid and Its Metabolites in Procambarus clarkii (Crayfish) Tissues" Molecules 26, no. 2: 274. https://doi.org/10.3390/molecules26020274